
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical fragment arrays for rapid druggability assessment†
J.
Aretz
ab,
Y.
Kondoh
cd,
K.
Honda
cd,
U. R.
Anumala
e,
M.
Nazaré
e,
N.
Watanabe
cdf,
H.
Osada
cdf and
C.
Rademacher
*ab
aDepartment of Biomolecular Systems, Max Planck Institute of Colloids and Interfaces, Potsdam, Germany. E-mail: Christoph.Rademacher@mpikg.mpg.de
bDepartment of Biology, Chemistry, and Pharmacy, Freie Universität Berlin, Berlin, Germany
cAntibiotics Laboratory, RIKEN, Wako, Japan
dChemical Biology Research Group, RIKEN Center for Sustainable Resource Science, Wako, Japan
eLeibniz Institut für Molekulare Pharmakologie (FMP), Berlin, Germany
fBioprobe Research Group, RIKEN-Max Planck Joint Research Center for Systems Chemical Biology, Japan
First published on 10th February 2016
Abstract
Incorporation of early druggability assessment in the drug discovery process provides a means to prioritize target proteins for high-throughput screening. We present chemical fragment arrays as a method that is capable of determining the druggability of a given target with low protein and compound consumption, enabling rapid decision making during early phases of drug discovery.
Failures during early phases of the drug discovery pipeline are major drivers of the costs.1 To minimize the risk before a drug discovery campaign is initiated, target proteins are evaluated thoroughly. This evaluation involves detailed insight into the molecular mechanisms of the target, its selective tissue expression, phenotypic data, and its ability to modulate a disease.2 Another important aspect is the potential of the targeted protein to harbour drug-like molecules that modulate its biological function. The presence of suitable binding sites for small molecule drugs is called druggability. Estimates consider about 10% of human genes encoding for druggable targets with only half of these being disease relevant proteins.3 Therefore, many drug discovery projects start with a druggability assessment to provide early predictions of success.4
Several approaches to calculate the druggability of a target protein have been developed. Among those, computational methods are very popular. Here, structural information of the binding pocket is used to calculate a druggability score.5 Accordingly, these methods rely on the availability of structural information. Moreover the majority of in silico druggability prediction programs do not account for conformational changes, flexible binding sites or allosteric pockets.6
Early experimental approaches to assess target druggability utilized protein cocrystallization with organic solvents. These solvents explore hydrophobic pockets, which are often targeted by drug-like molecules.7 This approach was later replaced by screening the fragments of drug-like molecules ranging between 150 and 250 Da in size.8 These low molecular weight compounds have low molecular complexity and increased likelihood for binding to druggable targets.8 During experimental druggability assessment, a high number of false negatives are considered to be the worst case because valuable targets are rejected.4 Therefore, due to its low false positive rate and high sensitivity to detect small-molecule binding, fragment-based NMR screening has emerged as the experimental method of choice for druggability assessment.4,9
