
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient hydrogen generation from formic acid using a reduced graphene oxide-supported AuPd nanoparticle catalyst†
Xinchun
Yang
ab,
Pradip
Pachfule
a,
Yao
Chen
a,
Nobuko
Tsumori
c and
Qiang
Xu
*ab
aNational Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka, Japan. E-mail: q.xu@aist.go.jp; qxuchem@hotmail.com; Fax: +81-72-751-9628; Tel: +81-72-751-9562
bGraduate School of Engineering, Kobe University, Nada Ku, Kobe, Hyogo, Japan
cToyama National College of Technology, 13, Hongo-machi, Toyama, 939-8630, Japan
First published on 17th February 2016
Abstract
Highly dispersed AuPd alloy nanoparticles have been successfully immobilized on reduced graphene oxide (rGO) using a facile non-noble metal sacrificial method, which exhibit the highest activity at 323 K (turnover frequency, 4840 h−1) for hydrogen generation without CO impurity from the formic acid/sodium formate system.
Hydrogen is of critical importance in clean energy applications although effective hydrogen storage technologies still need to be developed.1 Formic acid (FA) is a promising hydrogen carrier,2 which has several remarkable features such as (a) nontoxicity with high hydrogen content (4.4 wt%), (b) high stability as a liquid at room temperature, and (c) easy synthesis via a biomass process, CO2 reduction or methyl formate hydrolysis.3 Depending on the catalysts, however, FA can be decomposed in two ways: desirable dehydrogenation (HCOOH → H2 + CO2) and undesirable dehydration (HCOOH → H2O + CO), which produces CO impurities that are toxic to the fuel cell catalysts.4
So far, homogeneous catalysts have been extensively studied for hydrogen production from FA and very high catalytic activities have been reported.5 Along with the ongoing progress in FA dehydrogenation using homogeneous systems, heterogeneous catalysts have attracted tremendous attention due to their advantages of easy separation and recycling. A large number of metal nanoparticle (MNP) catalysts have been studied, most of which, however, suffer from severe aggregation and limited catalytic activities.6
Herein, we report a highly active AuPd alloy nanoparticle catalyst prepared using a non-noble metal sacrificial approach (NNMSA), which exhibits the highest turnover frequency (TOF) of 4840 h−1 at 323 K for hydrogen generation from the FA/sodium formate (SF) system.7
Cobalt was used as the sacrificial agent. Amorphous Co3(BO3)2 was formed from the reaction between Co(CH3COO)2 and NaBH4, which can be easily dissolved in H3PO4 (Fig. S1 and S2, for details see the ESI†).8 Graphite oxide (GO) was prepared from graphite using a modified Hummers' method.9 The rGO-supported AuPd nanoparticle (NP) catalyst was synthesized using a wet-chemical process, as illustrated in Scheme 1. Firstly, according to the stoichiometry, the aqueous solution containing HAuCl4, Co(CH3COO)2 and K2PdCl4 was added into 50 mL of GO suspension (0.1 wt%). Then the above mixture was reduced by a fresh aqueous solution of NaBH4. After continuous stirring for 2 h, the mixture was washed and re-dispersed in H3PO4 solution for etching (2 h). Finally, the catalyst, denoted as (Cox)EAuyPd1−y/rGO (ESI†), was obtained by centrifugation and washing with water.
