
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBrønsted acid-catalysed conjugate addition of photochemically generated α-amino radicals to alkenylpyridines†
Hamish B.
Hepburn
a and
Paolo
Melchiorre
*ab
aICIQ – Institute of Chemical Research of Catalonia, The Barcelona Institute of Science and Technology, Av. Països Catalans 16, 43007 Tarragona, Spain. E-mail: pmelchiorre@iciq.es
bICREA – Catalan Institution for Research and Advanced Studies, Pg. Lluís Companys 23, 08010 Barcelona, Spain
First published on 29th January 2016
Abstract
The conjugate addition of α-amino radicals to alkenylpyridines has been accomplished by the synergistic merger of Brønsted acid and visible light photoredox catalysis. Key to reaction development was the protonation of the alkenylpyridines that transiently generated a highly reactive, electrophilic pseudo-iminium ion intermediate. Initial investigations using chiral phosphoric acids provide clues on the feasibility of an enantioselective catalytic variant.
Nitrogen-containing six-membered heterocycles, such as pyridines, are essential structural motifs found in an array of pharmaceuticals, agrochemicals, and natural products.1 This explains why synthetic methods that facilitate the construction of structurally complex pyridine-containing molecules are highly sought after. Although the direct functionalisation of the heterocyclic ring continues to be the main approach,2 synthetic strategies that feature functionalisation at a remote position have also garnered attention.3 In particular, alkenylpyridines 1 have long been known as good Michael acceptors,4 since the embedded C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N imine functionality can act in analogy to a carbonyl compound, facilitating nucleophilic attack at the β-position. The vast majority of these reactions involve 2-vinylpyridine (R = H in 1, Scheme 1), as the introduction of a β-substituent substantially reduces the reactivity of 1. Recently, transition metal catalysis has been identified as a suitable activation strategy for promoting the conjugate addition of carbon-centred nucleophiles to β-substituted alkenylpyridines.5
N imine functionality can act in analogy to a carbonyl compound, facilitating nucleophilic attack at the β-position. The vast majority of these reactions involve 2-vinylpyridine (R = H in 1, Scheme 1), as the introduction of a β-substituent substantially reduces the reactivity of 1. Recently, transition metal catalysis has been identified as a suitable activation strategy for promoting the conjugate addition of carbon-centred nucleophiles to β-substituted alkenylpyridines.5
 | ||
| Scheme 1 Our proposed Brønsted acid-mediated LUMO-lowering activation strategy. | ||
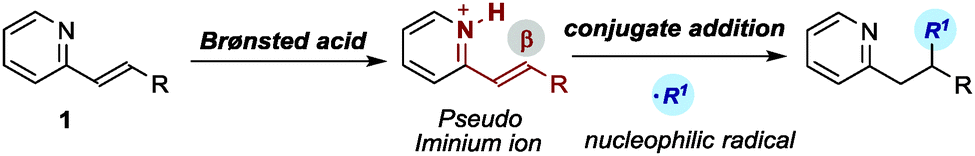
Given our interest in the field of organocatalysis,6 we were aware of the power of iminium ion activation for facilitating conjugate additions.7 We wondered if this activation strategy, which is classically applied to α,β-unsaturated aldehydes and ketones, could be further expanded to include alkenylpyridines 1. We reasoned that the protonation of the basic pyridine nitrogen in 1 with a Brønsted acid would result in the embedded CN imine functioning as a pseudo iminium ion, thus reducing the energy of the LUMO orbital at the β-carbon atom while facilitating a metal-free conjugate addition (Scheme 1). While Brønsted acids have been used to activate pyridines toward the reduction of the aromatic ring,8 to the best of our knowledge their use to activate alkenylpyridines towards nucleophilic addition has not been reported so far.9 Herein, we detail the realisation of this strategy.
