
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Insights into metal–ligand hydrogen transfer: a square-planar ruthenate complex supported by a tetradentate amino–amido-diolefin ligand†
Demyan E.
Prokopchuk
*ab,
Alan J.
Lough
a,
Rafael E.
Rodriguez-Lugo‡
b,
Robert H.
Morris
*a and
Hansjörg
Grützmacher
*b
aDepartment of Chemistry, University of Toronto, 80 Saint George St., Toronto, Ontario M5S3H6, Canada. E-mail: rmorris@chem.utoronto.ca
bDepartment of Chemistry and Applied Biosciences, ETH Zürich, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch
First published on 1st April 2016
Abstract
A four-coordinate, sixteen-electron Ru(0) complex containing the tetradentate diamino-diolefin ligand (±)-trans-N,N-bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,2-diaminocyclohexane (trop2dach) has been synthesised. Deprotonation of one amino N–H functional group generates an unprecedented four-coordinate ruthenate species which has been characterised in solution and in the solid state. The newly formed ruthenate complex undergoes intramolecular metal–ligand N–H addition/elimination in solution to generate a transient diamido ruthenium hydride species, as supported by NMR spectroscopy and density functional theory.
In organometallic chemistry, the η2-5H-dibenzo[a,d]-cycloheptenyl (η2-trop) moiety serves as a two-electron π-donor that can be functionalised with nitrogen or phosphorus atoms bound to the sp3 carbon within the seven-membered ring.1,2 In the presence of strong base, amine functionalised trop ligands have been shown to stabilise rhodium,3–8 iridium,9–11 and ruthenium12–14 compounds, much owing to the robust π-accepting nature of the olefin ligands (Scheme 1). In addition, the η2-olefin CH nuclei are sensitive to changes in their electronic environment, making them useful probes into the overall electronic structure.
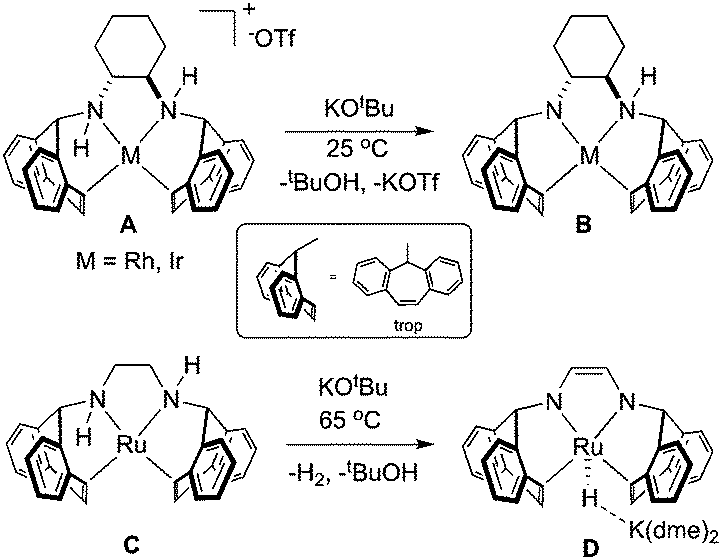 | ||
| Scheme 1 Reactivity of rhodium and ruthenium amino-olefin ligands with one equivalent strong base. dme = 1,2-dimethoxyethane. | ||
Complexes containing the trop2dach (dach = (R,R)-1,2-diaminocyclohexane) and trop2dae (dae = 1,2-diaminoethane) ligands have been used as catalysts for “green” chemical transformations (Scheme 1).15 The iridium complex B, generated by deprotonating A with strong base, is proposed as an intermediate in the iridium–nitrogen-radical catalyzed oxidation of alcohols, mimicking radical-based metalloenzyme catalysts.11 Recently, the anionic ruthenium hydride complex D was found to be an active catalyst for the formation of CO2/H2 from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 methanol:water mixtures, with complexes C and D being proposed as key intermediates within the catalytic cycle.12 In the presence of base, it is proposed that the ligand backbone of zero-valent Ru complex C becomes dehydrogenated and this metal–ligand reactivity/cooperativity plays a key role in the catalytic methanol dehydrogenation process. In related cases, hydrogenation or dehydrogenation of the PNNP ligand backbone of catalysts can lead to catalyst activation,16–18 deactivation,19–21 or side reaction22 while dehydrogenation of a tridentate PNP ligand allows for the reversible storage/release of two equivalents of hydrogen and change of the spin state.23–26
:1 methanol:water mixtures, with complexes C and D being proposed as key intermediates within the catalytic cycle.12 In the presence of base, it is proposed that the ligand backbone of zero-valent Ru complex C becomes dehydrogenated and this metal–ligand reactivity/cooperativity plays a key role in the catalytic methanol dehydrogenation process. In related cases, hydrogenation or dehydrogenation of the PNNP ligand backbone of catalysts can lead to catalyst activation,16–18 deactivation,19–21 or side reaction22 while dehydrogenation of a tridentate PNP ligand allows for the reversible storage/release of two equivalents of hydrogen and change of the spin state.23–26
Four coordinate, sixteen-electron Ru(0) complexes such as C remain quite rare and related compounds contain bulky and basic phosphines paired with carbonyl or nitrosyl ligands.27–33 In our efforts to expand the coordination chemistry and reactivity of ruthenium amino-olefin complexes, we report the synthesis and characterization of tetradentate amino-olefin Ru(0) and amido-olefin ruthenate complexes (Scheme 2). Our experimental observations and calculations support the ease with which hydrogen can migrate between metal and ligand, which might be operative in the mechanism of methanol dehydrogenation using complexes C and/or D.
