
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Photoswitchable basicity through the use of azoheteroarenes†
Claire E.
Weston
,
Robert D.
Richardson
and
Matthew J.
Fuchter
*
Department of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: m.fuchter@imperial.ac.uk
First published on 26th February 2016
Abstract
Azoheteroarene photoswitches offer functional advantages over their more conventional azobenzene counterparts by virtue of their heteroaromatic ring(s). Here we report that azobis(2-imidazole) functions as a photoswitchable base due to the additional proton stabilisation that is possible in the protonated Z isomer, facilitated by the basic imidazole nitrogens. This thermodynamic difference in stability corresponds to a 1.3 unit difference in pKa values between the E and Z isomers. This pKa difference can be used to reversibly control solution pH.
Using light as a means of modulating a system is extremely attractive, as it is non-invasive and is capable of providing excellent spatial and temporal precision. As such, numerous attempts have been reported that aim to use light to control a huge range of processes, from photopharmacology and optochemical genetics to spin-state switching and optomechanical devices; all mediated by photoresponsive molecules.1–7 One way in which light can be used to perturb a system is to modulate pH. This opportunity can, for example, lead to external control of the reaction rate of pH-dependent chemical/biochemical processes;8–10 however thus far this is limited by the availability of suitable photoresponsive acids or bases. The change in acidity/basicity of a photoactive molecule could be an irreversible process, with examples reported for the release of a ‘caged’ proton upon irradiation with light.10,11 Alternatively (and preferably), the process could be reversible, through use of a suitable photoswitchable acid or base.8,9,12–16
Perhaps unsurprisingly, given their privileged nature in this field (synthetic versatility, high extinction coefficients and quantum yields, high fatigue resistance, etc.), a number of the photoswitchable acids and bases reported are azobenzenes,8,9,14 which can be reversibly switched between the thermally stable E and metastable Z isomers. Previously, differences in ground state acidity/basicity between azobenzene E and Z isomers has been demonstrated for 2-hydroxyazobenzene, where the E acid is stabilised by hydrogen bonding to the azo unit, whereas the Z isomer is not; translating to an increase in acidity of the Z isomer (or conversely an increase in basicity of the deprotonated E isomer).13,14 Similarly, the azonium p-aminoazobenzenes reported by Woolley and co-workers, exhibit a pKa shift of approximately 1.5 units between isomers, due to hydrogen bonding between an ortho oxygen functional group on the azobenzene ring to the protonated azo unit in the E isomer (Fig. 1A).15,17 Hydrogen bonding to the azo unit is not the only plausible strategy for modifying azobenzene acidity/basicity however. Hecht and co-workers, for example, have modulated the basicity of piperidine-containing azobenzenes through steric shielding in one isomer over the other (Fig. 1B).8,18,19
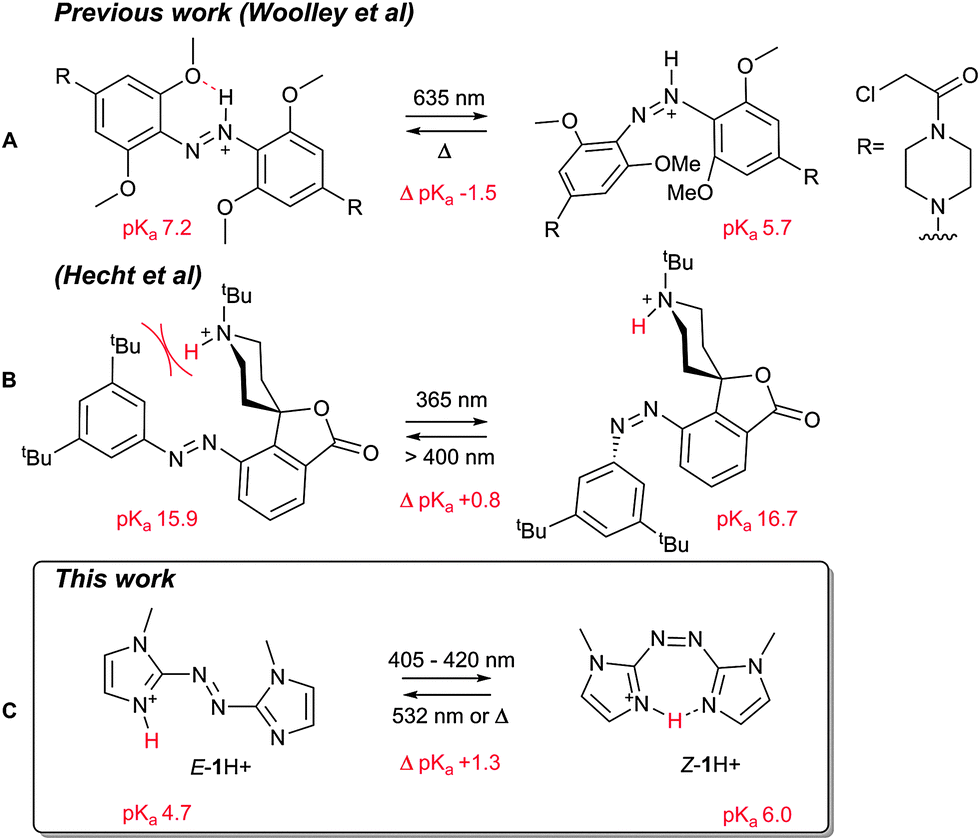 | ||
| Fig. 1 Previous azo photoswitchable bases reported by (A) Woolley, and; (B) Hecht, and; (C) our work. | ||