The emerging field of light-mediated photoredox catalysis10 had recently provided an effective way of generating radical intermediates from readily available, bench-stable precursors and under mild conditions. Using this photoredox strategy, the addition of nucleophilic open-shell species to simple 2-vinylpyridine have been recently reported.11 However, there have been no reports describing the addition of such photochemically-generated radicals to substituted alkenylpyridines 1. We sought to combine this effective radical generation strategy with Brønsted acid catalysis to develop a radical conjugate addition to alkenylpyridines.
To test the feasibility of our hypothesis, we investigated the ability of a variety of Brønsted acids (10 mol%) to catalyse the light-initiated conjugate addition of dimethylaniline122a to (E)-2-(p-tolylstyryl)pyridine 1a. We used the commercially available photocatalyst Ir[dF(CF3)ppy]2(dtbbpy)PF6 (A) which, upon absorption of visible light, can generate nucleophilic α-amino radicals directly from aniline 2avia single electron transfer (SET) oxidation.12 The experiments were conducted over 14 hours in toluene and under irradiation by a strip of blue light-emitting diodes (blue LEDs strip, λmax = 465 nm).
In the absence of any Brønsted acid, only 9% of the radical conjugate addition product 3a was observed after 14 hours, indicating that the background reaction did not occur readily (entry 1). The use of the weakly acidic pyridinium p-toluenesulfonate (PPTS) provided a negligible improvement of reactivity (entry 2). However, when stronger acids were used, higher yields of 3a were obtained (entries 3–7). Diphenylphosphoric acid proved as the optimal catalyst since it afforded a yield as high as 82% (entry 4). Similar results were observed reducing the acid and the photocatalyst loading to 5 and 1 mol%, respectively (entry 5). Interestingly, when using a very strong acid in the form of CSA the reaction was completely inhibited and a similar yield to the background process was observed (entry 8). Finally, no product formation was detected in the absence of the iridium photocatalyst A, or light, demonstrating that all these components are needed for this catalytic protocol (entry 9).
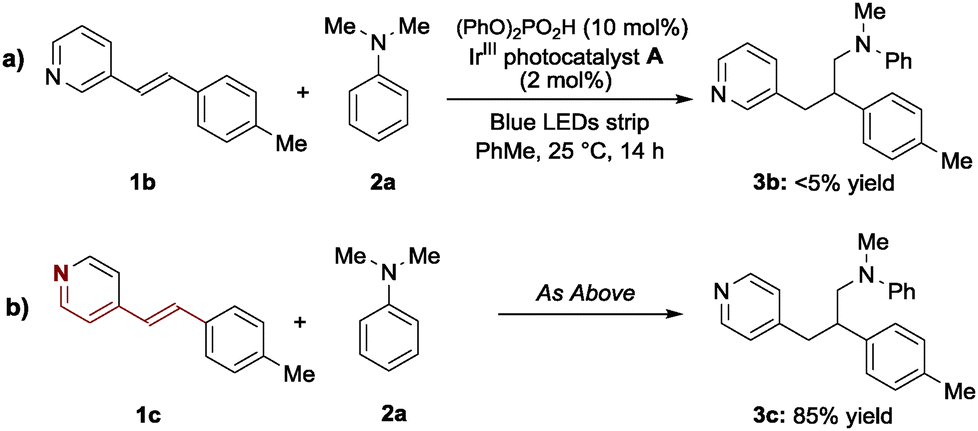
Our activation strategy is based on the formation of a pseudo-iminium ion, which is generated upon protonation of the pyridyl nitrogen within 1 (Scheme 1). This means that the position of the pyridine nitrogen relative to the alkenyl substitution pattern should have a great influence on the reactivity of the conjugate addition. Pleasingly, the corresponding 3-alkenylpyridine 1b, a substrate where the nitrogen is no longer in conjugation with the olefin, resulted in no product formation when subjected to the optimal conditions (Scheme 2a). This is because, upon protonation of 1b by diphenylphosphoric acid, the lack of conjugation hampered the LUMO-lowering activating effect at the β-carbon atom. Conversely, the conjugated 4-alkenylpyridine 1c reacted smoothly to provide the expected product 3c in 85% yield akin to a vinylogous 1,6-iminium ion addition.13 These results are consonant with the protonation of a conjugated nitrogen being essential for facilitating the radical conjugate addition.