However, NMR approaches have several limitations such as low sensitivity and consequently high protein consumption, high costs of instrumentation and the partial necessity for stable isotope labelling. Thus, a method for druggability assessment with low protein consumption and fast turnover evaluating binding to a large panel of fragments is highly desirable. Here, we present chemical fragment arrays to assess protein druggability. Fragments were immobilized on glass slides and probed with fluorescently labelled protein. We used this platform to analyse the prerequisites for the detection of fragment binding with respect to their affinity and molecular weight (MW). Next, hit rates of a diverse fragment library tested against five target proteins were compared using a sensitive, state-of-the-art NMR screening method.10
As a first step, we used a well-defined set of fragments to evaluate the printing and screening on microarrays. For this purpose, we immobilized ligands for two C-type lectin receptors, which are current targets investigated in our laboratory (Aretz et al., unpublished data): murine Langerin and human dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN). Overall, 13 natural ligands and 69 fragments previously identified by saturation transfer difference (STD) NMR and characterized by surface plasmon resonance (SPR) (Aretz et al., unpublished data) were immobilized on a photoaffinity-linker-coated (PALC) glass slide.11 For Langerin and DC-SIGN, ten and twelve fragments were included in the analysis, respectively, which did not show interaction in an SPR assay. For immobilization on the PALC glass slide, ligands were dissolved in DMSO and printed in duplicate of quadruplicate of concentration values of 10 mM, 5 mM and 2.5 mM. Two different linker chemistries consisting of a flexible PEG linker and a proline linker that were designed by inserting a rigid proline helix into the root of the PEG linker were used for immobilization (Fig. 1A).12
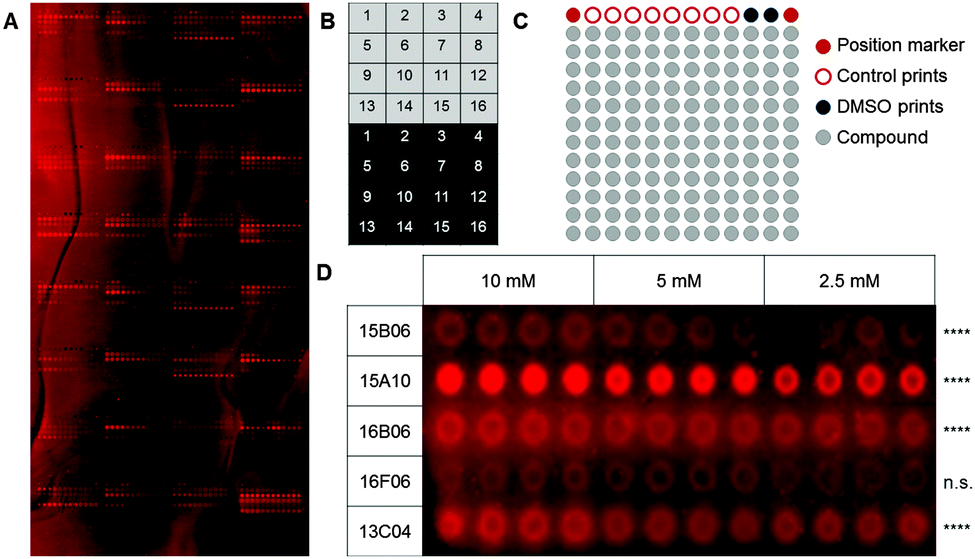 | ||
| Fig. 1 Fragments on PALC glass slides used to probe murine Langerin binding. (A) 82 characterized ligands for murine Langerin and human DC-SIGN were printed on a photoaffinity-linker-coated glass slide. This array was used to optimize binding and washing conditions for fragment arrays using Chromeo642-labelled Langerin. (B) One array consists of different blocks that are printed as copies on the upper (light grey) and lower half (dark grey) of the glass slide. (C) Every block consists of 156 spots including 12 control spots in the upper row and 144 spots to print compounds (grey). (D) Compounds that showed a significantly enhanced signal compared to the DMSO controls after incubation of the array with a fluorescently labelled protein sample were considered as hits. An expansion of five results for Langerin binding to five fragments immobilized at varying concentrations using a proline linker is shown (Dunnett's test, **** = p < 0.001, n.s. = p > 0.05). | ||
As fragments have rather low binding affinities for their targets, we optimized the binding, incubation and washing conditions (for details see the ESI†). This optimization aimed to detect as many hits as possible among the known binding fragments. A hit was defined as a compound that showed significantly enhanced signals compared to the DMSO control prints (p < 0.05, Dunnett's test). Ultimately, the glass slides were blocked with 1% BSA and incubated with the 0.2 μM Chromeo642-labelled protein overnight at room temperature under constant agitation followed by three short washing steps using cold buffer. Interestingly, the rigid proline linker outperformed the flexible PEG linker with respect to the background signal as well as the signal to noise level. Moreover, fragments were immobilized more efficiently at 10 mM (Fig. 1D). In summary, the immobilization, incubation and washing conditions were optimized to detect fragments with millimolar affinities for their targets employing a chemical fragment array.