We have previously reported novel azoheteroaryl photoswitches based on pyrroles and pyrazoles,20 where the type, positioning, and substitution of the heteroaryl ring impacts key switching properties such as addressability, efficiency and thermal stability. However the presence of heteroatoms on the heteroaryl ring system presents other functional opportunities.
For example, Herges and co-workers have used the pyridine nitrogen of azopyridines as a ligating unit to access photodissociable ligands,3,21,22 and have recently demonstrated an improvement in ligand affinity upon moving to arylazoimidazoles.23 Herein we report the first photoswitchable Brønsted base of this class, utilising an azobisimidazole. Importantly with respect to the state of the art (Fig. 1), the protonation event occurs on one of the imidazole rings and is stabilised by the neighbouring ring, rather than through participation of the azo moiety. This results in a compound in which the Z isomer is over an order of magnitude more basic than the E isomer. Furthermore, by avoiding protonation of the azo linkage, the thermal half-life of the protonated Z isomer is increased relative to the neutral form.
Imidazole was chosen as the heteroaromatic ring for this study, due to its high basicity relative to other heteroarenes. Both azobis(2-imidazole) 1 and azobis(4-imidazole) 2 (see Fig. 2) were considered to have the correct connectivity to enable proton stabilisation in the Z isomer. Energy minimised conformations and calculated UV/vis spectra were obtained for both these photoswitches in their neutral states using B3LYP/6-31G(d,p) and CAM-B3LYP/6-311G(2df,2p) respectively (Fig. 2). Azobisimidazole 1, which has the azo moiety attached at the 2-position of both imidazole moieties, was calculated to have a large n–π* absorbance in the Z isomer, and hence good separation of absorbances between the E and Z isomers, which should allow for excellent photoconversion between isomers at different wavelengths (good addressability24). This large n–π* absorbance is due to the twisted conformation adopted by the Z isomer (Fig. 2B), which minimises unfavourable steric interactions between a given N-methyl and the neighbouring azoaryl ring, and unfavourable lone pair interactions between the basic imidazole nitrogens and the azo nitrogens. Conversely, the energy minimised conformation of 2 predicts a planar conformation for the Z isomer and hence a negligible n–π* absorbance is predicted on symmetry grounds (Fig. 2B). These calculations are consistent with our findings on the conformational preferences and spectroscopic properties of the arylazopyrazoles and arylazopyrroles20 – when there is a mirror plane through the molecule, as with the planar E isomers and Z-2, the n–π* transition is symmetry forbidden and no absorbance is predicted (although experimentally a weak absorbance is seen due to vibrational and rotational motion). The calculated spectrum for E-1 also had a significantly red-shifted π–π* absorbance compared to E-2, suggesting that visible light rather than UV light could be used for photoswitching. On the basis of these two features, azobisimidazole 1 was chosen for further study. Energy minimised conformations of protonated 1 confirmed that in the Z isomer, the proton would be stabilised by interacting with both basic nitrogens in the photoswitch (as shown in Fig. 4B) resulting in a planar conformation and, in turn, a decreased n–π* absorbance (Fig. 4A).