 | ||
| Scheme 2 The importance of the pyridine nitrogen atom. | ||
Adopting the optimised conditions described in Table 1, entry 5, we demonstrated the generality of the diphenylphosphoric acid-catalysed radical conjugate addition by evaluating a variety of alkenylpyridines 1 (Table 2).
|
|
||
|---|---|---|
| Entry | Brensted acid | Yieldb (%) |
| a Reaction ran for 14 hours on 0.05 mmol scale of 1, using 1.2 equiv. of amine 2a, 2 mol% photocatalyst A and 10 mol% of Brønsted acid, under irridation by a blue LEDs strip (λmax = 465 nm). b Determined by 1H NMR analysis using 1,3,5-trimethoxybenzene as an internal standard. c Performed using 5 mol% of acid and 1 mol% of A. d Performed in the absence of A or light illumination. PPTS = pyridinium p-toluenesulfonate; (PhO)2PO2H = diphenylphosphoric acid; TFA = trifluoroacetic acid; pTSA = p-toluenesulfonic acid; CSA = camphorsulfonic acid. | ||
| 1 | None | 9 |
| 2 | PPTS | 13 |
| 3 | Benzoic acid | 31 |
| 4 | (PhO)2PO2H | 82 |
| 5 | (PhO)2PO2H | 82c |
| 6 | TFA | 65 |
| 7 | pTSA | 34 |
| 8 | CSA | 9 |
| 9 | (PhO)2PO2H | <5d |

|
|
|||
|---|---|---|---|
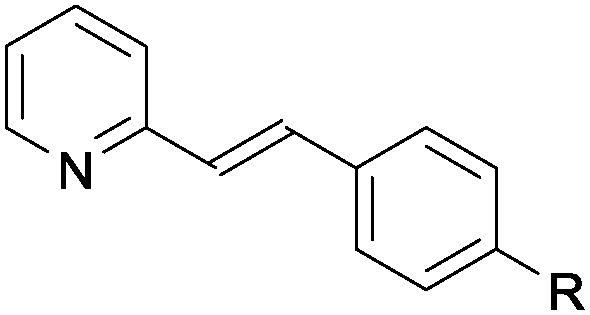
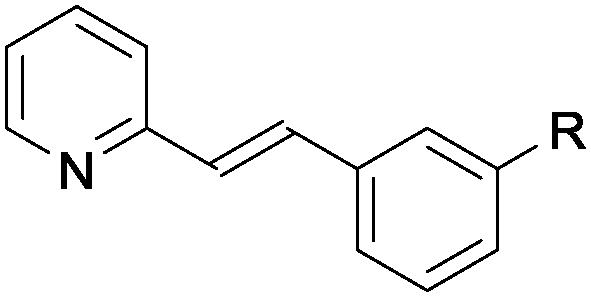
| Entry | Substrate 1 | Product 3 yieldb (%) | |
| a Reaction performed on 0.3 mmol scale, using 2.0 equiv. of 2a, 1 mol% photocatalyst A and 5 mol% of diphenylphosphoric acid, under irridation by a blue LEDs strip (λmax = 465 nm). b Yield of 3 after isolation by chromatography. c Using (Z)-1q rather than (E)-1q. | |||
| 1 |
|
1a R = Me | 87 |
| 2 | 1d R = H | 63 | |
| 3 | 1e R = OMe | 64 | |
| 4 | 1f R = t-Bu | 55 | |
| 5 | 1g R = Br | 84 | |