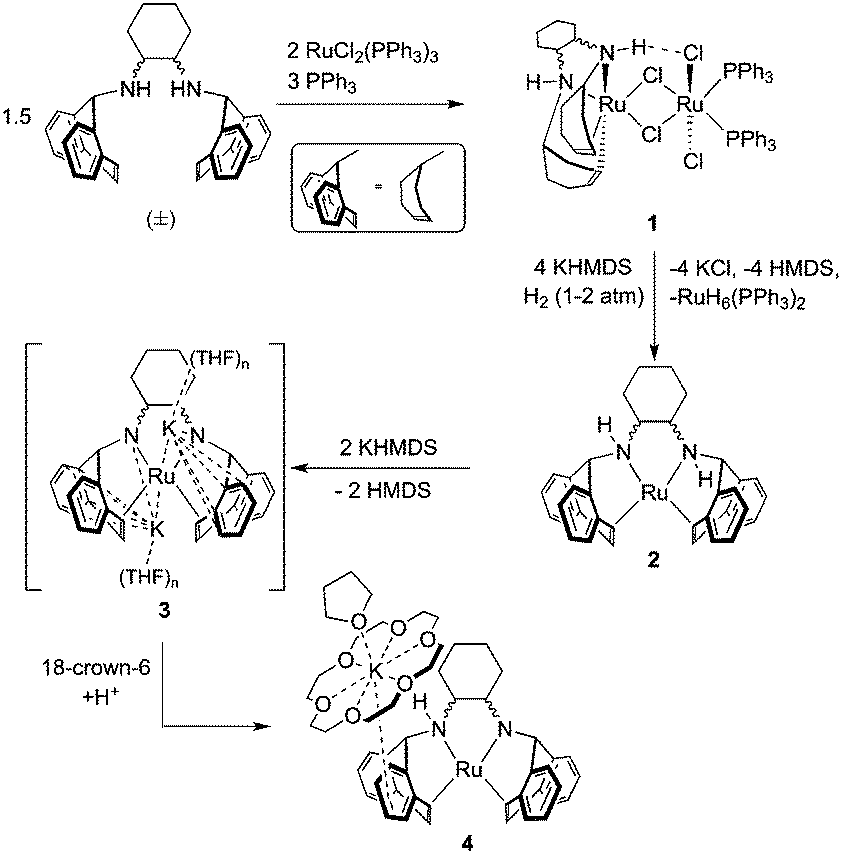 | ||
| Scheme 2 Synthesis of compounds 1–4. | ||
Upon reaction of two equivalents of RuCl2(PPh3)3 with an excess of (±)-trans-N,N-bis(5H-dibenzo[a,d]cyclohepten-5-yl)-1,2-diaminocyclohexane ((±)trop2dach)34 and PPh3, the chloro-bridged bimetallic species 1 precipitates from solution as an analytically pure powder which is insoluble in common organic solvents (Scheme 2). In lieu of solution-state analysis, 1 was characterised by solid-state CP/MAS NMR and single crystal X-ray diffraction (Scheme 1 and Fig. S1, ESI†). The solid-state 13C{1H} NMR spectrum of 1 reveals three groups of signals consistent with a coordinated trop2dach ligand while the 31P NMR contains a broad resonance centred at 53.8 ppm, also consistent with coordinated PPh3 (see the ESI†).