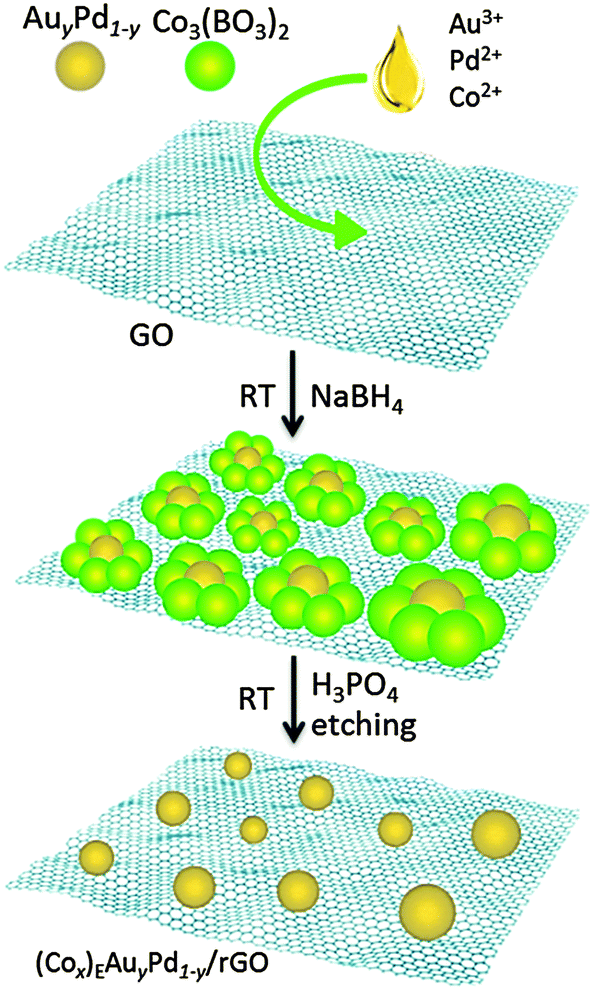 | ||
| Scheme 1 Schematic illustration of the synthesis of the (Cox)EAuyPd1−y/rGO catalyst via a non-noble metal (cobalt) sacrificial approach (NNMSA). | ||
Fig. 1a shows the powder X-ray diffraction (PXRD) pattern of the (Co3)EAu0.6Pd0.4/rGO catalyst. Except for a low and broad diffraction of rGO at 24°, the diffraction peaks, which are located between the fcc Au (JCPDS No. 65-8601) and Pd (JCPDS No. 65-2867), indicate the formation of an AuPd alloy.10 For comparison, PXRD patterns of the non-etched counterparts Co3Au0.6Pd0.4 and Co3Au0.6Pd0.4/rGO (Fig. S3, ESI†) show an additional broad band for Co3(BO3)2. After etching with an acid, the characteristic peaks corresponding to AuPd are clearly visible and match well with the directly deposited counterpart of Au0.6Pd0.4/rGO (Fig. S3, ESI†), confirming the successful removal of cobalt and the formation of AuPd. The N2 sorption isotherms for (Co3)EAu0.6Pd0.4/rGO, Co3Au0.6Pd0.4/rGO and Au0.6Pd0.4/rGO are shown in Fig. 1b. Generally, the restacking of rGO can be partially overcome by the direct deposition of NPs.11 Hence, the Au0.6Pd0.4/rGO and Co3Au0.6Pd0.4/rGO possess a type-IV N2 sorption curve with Brunauer–Emmett–Teller (BET) surface areas of 189 and 219 m2 g−1, respectively. Notably, the surface area of (Co3)EAu0.6Pd0.4/rGO (321 m2 g−1) increases drastically, illustrating that the removal of cobalt by the acid etching has resulted in high porosity, which will favour the diffusion of reactants to metal NPs in catalysis.