| 6 | 1h R = CN | 62 | |
| 7 | 1i R = F | 77 | |
| 8 |
|
1j R = Cl | 44 |
| 9 | 1k R = OMe | 89 | |
| 10 |

|
1l R = Me | 52 |
| 11 | 1m R = F | 86 | |
| 12 |
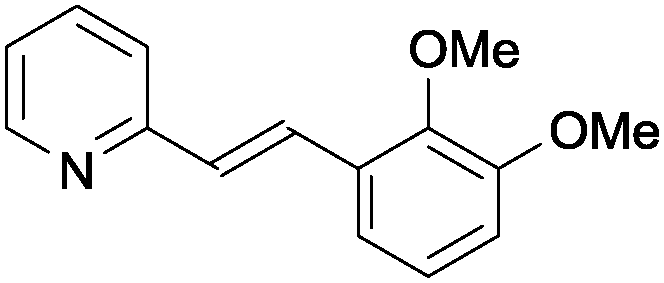
|
1n | 75 |
| 13 |
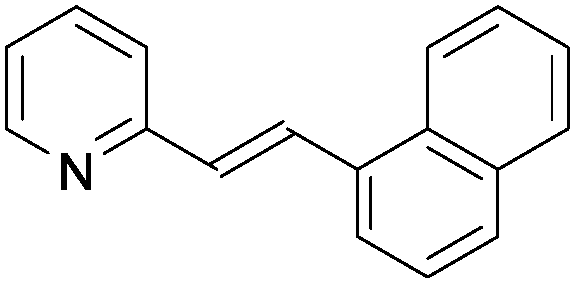
|
1o | 75 |
| 14 |
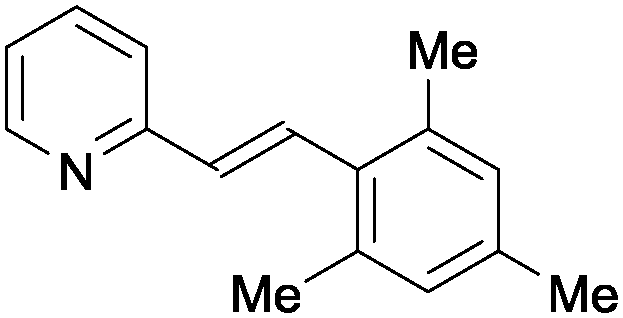
|
1p | 57 |
| 15 |

|
1q R = n-propyl | 71c |
| 16 | 1r R = CH2CH2Ph | 73 | |
| 17 | 1s R = –(CH2)6CH3 | 85 | |
| 18 | 1t R = cyclohexyl | 84 | |
| 19 |

|
1u | 58 |
| 20 |
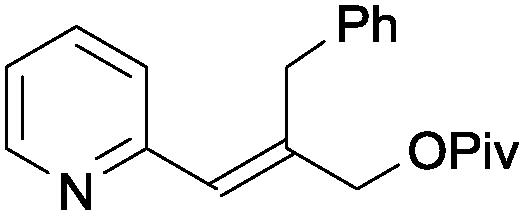
|
1v | <5 |

A wide range of aromatic substituents were tolerated at the β position, providing the corresponding products 3 in good yields. Both electron-donating (entries 1–4) and electron-withdrawing substituents (entries 5–7) at the para position were readily accommodated. A range of meta (entries 8 and 9) and ortho substituents (entries 10 and 11) were also tolerated with similar yields. Aryl groups featuring multiple substitution patterns were also successfully subjected to the reaction conditions (entry 12). The presence of sterically demanding groups, such as naphthyl and mesityl, did not affect the reactivity (entries 13 and 14). Importantly, alkyl substituted alkenylpyridines, including short chain (entry 15), longer chain (entries 16 and 17) and sterically demanding alkyl species (entry 18), also performed well under the optimised conditions. A quinoline substrate was also successfully used, but provided a slightly reduced yield (entry 19). A limitation of the methodology is that disubstituted alkenylpyridines did not react under the optimal conditions (entry 20).