When dimer 1 is heated in the presence of four equivalents of the strong base potassium hexamethyldisilazide (KHMDS) under a hydrogen atmosphere, the pale pink suspension turns deep orange. Analysis of the crude products by 1H and 31P NMR indicates the presence of two compounds, one whose hydride chemical shift matches the known polyhydride species RuH6(PPh3)235–37 and the other being the zero-valent Ru complex Ru(trop2dach) (2, Scheme 1). These products form by a series of steps involving, in no proven order, the precipitation of potassium chloride, the heterolytic splitting of dihydrogen at ruthenium, formation of ruthenium hydrides and hexamethyldisilazane, and the cleavage of the dimer to give the two monometallic products.
Complex 2 was characterised by NMR spectroscopy and single crystal X-ray diffraction (Fig. 1, left). The complex resides in a distorted square planar geometry with one molecule of THF hydrogen bonded to an amine proton (O1⋯H1 2.100 Å). The Ru-centroid bond distances (Ru1–ct1 = 1.980(4), Ru1–ct2 = 1.973(5) Å) are significantly shorter than in 1 and the metrical parameters are similar to those of complex C.12
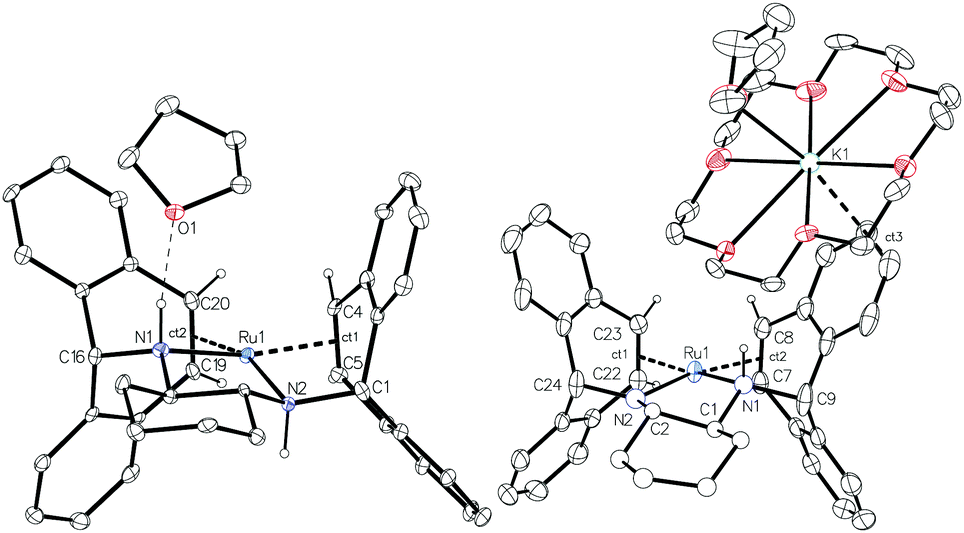 | ||
| Fig. 1 Molecular structures of 2 (left) and 4 (right) with 30% probability ellipsoids. Only a single enantiomer is shown and most hydrogen atoms have been omitted for clarity. Distances (Å) and angles (°) 2: Ru1–N1 2.131(4), Ru1–N2 2.129(3), Ru1–ct1 1.980(4), Ru1–ct2 1.973(5), C4–C5 1.423(7), C19–C20 1.435(7), O1⋯H1 2.100; N1–Ru1–N2 82.0(2), ct1–Ru1–ct2 98.30(2), N1–Ru1–ct2 92.18(2), N2–Ru1–ct1 92.18(2), N1–Ru1–ct1 163.51(2), N2–Ru1–ct2 162.37(2). 4: Ru1–N1 2.091(5), Ru1–N2 1.995(4), Ru1–ct1 1.992(4), Ru1–ct2 1.997(3), K1⋯ct3 3.221(5), C7–C8 1.445(5), C22–C23 1.435(5); N1–Ru1–N2 80.2(2), ct1–Ru1–ct2 98.73(2), N1–Ru1–ct2 90.90(2), N2–Ru1–ct1 90.98(2), N1–Ru1–ct1 167.65(2), N2–Ru1–ct2 169.07(2). | ||
In the presence of KHMDS and heat, complex 2 does not behave in a manner similar to C, which undergoes dehydrogenation at the ethylene bridge between the nitrogen atoms.12 With two or more equivalents strong base, we observe that the backbone of 2 remains intact, generating a symmetrical species containing CHdach NMR resonances at 2.09 ppm (1H) and 74.14 ppm (13C) (see ESI†). The same product is observed using the strong base KOtBu, however reactions were cleaner when using KHMDS. We propose that the solution state structure is the doubly deprotonated [K]2[Ru(trop2dach-2H)]·(THF)n (3), an intimate ion-paired structure that might prevent backbone dehydrogenation. Diamido-diolefin rhodium38 and nickel39 complexes containing similar potassium–nitrogen interactions have been structurally characterised while potassium–nitrogen ion pairing has also been observed in other metal–amido type systems.40–42 A square planar, dianionic Ru(0) complex [Li(THF)3]2[Ru(P2)2] with 2,2′-biphosphinine (P2) ligands was reported where both lithium cations sit directly on top of ruthenium.43