Following the optimization of the experimental conditions, the performance of the array was evaluated. In an ideal screening assay, signal intensity correlates with binding affinity, thus enabling the rank-ordering of hits. Ideally, screening results between two orthogonal assays do not differ. To test whether screening results from our chemical fragment array can be rank-ordered according to signal intensity, we evaluated a potential correlation with affinities measured by SPR (Fig. 2). While this correlation was not significant, the molecular weight of the immobilized fragments correlated significantly with signal intensity observed during array screening (Fig. 2). Many immobilized fragments show similar signal intensities for the two lectins, as they share high specificity and structural overlap.13 Moreover, higher molecular weight fragments experienced higher recovery rates, which were defined as percentage of compounds that were already detected by SPR and that also hit on the chemical array (Fig. 2). On average, these recovery rates for Langerin and DC-SIGN were 69% and 55%, respectively (Fig. 2). Moreover, from the fragments that did not bind in the SPR assay, 20% and 55% bound to Langerin and DC-SIGN on the array, respectively. These numbers are not unusual when comparing SPR results with other biophysical screening techniques in particular14 and for a comparison between different screening techniques in general.15,16
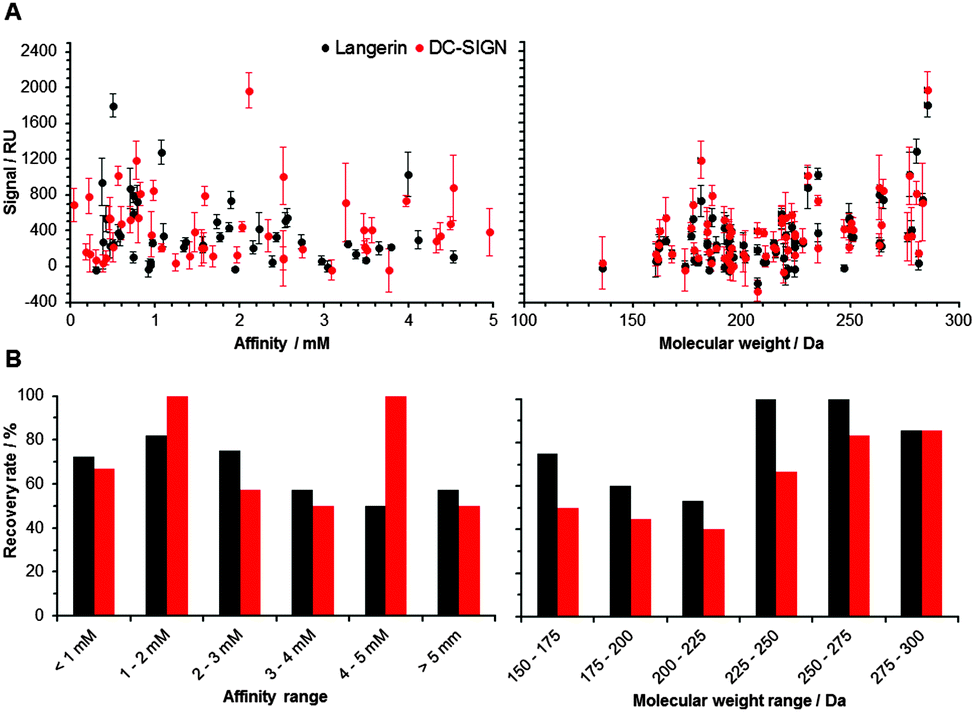 | ||
| Fig. 2 Fragment binding correlates with molecular weight (right), not with affinity (left). (A) Data for murine Langerin (black) and DC-SIGN (red) show a significant correlation between the signal observed from array screening and the molecular weight of the corresponding fragment (Spearman's correlation coefficient, p < 0.05). Fragments were immobilized at 10 mM concentration. (B) Recovery rates for murine Langerin (black) and DC-SIGN (red) were slightly higher for compounds with higher affinity and higher molecular weight. | ||
Finally, we investigated the performance and robustness of our assay. We examined if the detection levels are affected by the natural multimerization of C-type lectin receptors. Thus far, data were obtained by using the DC-SIGN tetramer (166 kD). Applying the directly labelled DC-SIGN monomer (20 kD), we observed a significant linear correlation (Pearson, p < 0.0001) with the results for the multimeric protein, suggesting that our method is not limited to oligomeric receptors (Fig. S3, ESI†). Overall, these results indicate that fragments can be printed and screened on chemical arrays against monomeric and oligomeric protein targets.