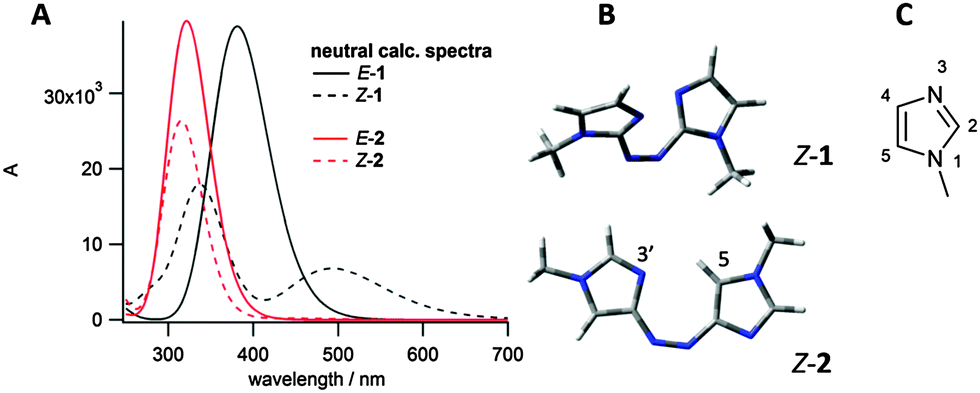 | ||
| Fig. 2 (A) Calculated spectra for neutral 1 and 2 using CAM-B3LYP/6-311G(2df,2p) level of theory; (B) energy minimised conformations of Z-1 and Z-2 using B3LYP/6-31G(d,p); (C) numbering of the imidazole ring. | ||
Azobisimidazole 1 was prepared in two steps from commercially available 2-nitroimidazole 3 (Scheme 1). First, N-methylation was performed using methyl iodide to give compound 4. In addition to impacting the conformation of these systems (vide supra), N-methylation of azole azoheteroaryl photoswitches is required to avoid very fast thermal isomerisation of the Z isomer through tautomerisation.25,26 Reductive coupling of 4 using zinc in aqueous ammonium chloride,27 gave the required photoswitch 1.
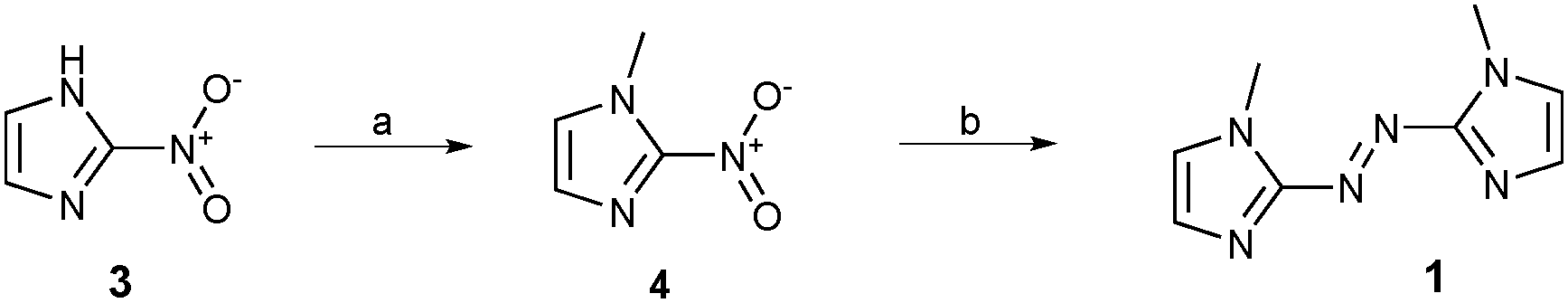 | ||
| Scheme 1 Reagents and conditions: (a) MeI, K2CO3, MeCN, 0–65 °C, 65%; (b) NH4Cl, Zn dust, H2O, 45 °C, 49%. | ||
Irradiation was initially carried out on the neutral compound 1 in aqueous buffer at pH 9 (where 1 was assumed to be not-significantly protonated; this was later shown to be the case, vide infra) using a range of wavelengths (Fig. 3A). The best E–Z photoconversion was achieved using a 408 nm laser diode, with an estimated photostationary state (PSS) of (90 ± 3)% Z (see ESI,† for estimation); however 415 nm irradation (generated by a dye laser), with an PSS of (79 ± 5)% Z, was chosen to carry out further photoswitching, due to the tunability of the power using this light source.‡ 532 nm irradiation of the n–π* absorbance of the Z isomer resulted in quantitative switching of the azobisimidazole back to the E isomer.§ Notably, Z-1 has an intense n–π* absorbance (λmax = 495 nm, ε = 8000 M−1 cm−1), much stronger than that seen for Z azobenzenes (λmax = ∼450 nm, ε = 1500 M−1 cm−1).28 This is in line with the DFT calculations that predict a twisted conformation (vide supra). The thermal isomerisation rate of neutral 1 was measured at 25 °C using UV/vis spectrometry in aqueous