Crystallization of solutions containing 3 in the presence of 18-crown-6 (18c6) consistently resulted in reprotonation of one amido nitrogen atom, generating large brown-black crystals of the four-coordinate amidoruthenate complex [K(18c6·THF)][Ru(trop2dach-H)]·THF (4, Fig. 1). The source of proton could be the hexamethyldisilazane that is generated in the preparation of 3, however its origin is still unclear. The molecular structure shows a shortening of the Ru–Namido bond by about 0.1 Å (1.995(4) Å) with respect to Ru–Namino (2.091(5) Å), with the Namido atom approaching a planar coordination geometry (Σ° = 354.8°). The complex [Ru(NO)(PtBu2CH2SiMe2NSiMe2CH2PtBu2)] has a Ru–Namido distance of 2.070(2) Å,44 significantly longer than Ru1–N2 1.995(4) in 4. The K(18c6) cation is capped with one THF molecule and weakly bound to one of the benzo groups on the inside of the trop ligand (K1⋯ct3 = 3.221(5) Å).
DFT-calculated (M06/TZVP/TZVPFit)45 molecular orbital (MO) and Atomic Polar Tensor (APT) charge analysis of 2 and 4 are presented in Fig. 2. In line with the Dewar–Chatt–Duncanson model for metal–olefin binding,46 the degenerate MOs at −0.20 eV for complex 2 predominantly consist of stabilizing interactions between the dxz/dyz and π* atomic orbitals (AOs) of the η2-trop olefin ligands. The next two MOs at −0.18 and −0.16 eV consist of nonbonding electrons of dxy and dz2 parentage. Evaluation of the APT charges on the ruthenium, nitrogen, and η2-trop carbon atoms of 2 reveals that the N atoms bear the most negative atomic charges (−0.68 and −0.65 ESU). The calculated and experimental metrical parameters for 2 and 4 are in good agreement (see ESI†).
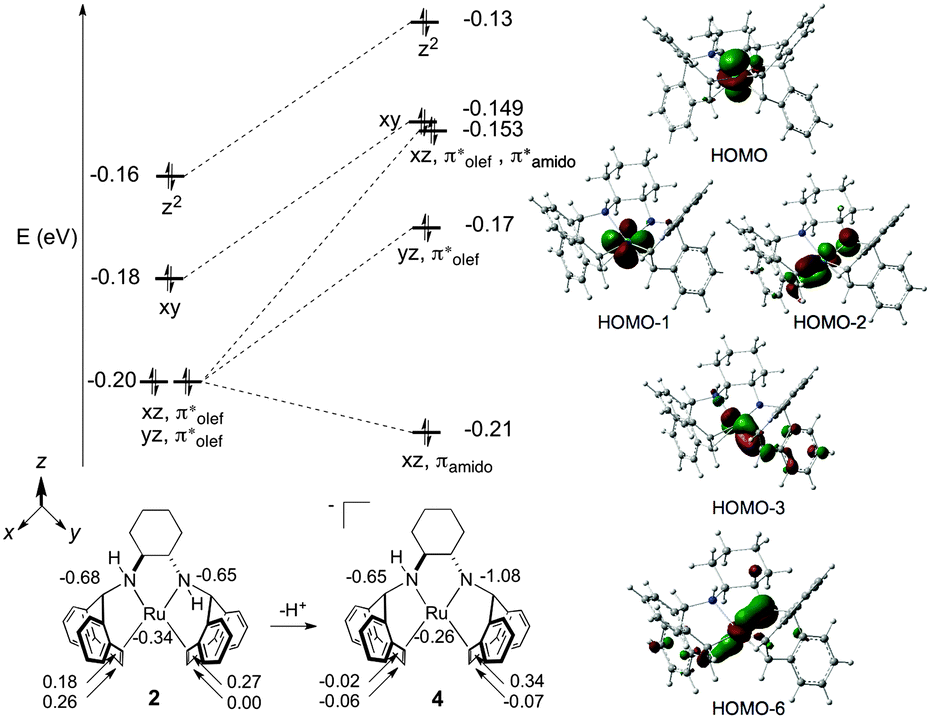 | ||
| Fig. 2 Top left: Partial orbital energy diagram on going from 2 to 4 with relative orbital energies given in eV. Right: Kohn–Sham orbitals of 4 (isovalue = 0.06). Bottom left: Selected APT charges of 2 and 4 given in ESU. | ||
Amino–amido complex 4 has two occupied MOs, HOMO−2 and HOMO−6, that participate in a three-centre-four-electron Namido–Ru–olefin “push–pull” π–π* interaction (Fig. 2, right).38,47,48 The electron pair in HOMO−6 is stabilised by an Namido bonding interaction with the dxz AO, and HOMO−2 consists of a backbonding interaction between the dxz and η2-trop antibonding orbitals with a strong Namido–dxz antibonding contribution. The APT charges show that there is significant charge localization on Namido (−1.08 ESU), which is accompanied by an increase in negative charge at the η2-trop carbon atoms trans to the Namido moiety (−0.02 and −0.06, respectively). Note that there is little change in the atomic charge at ruthenium on going from 2 to 4, indicating that the ligand framework absorbs the additional charge upon deprotonation.49