Encouraged by our results, we explored the potential use of our fragment array for druggability assessment of target proteins. We expanded our analysis to 281 fluorinated fragments, which previously were subjected to a 19F NMR-based druggability screening.10 These fragments were printed at 10 mM concentrations and to increase the target scope we included three other proteins in our analysis: human Langerin, human N-acetylmannosamine kinase (MNK) and bovine carbonic anhydrase II (CA2). These proteins cover a range of hit rates from 3 to 19% as previously determined10,20 (Aretz et al., unpublished data) and hence resemble a representative set of druggabilities. Hits were identified using DMSO spots as reference (Dunnett's test, p < 0.001, data not shown). Two major findings arose from this screening. Firstly, hit rates between 13 and 34% for our five target proteins were observed. These rates are elevated compared to results from NMR screening. We attribute this to the NMR method being able to identify non-specific binding more thoroughly, an effect reported previously.16 Similar to alternative screening techniques such as thermal shift assays, microscale thermophoresis or SPR, missing binding site information of the chemical fragment array method renders appropriate follow-up experiments a requirement to ensure modulation of the biological activity. The other major finding was that, compared to the 19F NMR screening results, the array screening identified between 25% and 50% of the known hits with an average recovery rate of 37% (Fig. 3A). Similar to the array screening using previously identified hits from SPR (Fig. 2), these recovery rates are in the expected range of fragment hit rates from orthogonal assays.15,16
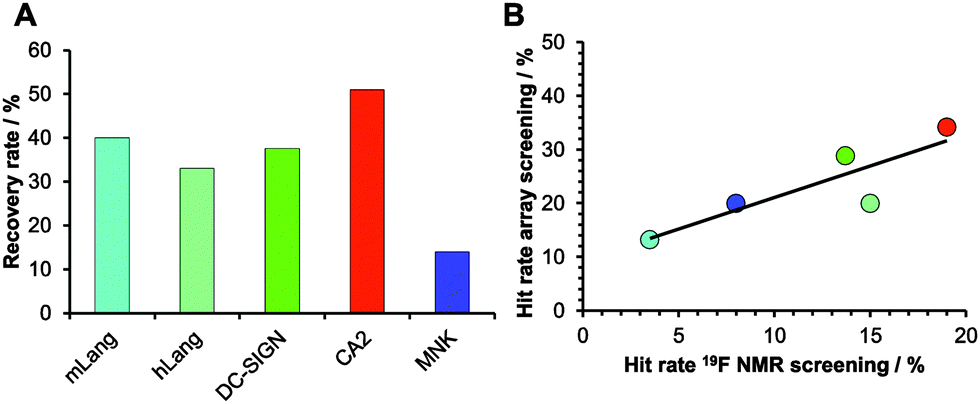 | ||
| Fig. 3 Comparison of NMR and chemical fragment array screening for hit identification. 281 fragments were immobilized on glass slides using a proline photoaffinity linker. (A) Recovery rates of hits identified from array screening compared to 19F NMR screenings. (B) Linear correlation of hit rates from fragment screening using chemical arrays and 19F NMR. | ||
With these data in hand, we were then able to ask whether hit rates from 19F NMR and array screening correlate. NMR fragment-screening is particularly well suited for this comparison, as it can offer binding site information and thus is one of the prime methods for druggability assessment.4,9,17 We found that hit-rates of both methods correlated well (R2 = 0.75, Fig. 3B). Additional evidence for this correlation came from experiments using a PEG linker for fragment immobilization in which we found a similar trend (Fig. S4 and S5, ESI†). Taken together, fragment array screening was sensitive enough to detect hits for challenging proteins with low 19F NMR hit rates (murine Langerin) as well as in prioritizing CA2 as the most druggable protein for further development in a hypothetical drug discovery campaign.