buffer (at pH 9) and the half-life was found to be 16 seconds. It is interesting that the Z isomer of the unsymmetrical azole photoswitch phenylazo-2-imidazole has been previously reported to have a thermal half-life of 9 hours (albeit in a different solvent).29 Clearly, the move to a symmetric bisimidazole azo compound has a large impact on the energetics of thermal stability in imidazole-based photoswitches. The origins of thermal stability in such heteroaromatic azo compounds is under further study and will be reported in due course.
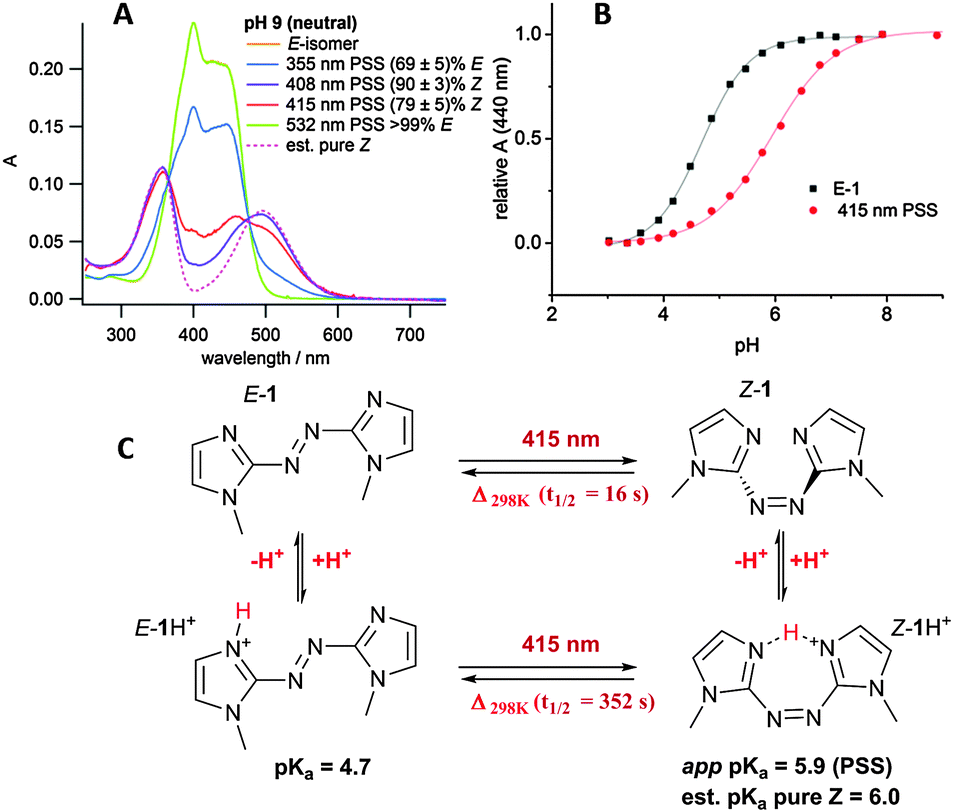 | ||
| Fig. 3 (A) UV/vis spectra of the different PSS of 1 at different irradiation wavelengths, and estimated pure Z spectrum (at 10−5 M); (B) pH titration of E-1 and the 415 nm PSS; (C) the photoswitch species present at different pHs and irradiation wavelengths. Thermal isomerisation rates and pKa values are included. | ||
In order to determine the pKa of both the E isomer and the 415 nm PSS (79% Z), a pH titration was carried out on a buffered aqueous sample of 1. At each pH increment the UV/vis spectra of the E isomer and PSS were measured. Due to the reasonably fast thermal isomerisation rate, the irradiated sample was allowed to thermally isomerise back to the E isomer before adjusting the pH. The π–π* absorption maximum of the E isomer at 440 nm (Fig. 3A) was plotted against pH in order to obtain the corresponding titration curves (Fig. 3B). The pH titration confirmed that at pH 9 azobisimidazole 1 was indeed neutral. The pKa was found to be 4.7 for the E isomer and the apparent pKa was 5.9 for the E/Z mixture present in the 415 nm PSS. By considering the PSS and the equilibria present in the system (Fig. 3C), the pKa of the Z isomer was estimated as 6.0 (see ESI†). A rough titration of the E isomer showed a second protonation event (assigned to protonation of the second imidazole ring) with an approximate pKa of 1.6 (see ESI†). No second protonation event was observed for the Z isomer above pH 3.