Complex 4 is highly sensitive to solvent conditions – it decomposes in pure DMSO and is insoluble in ethereal and hydrocarbon solvents. However, the addition of DMSO-d6 to crystals of 4 immersed in THF-d8 resulted in its dissolution and allowed for its complete 1D and 2D NMR characterization. The 1H NMR chemical shifts of the η2-trop CHolef protons in 4 appear as four doublets ranging from 2.6 to 0.9 ppm, far upfield with respect to the free trop2dach ligand (δ = 6.8, 6.6 ppm)34 and Rh/Ir complexes A and B (see Table S1, ESI† for comparison). The N–H proton resonance, which would be expected between 4 and 6 ppm,11,38 could not be located anywhere between +14 and −30 ppm. Furthermore, 1H–1H NOESY NMR experiments reveal strong and well-resolved EXSY cross-peaks, indicating dynamic chemical exchange in solution (see ESI†). In addition to aromatic region exchange cross-peaks, there are an additional eight pairs of exchange cross-peaks in total, arising from the sixteen protons that belong to the cyclohexyl ring, trop-CHbenzyl positions, and η2-trop olefinic protons. Further studies of 4 by variable temperature NMR were hindered by its high reactivity and the lack of a suitable solvent. However, an estimate of the rate constant based on a simple two-site chemical exchange process50,51 is 2.7 ± 0.4 s−1, with ΔG‡ = 16.9 ± 0.1 kcal mol−1 (see the ESI†).
As determined by DFT calculations, we propose that complex 4 is a chemically non-innocent52 species undergoing dynamic N–H addition/elimination, generating a transient diamido ruthenium hydride intermediate (Scheme 3). A cyclic three-membered transition state has been located (ΔG‡(TS4,5) = 22.6 kcal mol−1), generating the distorted square pyramidal complex 5 which can be formally viewed as a Ru(II) hydrido–diamido complex. Similar to 4, APT analysis of 5 shows that the charge at ruthenium remains unperturbed while the amido nitrogens absorb the negative charge, further indicating the “spectator-like” nature of the metal centre. Although the calculated free energy is substantially higher than the estimated free energy from experiment, all other calculated transition state structures were even higher in energy (Scheme S1, ESI†). Inclusion of one molecule of THF or DMSO increases the ground state energies and transition state barriers for insertion/elimination, suggesting that solvent plays no significant role within the