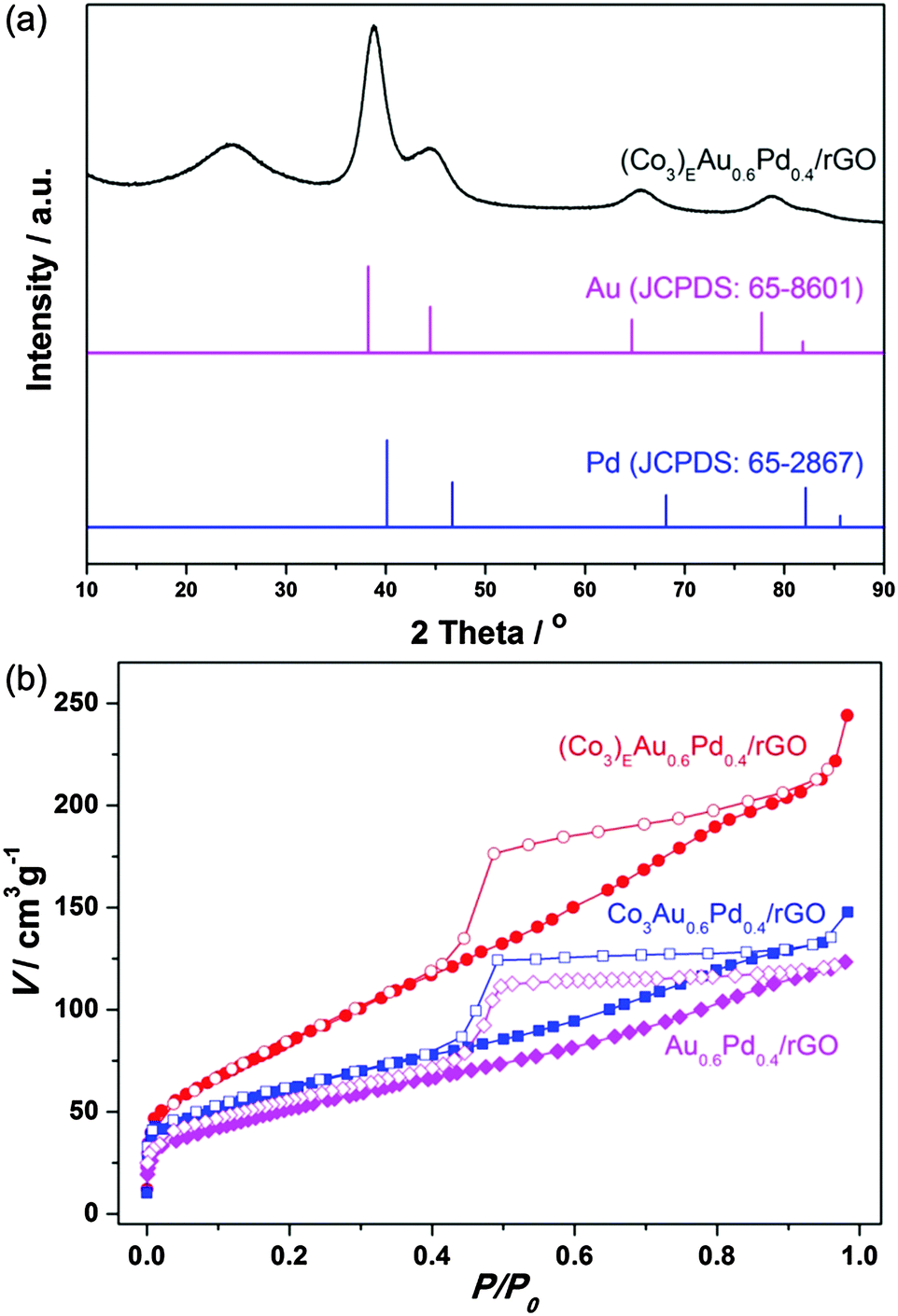 | ||
| Fig. 1 (a) XRD pattern of (Co3)EAu0.6Pd0.4/rGO and (b) N2 sorption analyses (77 K) of (Co3)EAu0.6Pd0.4/rGO, Co3Au0.6Pd0.4/rGO and Au0.6Pd0.4/rGO. | ||
The morphologies of the Au0.6Pd0.4/rGO, Co3Au0.6Pd0.4/rGO and (Co3)EAu0.6Pd0.4/rGO catalysts were analysed by transmission electron microscopy (TEM). As shown in Fig. S4 and S5 (ESI†), Au0.6Pd0.4/rGO and Co3Au0.6Pd0.4/rGO possess larger particles with severe aggregation (ESI†). In contrast, the AuPd NPs in the (Co3)EAu0.6Pd0.4/rGO catalyst are well dispersed and immobilized on the rGO support (Fig. 2a). The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) images further reveal the uniform distribution of AuPd NPs in the (Co3)EAu0.6Pd0.4/rGO catalyst with an average particle size of 3.9 ± 0.9 nm (Fig. 2b and c), indicating that the sacrifice of the cobalt compound can prevent the primary AuPd NPs from aggregation, which will benefit their catalytic performance. Energy dispersive X-ray (EDX) analyses (Fig. S6–S8, ESI†) show that Co3Au0.6Pd0.4/rGO is rich in cobalt, whereas no signals for cobalt are detected in (Co3)EAu0.6Pd0.4/rGO, clearly confirming that cobalt has been eliminated by the H3PO4 etching. The inductively coupled plasma optical emission spectroscopic analysis reveals that the molar ratio of Co![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Au:Pd in (Co3)EAu0.6Pd0.4/rGO to be 0.04:0.6:0.4, indicating that almost all the co-precipitated Co3(BO3)2 can be removed by H3PO4. The X-ray photoelectron spectroscopic (XPS) measurements show no signals for Co in (Co3)EAu0.6Pd0.4/rGO before and after Ar sputtering (Fig. 3a). The observed Au 4f and Pd 3d spectra reveals that (Co3)EAu0.6Pd0.4/rGO is composed of metallic Au and Pd (Fig. 3b and c), further confirming the successful formation of AuPd alloy NPs.