Given this data, the UV/vis spectrum for E-1 was recorded in acidic aqueous buffer at pH 3, to measure the photoswitch parameters of the protonated compound 1H+. Irradiation at 415 nm led to a PSS with a very similar spectrum to that of the E isomer (Fig. 4A). Although at first this could be considered to be a result of a PSS containing predominantly the E isomer, we instead attribute it to the fact that the protonated E and Z isomers E-1H+ and Z-1H+ have very similar UV/vis spectra. This assignment is supported by TDDFT calculated spectra for the protonated E and Z isomers (Fig. 4A); the spectra of both isomers is highly similar due to their planar conformation (Fig. 4B), as opposed to the neutral compound 1 (vide supra). As was the case for the neutral compound 1, the protonated compound 1H+ could be switched between two PSS upon irradiation with 415 and 532 nm light, however due to the similarity in the E and Z spectra, the PSS compositions could not be obtained.¶ The thermal isomerisation rate of protonated 1H+ at 25 °C was found to be approximately 20-fold slower than for the neutral compound in aqueous buffer (t1/2 = 352 s for protonated vs. 16 s for neutral). This increased half-life of the Z isomer on protonation is attributed to the requirement to (partially) break the intramolecular hydrogen bond present in Z-1H+ in either the rotation or inversion transition states for thermal Z–E isomerisation. This trend is opposite to that observed by Woolley and co-workers for their tetra-o-methoxy substituted aminoazobenzene photoswitches upon protonation (Fig. 1A); where the protonated Z isomer has a much shorter half life than the neutral species.15,17 This difference is explained by the site of protonation for the different photoswitches; protonation of the azo linkage in the substituted aminoazobenzenes (Fig. 1A) increases the thermal Z–E isomerisation rate, whereas in our case, protonation of 1 does not involve participation of the azo linkage.
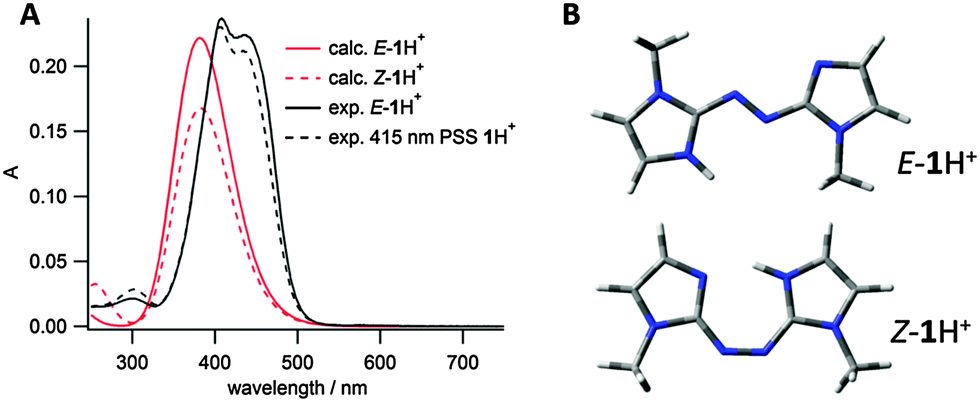 | ||
| Fig. 4 (A) Comparison of experimental and calculated (CAM-B3LYP//6-311G(2df,2p)) E-1H+ and Z-1H+ spectra; (B) energy minimised conformations (B3LYP/6-31G(d,p)) of E-1H+ and Z-1H+. | ||
Energy minimised conformations of E-1H+ support our assumption that protonation occurs on the imidazole nitrogen rather than the azo. The global minimum for the imidazolium is significantly lower energy than for the azonium (ΔG(azonium − imidazolium) = 44.4 kJ mol−1 – see ESI†). In order to further characterise the protonated compounds, the carbon NMR chemical shifts (δexp) for deuteronated E-1 were compared to calculated chemical shifts (δcalc) for E-1H+ protonated on either the imidazole or the azo (see ESI†). The difference in chemical shift between δexp and δcalc was much smaller for the imidazolium calculated structure than the azonium. Experimentally, pKa values for azonium cations are generally several units lower than imidazolium cations,30–33 with the exception being when the azonium proton can be additionally stabilised by hydrogen bonding as in the systems reported by Woolley and co-workers (e.g.Fig. 1A).15,17 Together the computational and experimental data supports our assumption that the protonation occurs on the imidazole rather than the azo nitrogen.