Next, we were concerned whether the immobilization of the fragments on the array would have rendered certain chemotypes prone for binding, while other recognition motifs would have been impaired. Twelve fragments gave rise to signals during screening of almost every array (‘frequent hitters’) and other 108 compounds were never identified using the chemical array (‘non-hitters’). We conducted a chemoinformatic analysis using molecular descriptors and MACCS fingerprints to identify chemotypes and features significantly altered in their likelihood to be identified by our chemical fragment array (Fig. S6, discussed in detail in the ESI†). Non-hitters had significantly fewer hydroxyl groups compared to the other compounds in the library (t-test, equal variances not assumed, p < 0.001), which is in line with a previous study regarding preferred reactivity of photo-cross-linkers.19 Conversely, fragments with less reactive groups may have a lower immobilization efficiency. In addition, non-hitters were significantly smaller than regular hitters (21 Da and 2 HA, t-test, equal variances not assumed, p < 0.001), a trend that was already observed in the initial tests (Fig. 2). We hypothesize that a higher molecular weight decreases the likelihood of the photochemical immobilization to unfavorably affect the essential binding epitopes of the fragment recognition. Importantly, the chemical diversity of fragment hits identified by arrays did not differ from the complete fragment library (Fig. S7, ESI†). In sum, while we observed a low recovery rate of previously identified hits, which may be caused by a bias towards certain chemotypes as a result of the photo-crosslinking, the overall diversity of hits was not altered. This is an important prerequisite for the applicability of the fragment array for druggability screening.
Finally, we investigated whether hits from the chemical fragment array would serve as valuable starting points for fragment evolution. For CA2, many nanomolar inhibitors carry a benzenesulfonamide scaffold.18 Nine fragments on the array shared this privileged substructure and four were identified as hits during our screening. The close structural analogy to already developed nanomolar inhibitors of CA2 exemplifies the potential of our array screening (Fig. S8, ESI†). Furthermore, for MNK we recently identified picolinic acid scaffolds as starting points for inhibitor design.20 One out of four inhibitors was detected during the array screening (Fig. S9, ESI†). While both examples clearly show the value of initial hit identification from chemical fragment array screening, some actives were not identified during the screening. We suspect that either the immobilization was unsuccessful or the photo-chemical conjugation altered the recognition epitopes of the fragments. Conversely, most active or potentially active compounds were identified from the regular or frequent hitter groups. On the one hand, this stresses the need to develop suitable fragment libraries for immobilization. On the other hand, it demonstrates the ability of the fragment array to identify enzyme inhibitors. To expand the applicability of the chemical fragment array even further we explored its performance in the presence of cell lysates, since a druggability assessment of non-purified proteins directly from cell lysates significantly increases the scope and throughput of the method. To test our hypothesis, directly labeled proteins were tested in the presence of lysates from the human cell line HEK-293T, a commonly used cell line for protein production. Notably, four out of the five proteins tested remained active (Fig. S10, ESI†). Overall, these data suggest that fragment arrays are suitable for target druggability evaluation. With more than a 1000-fold reduced protein consumption compared to an NMR assay it is a rapid and attractive alternative. Additionally, small molecule arrays outperform NMR as a screening tool also with respect to throughput: up to 864 compounds can be printed on one array in duplicate of quadruplicate and a single person can screen and analyze at least ten arrays in two days. With an appropriate device, one person can prepare 200 glass slides in 27 h.
Taken together, these preliminary results indicate that fragment arrays can be introduced into drug discovery at a very early point to estimate druggability and prioritize targets before protein expression is optimized or structural data are available.
To iteratively improve chemical fragment arrays for screening and druggability assessment, the number of fragments should be increased to provide more insight into the reactivity of the fragments during photo-chemical immobilization and at the same time to remove false-negatives. For the latter, the scope of the target protein families should also be expanded. Optimizing chemical fragment arrays for the application of whole cells or lysates provides an opportunity to screen fluorescently tagged intracellular or transmembrane proteins in their native environment. In the future, chemical fragment arrays could enable large scale experimental druggability analyses of human proteins.
Fragments printed on photoactivated glass slides are a fast and inexpensive method for screening and experimentally determining the druggability of a target protein at an early stage of the drug discovery process compared to other commonly used techniques. Only a few micrograms of labelled protein are sufficient to enable druggability assessment of potential drug targets even before expression is optimized or its protein structure is solved.
The authors thank the Max Planck Society, the RIKEN-Max Planck Joint Center for Systems Chemical Biology, the Berlin Institute of Health, (BIH) and the German Research Foundation (DFG, Emmy Noether program RA1944/2-1) for financial support. We thank Paul Robin Wratil and Prof. Dr Werner Reutter for kindly providing MNK, Jonas Hanske for providing human Langerin and Prof. Dr Peter H. Seeberger for support and helpful discussions.
Notes and references
- S. Laufer, U. Holzgrabe and D. Steinhilber, Angew. Chem., Int. Ed., 2013, 52, 4072–4076 CrossRef CAS PubMed.