:Au:Pd in (Co3)EAu0.6Pd0.4/rGO to be 0.04:0.6:0.4, indicating that almost all the co-precipitated Co3(BO3)2 can be removed by H3PO4. The X-ray photoelectron spectroscopic (XPS) measurements show no signals for Co in (Co3)EAu0.6Pd0.4/rGO before and after Ar sputtering (Fig. 3a). The observed Au 4f and Pd 3d spectra reveals that (Co3)EAu0.6Pd0.4/rGO is composed of metallic Au and Pd (Fig. 3b and c), further confirming the successful formation of AuPd alloy NPs.
 | ||
| Fig. 2 (a) TEM, (b and c) HAADF-STEM images and (d) particle size distribution histograms of AuPd NPs of (Co3)EAu0.6Pd0.4/rGO. | ||
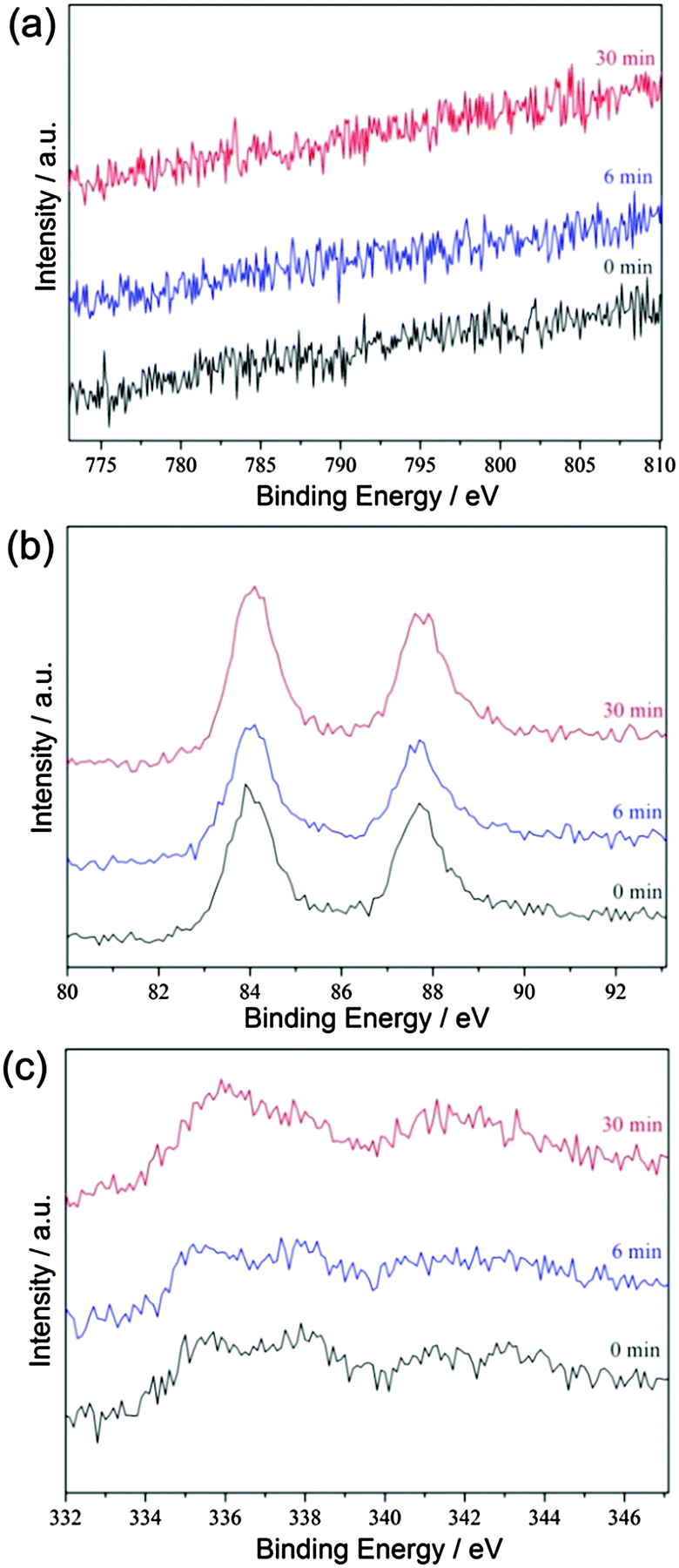 | ||
| Fig. 3 (a) Co 2p, (b) Au 4f, and (c) Pd 3d XPS spectra of (Co3)EAu0.6Pd0.4/rGO after argon sputtering for 0, 6 and 30 min. | ||
The catalytic activities of the (Cox)EAuyPd1−y/rGO catalysts in hydrogen generation from the FA/SF system were evaluated. Fig. 4a shows the gas generation from the FA/SF system in the presence of (Co3)EAu0.6Pd0.4/rGO and Au0.6Pd0.4/rGO. Unquestionably, the sacrificial agent plays a dominant role, by decreasing the size of NPs, in the promotion of catalytic performance. Under our evaluation conditions, the (Co3)EAu0.6Pd0.4/rGO catalyst shows an extremely high catalytic activity for the complete dehydrogenation of FA with the release of 146 mL gas of H2 + CO2 in 0.62 min (nAuPd/nFA = 0.02 and nSF/nFA = 2.5) at 323 K, giving a TOF as high as 4840 h−1 (Fig. S9, ESI†), the highest value reported thus far (Table S1, ESI†).7 After that, the generated gas with a slow speed is attributed to the decomposition of SF. The catalytic activities of the (Co3)EAu0.6Pd0.4/rGO catalysts with lower metal loadings have not shown significant changes (Fig. S10, ESI†).