Given the significant pKa difference between the E and Z isomers of azobisimidazole 1, we sought to achieve a reversible pH ‘jump’ upon photoswitching. In principle, switching solution pH could have many uses, such as controlling the rate of a specific base catalysed reaction, or modulating enzyme activity.10 In order to measure the pH of the solution a pH indicator was used.34 Bromocresol purple (BCP) was chosen as it has a similar pKa value (vide infra) to that of the photoswitch, and it has an absorption maximum at a wavelength where the azobisimidazole has a negligible absorbance. A pH titration was carried out on BCP (Fig. S5, ESI†) and the pKa value obtained (6.8) was similar to the literature value of 6.2–6.4.35 Prior to attempting the pH ‘jump’, pH titrations of both E-1 and the 405 nm PSS with equimolar BCP were carried out (Fig. 5A). From this a graph of Amax(1)/Amax(BCP) against pH was plotted (for both E isomer and PSS) and the exponential fit obtained (Fig. S6, ESI†). Using this graph the pH value could be extracted from the UV/vis spectrum of a given sample of 1 and equimolar BCP. Irradiation of a 2.7 × 10−4 M aqueous solution of 1 at 405 nm led to an immediate change in pH (Fig. 5B). A change of 0.29 pH units was observed, equating to a two-fold reduction in proton concentration in the PSS. In theory, using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of E-1 and E-1H+ should enable a pH jump equal to the ΔpKa of the photoswitch (approximately 1.3 in this case). However, limitations on the concentrations used in this experiment and buffering by the indicator and potentially by carbonate is likely to decrease the range that can be obtained. Decreasing the concentration from 10−4 to 10−5 M resulted in a decrease in the pH ‘jump’ observed. While an increase in concentration would be expected to result in an increase in the pH jump, we were unable to assess this due to limitations in the method used for pH detection and the fast thermal isomerisation rates of the photoswitch. Nonetheless, several repeated cycles of photoswitching were carried out to ensure that the pH jump was reproducible over multiple irradiation cycles (Fig. S7, ESI†). The pH jump was consistent each time, within 3% of the mean value, suggesting that no irreversible photochemistry involving the indicator was occurring.
:1 mixture of E-1 and E-1H+ should enable a pH jump equal to the ΔpKa of the photoswitch (approximately 1.3 in this case). However, limitations on the concentrations used in this experiment and buffering by the indicator and potentially by carbonate is likely to decrease the range that can be obtained. Decreasing the concentration from 10−4 to 10−5 M resulted in a decrease in the pH ‘jump’ observed. While an increase in concentration would be expected to result in an increase in the pH jump, we were unable to assess this due to limitations in the method used for pH detection and the fast thermal isomerisation rates of the photoswitch. Nonetheless, several repeated cycles of photoswitching were carried out to ensure that the pH jump was reproducible over multiple irradiation cycles (Fig. S7, ESI†). The pH jump was consistent each time, within 3% of the mean value, suggesting that no irreversible photochemistry involving the indicator was occurring.