It was also possible to successfully vary the amine component of the reaction (Scheme 3). We found that both mixed N-alkyl-N-aryl (adducts 4a–d) and N,N-diaryl tertiary amines (4e) efficiently participated in the radical conjugate addition. Substituents of different electronic nature were easily accommodated at the aryl para (4a,b) or meta position (4c,d), without affecting the efficiency of the process, while the use of cyclic amines afforded compounds 4f and 4g, bearing two vicinal stereogenic centres.
 | ||
| Scheme 3 Scope of amine. Reaction performed on 0.3 mmol scale of 1a, using 2.0 equiv. of amine 2, 1 mol% of photocatalyst A and 5 mol% of diphenylphosphoric acid, under irridation by a blue LEDs strip (λmax = 465 nm). Yield of 4 after isolation by chromatography. | ||
We found that other electron deficient alkenyl heterocycles 5 readily participated in the radical conjugate addition (Scheme 4). Although a longer irradiation time was required (24 h), the products 6 were achieved in good yields. Interestingly, benzothiazoles were not suitable substrates (6e).
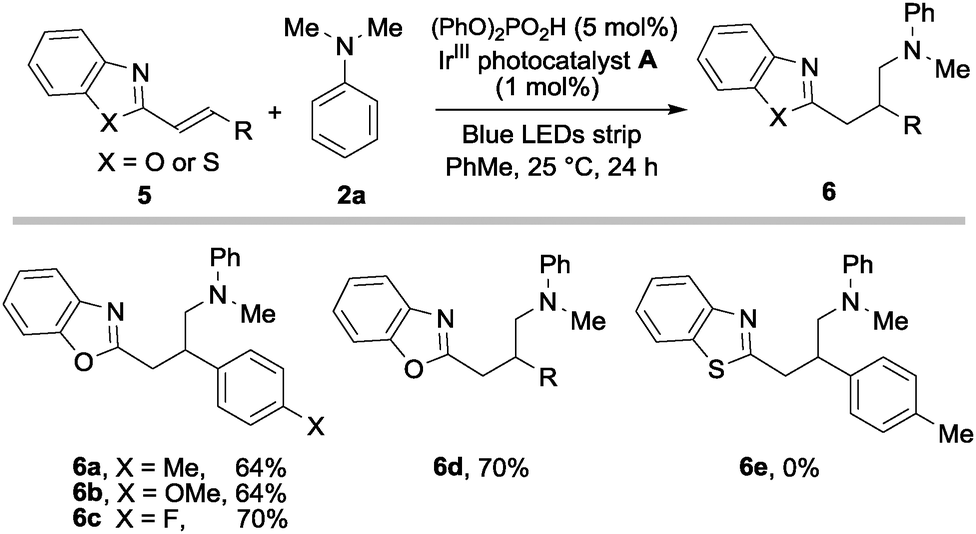 | ||
| Scheme 4 Other alkenylheterocycles that participate in the process. Reaction performed on 0.3 mmol scale of 1a, using 2.0 equiv. of amine 2, 1 mol% of A and 5 mol% of diphenylphosphoric acid. Yield of 6 after isolation by chromatography. | ||
Fig. 1 details our proposed mechanism, which features both an organocatalytic and a photoredox catalytic cycle working in concert to furnish the product 3. Protonation of the pyridine 1 generates the activated pyridinium intermediate 7. Simultaneously, irradiation of the photoredox catalyst leads to the formation of an excited Ir(III) species, which generates a nucleophilic α-amino radical 8 from dimethylaniline by means of single electron transfer (SET) oxidation followed by deprotonation. Upon radical conjugate addition to 7, the resulting radical intermediate 9 participates in a second SET event with the reduced Ir(II) species to regenerate the photocatalyst and, after protonation of 10, liberate the product 3 and the organocatalyst. When performing our studies, we noticed that any unreacted 1a was always recovered as a mixture of the E- and Z-isomers, despite the starting material having exclusively an E geometry. Intrigued by this observation further analysis was undertaken. We found that simple irradiation of E-1a did not lead to isomerisation, which occurred instead upon addition of the iridium photocatalyst A. Stern–Volmer fluorescence quenching experiments, detailed in the ESI,† have demonstrated that the excited state of the photocatalyst A is quenched by both the amine 2a and the pyridine substrate 1a.14 Thus, the excited A participates in two different pathways: a non-productive energy transfer mechanism, which only leads to the isomerisation of the alkenylpyridine, and an electron transfer mechanism with the dimethylaniline 2a, leading to the formation of product 3.