- I. Gashaw, P. Ellinghaus, A. Sommer and K. Asadullah, Drug Discovery Today, 2012, 17, S24–S30 CrossRef CAS PubMed.
- A. L. Hopkins and C. R. Groom, Nat. Rev. Drug Discovery, 2002, 1, 727–730 CrossRef CAS PubMed.
- F. N. Edfeldt, R. H. Folmer and A. L. Breeze, Drug Discovery Today, 2011, 16, 284–287 CrossRef CAS PubMed.
- A. Volkamer and M. Rarey, Future Med. Chem., 2014, 6, 319–331 CrossRef CAS PubMed.
- A. C. Cheng, R. G. Coleman, K. T. Smyth, Q. Cao, P. Soulard, D. R. Caffrey, A. C. Salzberg and E. S. Huang, Nat. Biotechnol., 2007, 25, 71–75 CrossRef CAS PubMed.
- D. Ringe, Curr. Opin. Struct. Biol., 1995, 5, 825–829 CrossRef CAS PubMed.
- S. Barelier and I. Krimm, Curr. Opin. Chem. Biol., 2011, 15, 469–474 CrossRef CAS PubMed.
- P. J. Hajduk, J. R. Huth and S. W. Fesik, J. Med. Chem., 2005, 48, 2518–2525 CrossRef CAS PubMed.
- J. Aretz, E. C. Wamhoff, J. Hanske, D. Heymann and C. Rademacher, Front. Immunol., 2014, 5, 323 Search PubMed.
- N. Kanoh, S. Kumashiro, S. Simizu, Y. Kondoh, S. Hatakeyama, H. Tashiro and H. Osada, Angew. Chem., Int. Ed., 2003, 42, 5584–5587 CrossRef CAS PubMed.
- Y. Kondoh, K. Honda and H. Osada, Methods Mol. Biol., 2015, 1263, 29–41 Search PubMed.
- L. Chatwell, A. Holla, B. B. Kaufer and A. Skerra, Mol. Immunol., 2008, 45, 1981–1994 CrossRef CAS PubMed.
- J. Wielens, S. J. Headey, D. I. Rhodes, R. J. Mulder, O. Dolezal, J. J. Deadman, J. Newman, D. K. Chalmers, M. W. Parker, T. S. Peat and M. J. Scanlon, J. Biomol. Screening, 2013, 18, 147–159 CrossRef PubMed.
- P. S. Kutchukian, A. M. Wassermann, M. K. Lindvall, S. K. Wright, J. Ottl, J. Jacob, C. Scheufler, A. Marzinzik, N. Brooijmans and M. Glick, J. Biomol. Screening, 2015, 20, 588–596 CrossRef CAS PubMed.
- J. Schiebel, N. Radeva, H. Koster, A. Metz, T. Krotzky, M. Kuhnert, W. E. Diederich, A. Heine, L. Neumann, C. Atmanene, D. Roecklin, V. Vivat-Hannah, J. P. Renaud, R. Meinecke, N. Schlinck, A. Sitte, F. Popp, M. Zeeb and G. Klebe, ChemMedChem, 2015, 10, 1511–1521 CrossRef CAS PubMed.
- I. J. Chen and R. E. Hubbard, J. Comput. – Aided Mol. Des., 2009, 23, 603–620 CrossRef CAS PubMed.
- V. M. Krishnamurthy, G. K. Kaufman, A. R. Urbach, I. Gitlin, K. L. Gudiksen, D. B. Weibel and G. M. Whitesides, Chem. Rev., 2008, 108, 946–1051 CrossRef CAS PubMed.
- T. Suzuki, T. Okamura, T. Tomohiro, Y. Iwabuchi and N. Kanoh, Bioconjugate Chem., 2015, 26, 389–395 CrossRef CAS PubMed.
- J. Aretz, P. R. Wratil, E. C. Wamhoff, H. G. Nguyen, W. Reutter and C. Rademacher, Can. J. Chem., 2016 DOI:10.1139/cjc-2015-0603.
Footnote |
| † Electronic supplementary information (ESI) available: Material and Methods, quality controls, additional data and discussion. See DOI: 10.1039/c5cc10457b |
| This journal is © The Royal Society of Chemistry 2016 |