 | ||
| Fig. 4 (a) Volume of the generated gas (H2 + CO2) versus time for the dehydrogenation of FA over the (Co3)EAu0.6Pd0.4/rGO and Au0.6Pd0.4/rGO catalysts (inset: the corresponding TOF values) and (b) durability test for the dehydrogenation of FA over (Co3)EAu0.6Pd0.4/rGO (nAuPd/nFA = 0.02, nSF/nFA = 2.5, 323 K). | ||
Gas chromatography (GC) measurements demonstrate that the produced gas consists of H2 and CO2 without a trace of CO (below 5 ppm), implying that the as-synthesized (CO3)EAu0.6Pd0.4/rGO catalyst has a high selectivity for the dehydrogenation of FA into H2 and CO2 (Fig. S11, ESI†). Such excellent catalytic performance for FA dehydrogenation can meet the requirements of practical applications. Herein, it should be noted that the molar ratio of SF/FA has an obvious influence on the gas release rate (Fig. S12, ESI†). The gas release rate increases rapidly until the molar ratio of SF/FA increases up to 2.5. The further increase, however, makes no actual impression on the FA decomposition. On the other hand, the catalytic performance of (Co3)EAuyPd1−y/rGO is affected by the molar ratio of Au/Pd (Fig. S13, ESI†). The mono-metallic (Co3)EAu/rGO has no activity while (Co3)EPd/rGO has a low activity. However, the introduction of Au to Pd leads to a dramatically enhanced efficiency, which reaches its best value at nAu/nPd = 6/4, indicative of the excellent synergistic effects between Au and Pd for FA decomposition.12 Additionally, rGO is an excellent support for the catalysis, which is demonstrated by the quite low activities of their support-free counterparts (Fig. S14, ESI†).