 | ||
| Fig. 1 Proposed mechanism. | ||
Finally, we envisaged that the use of a chiral phosphoric acid could potentially infer enantio-induction through the formation of a chiral ion pair of type 7.15 Pleasingly, after substantial optimization, detailed in the ESI,† we found that the BINOL-based chiral acid 10 catalysed the generation of 3a with 35% ee, indicating the feasibility of a stereocontrolled variant of the radical conjugate addition (Scheme 5).16 Notably, using either E-1a or Z-1a resulted in the formation of the same enantiomer of 3a, which suggests a Curtin–Hammett type kinetic situation, sustained by the energy transfer isomerisation process, where an isomer of the alkenylpyridine reacts faster than the other.
 | ||
| Scheme 5 Enantioselective variant. | ||
In summary, a Brønsted acid-catalysed conjugate addition of photochemically generated α-amino radicals to alkenylpyridines has been accomplished. We also demonstrated the feasibility of an enantioselective version of the chemistry.
This work was supported by the ICIQ Foundation, MINECO (project CTQ2013-45938-P), the AGAUR (2014 SGR 1059), and the European Research Council (ERC 278541 – ORGA-NAUT). H. H. thanks the Marie Curie COFUND action (2015-1-ICIQ-IPMP) and Severo Ochoa Excellence Accreditation 2014–2018, SEV-2013-0319 for a postdoctoral fellowship. Dr Suva Paria is thanked for assistance with the Stern–Volmer studies.
Notes and references
- A. E. Goetz and N. K. Garg, Nat. Chem., 2013, 5, 54–60 CrossRef CAS PubMed.
- J. Llaveria, D. Leonori and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 10958–10961 CrossRef CAS PubMed and references therein.
- D. Best and H. W. Lam, J. Org. Chem., 2014, 79, 831–845 CrossRef CAS PubMed.
- (a) A. Hoffman, M. W. Farlow and R. C. Fuson, J. Am. Chem. Soc., 1933, 55, 2000–2004 CrossRef CAS; (b) J. Michalski and H. Zajac, J. Chem. Soc., 1963, 593–597 RSC; (c) J. H. Nelson, P. N. Howells, G. C. DeLullo and G. L. Landen, J. Org. Chem., 1980, 45, 1246–1249 CrossRef CAS.
- For examples of rhodium-catalysed processes, see: (a) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed; (b) I. D. Roy, A. R. Burns, G. Pattison, B. Michel, A. J. Parker and H. W. Lam, Chem. Commun., 2015, 50, 2865–2868 RSC; (c) A. A. Friedman, J. Panteleev, J. Tsoung, V. Huynh and M. Lautens, Angew. Chem., Int. Ed., 2013, 125, 9937–9940 CrossRef . For examples of nickel-catalysed processes, see: ; (d) I. N. Houpis, J. Lee, I. Dorziotis, A. Molina, B. Reamer, R. P. Volante and P. J. Reider, Tetrahedron, 1998, 54, 1185–1195 CrossRef CAS . For cobalt-catalysed processes, see: ; (e) T. Kobayashi, H. Yorimitsu and K. Oshima, Chem. – Asian J., 2011, 6, 669–673 CrossRef CAS PubMed.