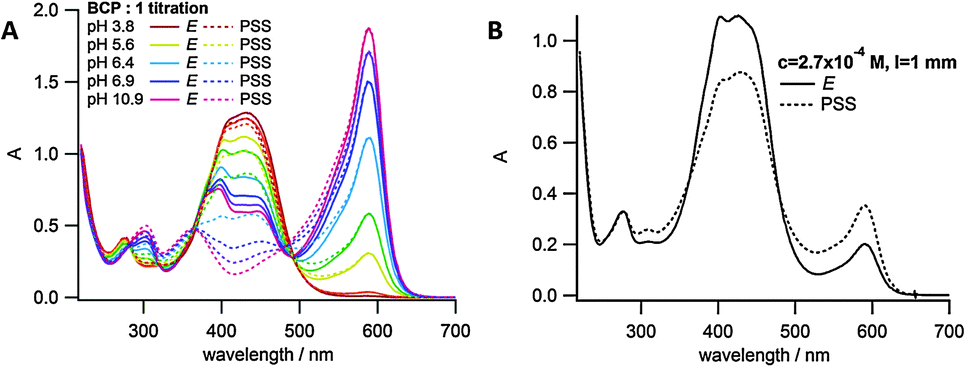 | ||
| Fig. 5 E-1 and 405 nm PSS with equimolar BCP; (A) pH titration; (B) pH ‘jump’ experiment at 2.7 × 10−4 M, 1 mm path length. | ||
In conclusion, we have designed an azoheteroaryl photoswitch that bears basic nitrogens on the two heteroaryl rings, correctly positioned to stabilise a proton in the Z isomer. This results in a significant and reversible shift in pKa values between the E and Z isomers. We believe extension of the concept developed in this paper may lead to related photochromic molecules with an increased difference in pKa between the isomers, and that such systems may lead to a wide range of applications. For example, photoswitchable bases have already been shown to have potential as switchable general base catalysts,8 but significant basicity differences between isomers may open up the possibility of switchable specific base catalysis and enzyme catalysis. Furthermore, light-driven proton concentration oscillations14 may be used to extract kinetic data on proton transfer events in a range of chemical and biological systems.
We thank the Engineering and Physical Sciences Research Council and the Leverhulme Trust (RPG-2012-441 to R. D. R.) for funding. We also thank Dr Kuimova for equipment access.
Notes and references
- T. Fehrentz, M. Schönberger and D. Trauner, Angew. Chem., Int. Ed., 2011, 50, 12156–12182 CrossRef CAS PubMed.
- W. A. Velema, W. Szymanski and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 2178–2191 CrossRef CAS PubMed.
- S. Thies, H. Sell, C. Bornholdt, C. Schütt, F. Köhler, F. Tuczek and R. Herges, Chem. – Eur. J., 2012, 18, 16358–16368 CrossRef CAS PubMed.
- T. Hugel, N. B. Holland, A. Cattani, L. Moroder, M. Seitz and H. E. Gaub, Science, 2002, 296, 1103–1106 CrossRef PubMed.
- W. R. Browne and B. L. Feringa, Nat. Nanotechnol., 2006, 1, 25–35 CrossRef CAS PubMed.
- K. Matsuda and M. Irie, Polyhedron, 2005, 24, 2477–2483 CrossRef CAS.
- M. Alemani, M. V. Peters, S. Hecht, K. H. Rieder, F. Moresco and L. Grill, J. Am. Chem. Soc., 2006, 128, 14446–14447 CrossRef CAS PubMed.
- M. V Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968–5972 CrossRef PubMed.
- M. Samanta, V. S. R. Krishna and S. Bandyopadhyay, Chem. Commun., 2014, 50, 10577–10579 RSC.
- S. Kohse, A. Neubauer, A. Pazidis, S. Lochbrunner and U. Kragl, J. Am. Chem. Soc., 2013, 135, 9407–9411 CrossRef CAS PubMed.
- S. Abbruzzetti, M. Carcelli, D. Rogolino and C. Viappiani, Photochem. Photobiol. Sci., 2003, 2, 796–800 CAS.
- S. H. Kawai, S. L. Gilat and J. Lehn, Eur. J. Org. Chem., 1999, 2359–2366 CrossRef CAS.