inner coordination sphere. The TS barrier on going from 5 to the tautomer of 4, formed by a hydrogen shift to the other nitrogen, is higher in free energy than TS4,5 by 5.5 kcal mol−1. However a fluxional rearrangement of 5 that brings the hydride to the opposite side of the square plane, followed by an H shift to the other nitrogen via the symmetry-related TS4,5 would explain the features of the EXSY spectrum. Two isomers resulting from β-hydride elimination were also ruled out due to their high ground state free energies. Furthermore, recent calculations show that a similar three-membered insertion transition state TS4,5 is a proposed key step during methanol dehydrogenation using catalyst C (Scheme 1).53
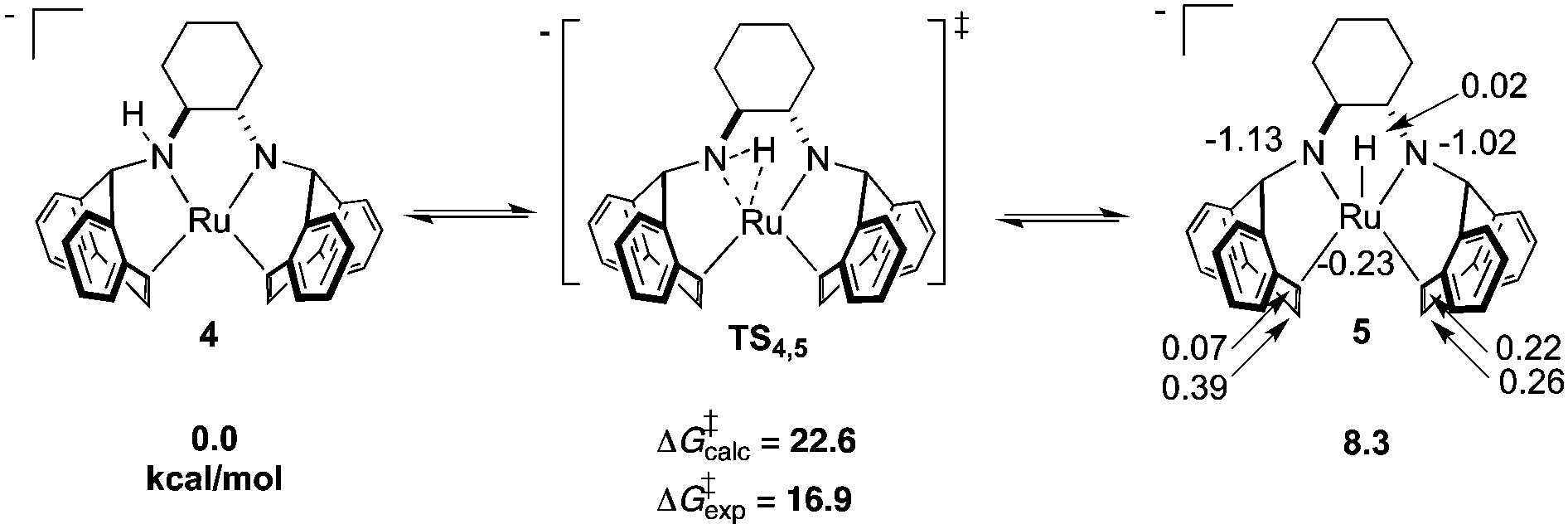 | ||
| Scheme 3 DFT calculated free energies (relative to 4) for proposed N–H addition/elimination reactions in solution. Energies are presented in kcal mol−1 and APT charges in ESU are also shown for 5. | ||
In summary, we have presented a low-coordinate Ru(0) complex based on a diamino-diolefin ligand with a functionalised diaminocyclohexyl backbone. In stark contrast to the trop2dae Ru(0) complex C, reactions with base only deprotonate at the amine positions, allowing for the isolation and structural characterization of an unprecedented four-coordinate ruthenate complex. Such a species is reminiscent of its valence isoelectronic rhodium6 and iridium11 analogues, but NMR and DFT experiments indicate that the N–H moiety is non-innocent in solution. The observed solution state behaviour of 4 provides some evidence into how C might participate in transferring hydrogen between the metal and ligand during catalytic methanol dehydrogenation. Future efforts will be directed towards synthesizing other functionalised trop2(diamine) derivatives and investigating these compounds for the catalytic dehydrogenation of alcohols.
D. E. P. thanks the NSERC Michael Smith Foreign Study Supplement (MSFSS) for funding and H. G. for hosting. R. H. M. thanks NSERC for a Discovery Grant. The authors wish to acknowledge the Canadian Foundation for Innovation, project number 19119, and the Ontario Research Fund for funding of the Centre for Spectroscopic Investigation of Complex Organic Molecules and Polymers. D. E. P. also thanks Dr Darcy Burns and Sergiy Nokhrin for assistance with NMR experiments, Molly Sung for sample preparation/analysis, and Dr Monica Trincado for useful discussions.
Notes and references
- J. Harmer, G. Frison, M. Rudolph, H. Schönberg, S. Deblon, P. Maire, S. Boulmaaz, F. Breher, C. Bohler, H. Ruegger, A. Schweiger and H. Grützmacher, Perspectives in Organometallic Chemistry, The Royal Society of Chemistry, 2003, pp. 222 Search PubMed.