- For our recent contributions, see: (a) G. Bergonzini and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 971–974 CrossRef CAS PubMed; (b) X. Tian, Y. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2012, 51, 6439–6442 CrossRef CAS PubMed; (c) A. Moran, A. Hamilton, C. Bo and P. Melchiorre, J. Am. Chem. Soc., 2013, 135, 9091–9098 CrossRef CAS PubMed; (d) M. Silvi, I. Chatterjee, Y. Liu and P. Melchiorre, Angew. Chem., Int. Ed., 2013, 52, 10780–10783 CrossRef CAS PubMed.
- (a) K. A. Ahrendt, C. J. Borths and D. W. C. MacMillan, J. Am. Chem. Soc., 2000, 122, 4243–4244 CrossRef CAS; (b) G. Lelais and D. W. C. MacMillan, Aldrichimica Acta, 2006, 39, 79–87 CAS.
- (a) M. Rueping, A. P. Antonchick and T. Theissmann, Angew. Chem., Int. Ed., 2006, 45, 3683–3686 CrossRef CAS PubMed; (b) M. Rueping and A. P. Antonchick, Angew. Chem., Int. Ed., 2007, 46, 4562–4565 CrossRef CAS PubMed.
- For a report on the addition of enamines to 4-vinylpyridine by a different Brønsted acid-catalysed strategy, see: (a) S. Wang, X. Li, H. Liu, L. Xu, J. Zhuang, J. Li, H. Li and W. Wang, J. Am. Chem. Soc., 2015, 137, 2303–2310 CrossRef CAS PubMed . During the preparation of this manuscript, a Brønsted acid activation of alkenyl benzimidazoles toward aza-Michael reactions was reported, see: ; (b) Y.-Y. Wang, K. Kanomata, T. Korenaga and M. Terada, Angew. Chem., Int. Ed., 2016, 55, 927–931 CrossRef CAS PubMed.
- For recent reviews, see: (a) J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617–1622 CrossRef CAS PubMed; (b) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed; (c) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 1239176 CrossRef PubMed.
- (a) K. Miyazawa, Y. Yasu, T. Koike and M. Akita, Chem. Commun., 2013, 49, 7249–7251 RSC; (b) K. Miyazawa, T. Koike and M. Akita, Adv. Synth. Catal., 2014, 356, 2749–2755 CrossRef CAS; (c) G. J. Choi and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 9226–9229 CrossRef CAS PubMed . For the 1,2-addition of α-amino radicals to iminium cations of glyoxylic oxime ethers, see: ; (d) Y. Yoshimi, K. Kobayashi, H. Kamakura, K. Nishikawa, Y. Haga, K. Maeda, T. Morita, T. Itou, Y. Okada and M. Hatanaka, Tetrahedron Lett., 2010, 51, 2332–2334 CrossRef CAS.
- Y. Miyake, K. Nakajima and Y. Nishibayashi, J. Am. Chem. Soc., 2012, 134, 3338–3341 CrossRef CAS PubMed.
- I. D. Jurberg, I. Chatterjee, R. Tannert and P. Melchiorre, Chem. Commun., 2013, 49, 4869–4883 RSC.
- K. Singh, S. Staig and J. Weaver, J. Am. Chem. Soc., 2014, 136, 5275–5278 CrossRef CAS PubMed.
- For reviews on chiral phosphoric acid catalysis, see: (a) T. Akiyama, Chem. Rev., 2007, 107, 5744–5758 CrossRef CAS PubMed; (b) M. Terada, Synthesis, 2010, 1929–1982 CrossRef CAS; (c) D. Parmar, E. Sugiono, S. Raja and M. Rueping, Chem. Rev., 2014, 114, 9047–9153 CrossRef CAS PubMed.
- (a) M. P. Sibi, J. Ji, J. H. Wu, S. Gürtler and N. A. Porter, J. Am. Chem. Soc., 1996, 118, 9200–9201 CrossRef CAS; (b) L. Ruiz Espelt, I. S. McPherson, E. M. Wiesnsch and T. P. Yoon, J. Am. Chem. Soc., 2015, 137, 2452–2455 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1450243. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc10401g |
| This journal is © The Royal Society of Chemistry 2016 |