Further, the increased hydrogen release rates from the FA/SF systems have been observed at elevated temperatures (Fig. S15, ESI†). The activation energy (Ea) in this process is calculated to be 39.77 kJ mol−1, which is lower than most of the reported heterogeneous and homogeneous systems for FA dehydrogenation.5,6d,7,13 After the catalytic reaction, the catalyst was recollected by centrifugation, washed with water and recycled for further use. As shown in Fig. 4b and Table S2 (ESI†), no significant loss in activity was observed over five cycles. The structure and size distribution of metal NPs after the catalytic reactions have been found to be identical to those of the pristine catalyst (Fig. S16 and S17, ESI†). In contrast, the CO poisoned (Co3)EAu0.6Pd0.4/rGO, prepared by exposing the catalyst to a CO atmosphere showed no catalytic activity (Fig. S18, ESI†). It is clear that the (Co3)EAu0.6Pd0.4/rGO catalyst possesses high selectivity, durability and stability during the FA dehydrogenation.
In conclusion, a (Co3)EAu0.6Pd0.4/rGO catalyst has been synthesized using a simple non-noble metal sacrificial method. The obtained catalyst shows efficient catalytic activity for hydrogen generation from the FA/SF system with the highest TOF of 4840 h−1 at 323 K and possesses high selectivity, durability and stability over 5 cycles. It is believed that the way to the utilization of FA as a hydrogen carrier will be paved by the practical use of the presently provided catalyst.
The authors thank Dr Takeyuki Uchida for TEM measurements, and METI, AIST and Kobe University for financial support. X. C. Yang is grateful to the China Scholarship Council (CSC) and Ministry of Education, Culture, Sports, Science and Technology-Japan (MEXT) for a PhD scholarship.
Notes and references
- (a) L. Schlapbach and A. Züttel, Nature, 2001, 414, 353 CrossRef CAS PubMed; (b) P. E. de Jongh, Nat. Mater., 2011, 10, 265 CrossRef CAS PubMed; (c) A. Boddien, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge, G. Laurenczy and M. Beller, Energy Environ. Sci., 2012, 5, 8907 RSC.
- (a) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171 RSC; (b) A. F. Dalebrook, W. Gan, M. Grasemann, S. Moret and G. Laurenczy, Chem. Commun., 2013, 49, 8735 RSC.
- (a) M. Yadav and Q. Xu, Energy Environ. Sci., 2012, 5, 9698 RSC; (b) Q. Y. Bi, J. D. Lin, Y. M. Liu, X. L. Du, J. Q. Wang, H. Y. He and Y. Cao, Angew. Chem., Int. Ed., 2014, 53, 13583 CrossRef CAS PubMed.
- (a) S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496 CrossRef CAS PubMed; (b) Z. L. Wang, J. M. Yan, H. L. Wang, Y. Ping and Q. Jiang, J. Mater. Chem. A, 2013, 1, 12721 RSC.
- (a) C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966 CrossRef CAS PubMed; (b) Y. Himeda, Green Chem., 2009, 11, 2018 RSC; (c) A. Boddien, B. Loges, F. Gartner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924 CrossRef CAS PubMed; (d) A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733 CrossRef CAS PubMed; (e) Z. L. Wang, J. M. Yan, Y. Ping, H. L. Wang, W. T. Zheng and Q. Jiang, Angew. Chem., Int. Ed., 2013, 52, 4406 CrossRef CAS PubMed; (f) D. Mellmann, E. Barsch, M. Bauer, K. Grabow, A. Boddien, A. Kammer, P. Sponholz, U. Bentrup, R. Jackstell, H. Junge, G. Laurenczy, R. Ludwig and M. Beller, Chem. – Eur. J., 2014, 20, 13589 CrossRef CAS PubMed; (g) S. J. Li, Y. Ping, J. M. Yan, H. L. Wang, M. Wu and Q. Jiang, J. Mater. Chem. A, 2015, 3, 14535 RSC.