- M. Emond, J. Sun, J. Grégoire, S. Maurin, C. Tribet and L. Jullien, Phys. Chem. Chem. Phys., 2011, 13, 6493–6499 RSC.
- M. Emond, T. Le Saux, S. Maurin, J.-B. Baudin, R. Plasson and L. Jullien, Chem. – Eur. J., 2010, 16, 8822–8831 CrossRef CAS PubMed.
- S. Samanta, A. Babalhavaeji, M.-X. Dong and G. A. Woolley, Angew. Chem., Int. Ed., 2013, 52, 14127–14130 CrossRef CAS PubMed.
- N. Abeyrathna and Y. Liao, J. Am. Chem. Soc., 2015, 137, 11282–11284 CrossRef CAS PubMed.
- M. Dong, A. Babalhavaeji, M. J. Hansen, L. Kalman and G. A. Woolley, Chem. Commun., 2015, 51, 12981–12984 RSC.
- R. S. Stoll and S. Hecht, Org. Lett., 2009, 11, 4790–4793 CrossRef CAS PubMed.
- R. S. Stoll, M. V Peters, A. Kuhn, S. Heiles, R. Goddard, M. Buhl, C. M. Thiele and S. Hecht, J. Am. Chem. Soc., 2009, 131, 357–367 CrossRef CAS PubMed.
- C. Weston, R. Richardson, P. Haycock, A. White and M. Fuchter, J. Am. Chem. Soc., 2014, 136, 11878–11881 CrossRef CAS PubMed.
- F. Matino, G. Schull, U. Jana, F. Köhler, R. Berndt and R. Herges, Chem. Commun., 2010, 46, 6780–6782 RSC.
- S. Thies, H. Sell, C. Schütt, C. Bornholdt, C. Näther, F. Tuczek and R. Herges, J. Am. Chem. Soc., 2011, 133, 16243–16250 CrossRef CAS PubMed.
- C. Schütt, G. Heitmann, T. Wendler, B. Krahwinkel and R. Herges, J. Org. Chem., 2016, 81, 1206–1215 CrossRef PubMed.
- D. J. van Dijken, P. Kovaříček, S. P. Ihrig and S. Hecht, J. Am. Chem. Soc., 2015, 137, 14982–14991 CrossRef CAS PubMed.
- J. Otsuki, K. Suwa, K. K. Sarker and C. Sinha, J. Phys. Chem. A, 2007, 111, 1403–1409 CrossRef CAS PubMed.
- J. Chen and Z. Yin, Dyes Pigm., 2014, 102, 94–99 CrossRef CAS.
- E. Gattavecchia and D. Tonelli, J. Chem. Soc., Perkin Trans. 2, 1986, 689–693 RSC.
- H. M. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- J. Otsuki, K. Suwa, K. Narutaki, C. Sinha, I. Yoshikawa and K. Araki, J. Phys. Chem. A, 2005, 109, 8064–8069 CrossRef CAS PubMed.
- S. Stoyanov, L. Antonov, T. Stoyanova and V. Petrova, Dyes Pigm., 1996, 32, 171–185 CrossRef CAS.
- T. Stoyanova, S. Stoyanov, L. Antonov and V. Petrova, Dyes Pigm., 1996, 31, 1–12 CrossRef CAS.
- A. A. M. Prabhu, G. Venkatesh, R. K. Sankaranarayanan, S. Siva and N. Rajendiran, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2010, 49, 407–417 Search PubMed.
- J. A. Dean, Lang's Handbook of Chemistry, McGraw-Hill, New York, 12th edn, 1979 Search PubMed.
- R. M. D. Nunes, M. Pineiro and L. G. Arnaut, J. Am. Chem. Soc., 2009, 131, 9456–9462 CrossRef CAS PubMed.
- R. W. Sabnis, Handbook of acid–base indicators, CRC Press, Boca Raton, 2007 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc10380k. Raw data files are openly available, DOI: 10.6084/m9.figshare.2975923 |
| ‡ When Z–E thermal isomerisation is fast, higher power is needed to reach the PSS. |
| § This occurred at a faster rate than that of the Z–E thermal isomerisation. |
| ¶ The thermal isomerisation was too fast to allow quantification of the PSS by other spectroscopic methods (for example, by NMR). |
| This journal is © The Royal Society of Chemistry 2016 |