- C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482 CrossRef CAS PubMed.
- H. Schönberg, S. Boulmaâz, M. Wörle, L. Liesum, A. Schweiger and H. Grützmacher, Angew. Chem., Int. Ed., 1998, 37, 1423 CrossRef.
- T. Buttner, F. Breher and H. Grutzmacher, Chem. Commun., 2004, 2820 RSC.
- P. Maire, T. Büttner, F. Breher, P. Le Floch and H. Grützmacher, Angew. Chem., Int. Ed., 2005, 44, 6318 CrossRef CAS PubMed.
- P. Maire, M. Königsmann, A. Sreekanth, J. Harmer, A. Schweiger and H. Grützmacher, J. Am. Chem. Soc., 2006, 128, 6578 CrossRef CAS PubMed.
- G. Mora, S. van Zutphen, C. Thoumazet, X. F. Le Goff, L. Ricard, H. Grutzmacher and P. Le Floch, Organometallics, 2006, 25, 5528 CrossRef CAS.
- T. Zweifel, J.-V. Naubron and H. Grützmacher, Angew. Chem., Int. Ed., 2009, 48, 559 CrossRef CAS PubMed.
- P. Maire, S. Deblon, F. Breher, J. Geier, C. Böhler, H. Rüegger, H. Schönberg and H. Grützmacher, Chem. – Eur. J., 2004, 10, 4198 CrossRef CAS PubMed.
- E. Piras, F. Läng, H. Rüegger, D. Stein, M. Wörle and H. Grützmacher, Chem. – Eur. J., 2006, 12, 5849 CrossRef CAS PubMed.
- M. Königsmann, N. Donati, D. Stein, H. Schönberg, J. Harmer, A. Sreekanth and H. Grützmacher, Angew. Chem., Int. Ed., 2007, 46, 3567 CrossRef PubMed.
- R. E. Rodríguez-Lugo, M. Trincado, M. Vogt, F. Tewes, G. Santiso-Quinones and H. Grützmacher, Nat. Chem., 2013, 5, 342 CrossRef PubMed.
- G. Santiso-Quinones, R. Rodriguez-Lugo, V. Sacchetti and H. Grutzmacher, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1421 CAS.
- G. Santiso-Quinones and R. E. Rodriguez-Lugo, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 859 CAS.
- M. Trincado, D. Banerjee and H. Grutzmacher, Energy Environ. Sci., 2014, 7, 2464 CAS.
- A. A. Mikhailine, M. I. Maishan, A. J. Lough and R. H. Morris, J. Am. Chem. Soc., 2012, 134, 12266 CrossRef CAS PubMed.
- W. Zuo, A. J. Lough, Y. F. Li and R. H. Morris, Science, 2013, 342, 1080 CrossRef CAS PubMed.
- R. H. Morris, Acc. Chem. Res., 2015, 48, 1494 CrossRef CAS PubMed.
- F. N. Haque, A. J. Lough and R. H. Morris, Inorg. Chim. Acta, 2008, 361, 3149 CrossRef CAS.
- P. O. Lagaditis, P. E. Sues, A. J. Lough and R. H. Morris, Dalton Trans., 2015, 44, 12119 RSC.
- W. Zuo, D. E. Prokopchuk, A. J. Lough and R. H. Morris, ACS Catal., 2016, 6, 301 CrossRef CAS.
- R. Langer, I. Fuchs, M. Vogt, E. Balaraman, Y. Diskin-Posner, L. J. W. Shimon, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2013, 19, 3407 CrossRef CAS PubMed.
- S. Schneider, J. Meiners and B. Askevold, Eur. J. Inorg. Chem., 2012, 412 CrossRef CAS.
- B. Askevold, H. W. Roesky and S. Schneider, ChemCatChem, 2012, 4, 307 CrossRef CAS.
- M. Kinauer, M. G. Scheibel, J. Abbenseth, F. W. Heinemann, P. Stollberg, C. Wurtele and S. Schneider, Dalton Trans., 2014, 43, 4506 RSC.
- B. Askevold, M. M. Khusniyarov, W. Kroener, K. Gieb, P. Müller, E. Herdtweck, F. W. Heinemann, M. Diefenbach, M. C. Holthausen, V. Vieru, L. F. Chibotaru and S. Schneider, Chem. – Eur. J., 2015, 21, 579 CrossRef CAS PubMed.