- (a) D. A. Bulushev, S. Beloshapkin and J. R. H. Ross, Catal. Today, 2010, 154, 7 CrossRef CAS; (b) D. R. Dreyer, S. J. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228 RSC; (c) K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302 CrossRef CAS PubMed; (d) X. J. Gu, Z. H. Lu, H. L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822 CrossRef CAS PubMed; (e) Q. Y. Bi, X. L. Du, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Am. Chem. Soc., 2012, 134, 8926 CrossRef CAS PubMed; (f) N. Yi, H. Saltsburg and M. Flytzani-Stephanopoulos, ChemSusChem, 2013, 6, 816 CrossRef CAS PubMed; (g) K. Mori, M. Dojo and H. Yamashita, ACS Catal., 2013, 3, 1114 CrossRef CAS; (h) Z. L. Wang, Y. Ping, J. M. Yan, H. L. Wang and Q. Jiang, Int. J. Hydrogen Energy, 2014, 39, 4850 CrossRef CAS; (i) Q. L. Zhu, N. Tsumori and Q. Xu, Chem. Sci., 2014, 5, 195 RSC; (j) Q. L. Zhu and Q. Xu, Energy Environ. Sci., 2015, 8, 478 RSC; (k) K. Mandal, D. Bhattacharjee and S. Dasgupta, Int. J. Hydrogen Energy, 2015, 40, 4786 CrossRef CAS; (l) S. Wu, F. Yang, H. Wang, R. Chen, P. C. Sun and T. H. Chen, Chem. Commun., 2015, 51, 10887 RSC; (m) M. Hattor, H. Einaga, T. Daioc and M. Tsuji, J. Mater. Chem. A, 2015, 3, 4453 RSC; (n) A. Bulut, M. Yurderi, Y. Karatas, M. Zahmakiran, H. Kivrak, M. Gulcan and M. Kaya, Appl. Catal., B, 2015, 164, 324 CrossRef CAS.
- (a) J. M. Yan, Z. L. Wang, L. Gu, S. J. Li, H. L. Wang, W. T. Zheng and Q. Jiang, Adv. Energy Mater., 2015, 5, 1500107 Search PubMed; (b) Y. Chen, Q. L. Zhu, N. Tsumori and Q. Xu, J. Am. Chem. Soc., 2015, 137, 106 CrossRef CAS PubMed; (c) Q. G. Liu, X. F. Yang, Y. Q. Huang, S. T. Xu, X. Su, X. L. Pan, J. M. Xu, A. Q. Wang, C. H. Liang, X. K. Wang and T. Zhang, Energy Environ. Sci., 2015, 8, 3207 Search PubMed; (d) F. Z. Song, Q. L. Zhu, N. Tsumori and Q. Xu, ACS Catal., 2015, 5, 5141 CrossRef CAS.
- G. N. Glavee, K. J. Klabunde, C. M. Sorensen and G. C. Hadjapnayis, Langmuir, 1992, 8, 771 CrossRef CAS.
- (a) W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS; (b) Y. Chen, X. Zhang, P. Xu and Y. Ma, Chem. Commun., 2009, 4527 RSC.
- (a) T. García, S. Agouram, A. Dejoz, J. F. Sánchez-Royo, L. Torrente-Murciano and B. Solsona, Catal. Today, 2015, 248, 48 CrossRef; (b) C. C. Han, L. N. Wu, L. Ge, Y. J. Li and Z. Zhao, Carbon, 2015, 92, 32 CrossRef.
- (a) F. Ye, X. Cao, L. Yu, S. Chen and W. Lin, Int. J. Electrochem. Sci., 2012, 7, 1251 CAS; (b) Z. Huang, S. Wang, J. Wang, Y. Yu, J. Wen and R. Li, Electrochim. Acta, 2015, 152, 117 CrossRef CAS.
- (a) B. Pawelec, E. Cano-Serrano, J. M. Campos-Martin, R. M. Navarro, S. Thomas and J. L. G. Fierro, Appl. Catal., A, 2004, 275, 127 CrossRef CAS; (b) T. Déronzier, F. Morfin, M. Lomello and J. L. Rousset, J. Catal., 2014, 311, 221 CrossRef.
- (a) X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 3540 RSC; (b) D. A. Bulushev, L. Jia, S. Beloshapkin and J. R. H. Ross, Chem. Commun., 2012, 48, 4184 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, PXRD, TEM, EDX, GC and catalytic activity. See DOI: 10.1039/c5cc10311h |
| This journal is © The Royal Society of Chemistry 2016 |