- M. A. A. F. D. C. T. Carrondo, B. N. Chaudret, D. J. Cole-Hamilton, A. C. Skapski and G. Wilkinson, J. Chem. Soc., Chem. Commun., 1978, 463 RSC.
- M. Ogasawara, S. A. Macgregor, W. E. Streib, K. Folting, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1995, 117, 8869 CrossRef CAS.
- R. Flügel, B. Windmüller, O. Gevert and H. Werner, Chem. Ber., 1996, 129, 1007 CrossRef.
- M. Ogasawara, S. A. Macgregor, W. E. Streib, K. Folting, O. Eisenstein and K. G. Caulton, J. Am. Chem. Soc., 1996, 118, 10189 CrossRef CAS.
- D. Huang, W. E. Streib, E. Odile and K. G. Caulton, Organometallics, 2000, 19, 1967 CrossRef CAS.
- T. Gottschalk-Gaudig, J. C. Huffman, H. Gérard, O. Eisenstein and K. G. Caulton, Inorg. Chem., 2000, 39, 3957 CrossRef CAS PubMed.
- H. Salem, L. J. W. Shimon, Y. Diskin-Posner, G. Leitus, Y. Ben-David and D. Milstein, Organometallics, 2009, 28, 4791 CrossRef CAS.
- P. Maire, F. Breher, H. Schönberg and H. Grützmacher, Organometallics, 2005, 24, 3207 CrossRef CAS.
- B. Chaudret, J. Devillers and R. Poilblanc, J. Chem. Soc., Chem. Commun., 1983, 641 RSC.
- B. Chaudret, J. Devillers and R. Poilblanc, Organometallics, 1985, 4, 1727 CrossRef CAS.
- K. Abdur-Rashid, D. G. Gusev, A. J. Lough and R. H. Morris, Organometallics, 2000, 19, 1652 CrossRef CAS.
- P. Maire, F. Breher and H. Grützmacher, Angew. Chem., Int. Ed., 2005, 44, 6325 CrossRef CAS PubMed.
- M. Vogt, PhD Dissertation, ETH Zürich, Nr. 19295, 2010.
- D. Song and R. H. Morris, Organometallics, 2004, 23, 4406 CrossRef CAS.
- P. Cui, T. P. Spaniol and J. Okuda, Organometallics, 2013, 32, 1176 CrossRef CAS.
- J. M. John, S. Takebayashi, N. Dabral, M. Miskolzie and S. H. Bergens, J. Am. Chem. Soc., 2013, 135, 8578 CrossRef CAS PubMed.
- P. Rosa, N. Mézailles, L. Ricard, F. Mathey, P. Le Floch and Y. Jean, Angew. Chem., Int. Ed., 2001, 40, 1251 CrossRef CAS.
- A. Walstrom, M. Pink, H. Fan, J. Tomaszewski and K. G. Caulton, Inorg. Chem., 2007, 46, 7704 CrossRef CAS PubMed.
- For additional information on the DFT methods used, see the ESI†.
- D. M. P. Mingos, J. Organomet. Chem., 2001, 635, 1 CrossRef CAS.
- D. Conner, K. N. Jayaprakash, T. B. Gunnoe and P. D. Boyle, Inorg. Chem., 2002, 41, 3042 CrossRef CAS PubMed.
- B. Askevold, A. Friedrich, M. R. Buchner, B. Lewall, A. C. Filippou, E. Herdtweck and S. Schneider, J. Organomet. Chem., 2013, 744, 35 CrossRef CAS.
- Cyclic voltammetry of 2 and 4 revealed multiple weak and irreversible redox events.
- C. L. Perrin and T. J. Dwyer, Chem. Rev., 1990, 90, 935 CrossRef CAS.
- J. Jeener, B. H. Meier, P. Bachmann and R. R. Ernst, J. Chem. Phys., 1979, 71, 4546 CrossRef CAS.
- L. A. Berben, B. de Bruin and A. F. Heyduk, Chem. Commun., 2015, 51, 1553 RSC.
- H. Li and M. B. Hall, J. Am. Chem. Soc., 2015, 137, 12330 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, DFT calculations. CCDC 1444981–1444983. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6cc00041j |
| ‡ Current address: Laboratorio de Química Bioinorgánica, Centro de Química, Instituto Venezolano de Investigaciones Científicas (IVIC), Caracas 1020-A, Venezuela. E-mail: E-mail: rrodriguez@ivic.gob.ve |
| This journal is © The Royal Society of Chemistry 2016 |