
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRational design and diversity-oriented synthesis of peptoid-based selective HDAC6 inhibitors†
D.
Diedrich
a,
A.
Hamacher
a,
C. G. W.
Gertzen
a,
L. A.
Alves Avelar
a,
G. J.
Reiss
b,
T.
Kurz
a,
H.
Gohlke
a,
M. U.
Kassack
a and
F. K.
Hansen
*a
aInstitut für Pharmazeutische und Medizinische Chemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany. E-mail: finn.hansen@hhu.de
bInstitut für Anorganische Chemie und Strukturchemie, Heinrich-Heine-Universität Düsseldorf, Universitätsstr. 1, 40225 Düsseldorf, Germany
First published on 14th January 2016
Abstract
A mini library of HDAC inhibitors with peptoid-based cap groups was synthesized using an efficient multicomponent approach. Four compounds were identified as potent HDAC6 inhibitors with a selectivity over other HDAC isoforms. The most potent HDAC6 inhibitor revealed remarkable chemosensitizing properties and completely reverted the cisplatin resistance in Cal27 CisR cells.
Histone deacetylases (HDACs) catalyze the cleavage of acetyl groups from N-acetyl-lysine residues of histones and non-histone proteins. These posttranslational modifications (PTMs) are important for the regulation of gene transcription and protein function.1 HDAC classes I (HDACs 1–3,8), IIa (HDACs 4,5,7,9), IIb (HDACs 6,10), and IV (HDAC 11) contain zinc-dependent deacetylase domains and are considered “classical” HDACs.2,3 The class III HDACs (sirtuins) are structurally different and NAD+-dependent.3 Most inhibitors of “classical” HDACs are characterized by a widely accepted pharmacophore model comprising a zinc binding group (ZBG) chelating the zinc atom in the active site, a linker accommodating the tubular access of the active site, and a cap group interacting with the external surface (Fig. 1A).4 Currently, four HDAC inhibitors (HDACi) have been approved by the FDA for the treatment of cancer (vorinostat, romidepsin, belinostat and panobinostat). Furthermore, HDACi have attracted attention as potential therapeutic drugs for treating a variety of diseases beyond cancer including inflammation, diabetes, HIV, neurodegenerative diseases (e.g. Alzheimer, Huntington), and parasitic diseases (e.g. malaria).5
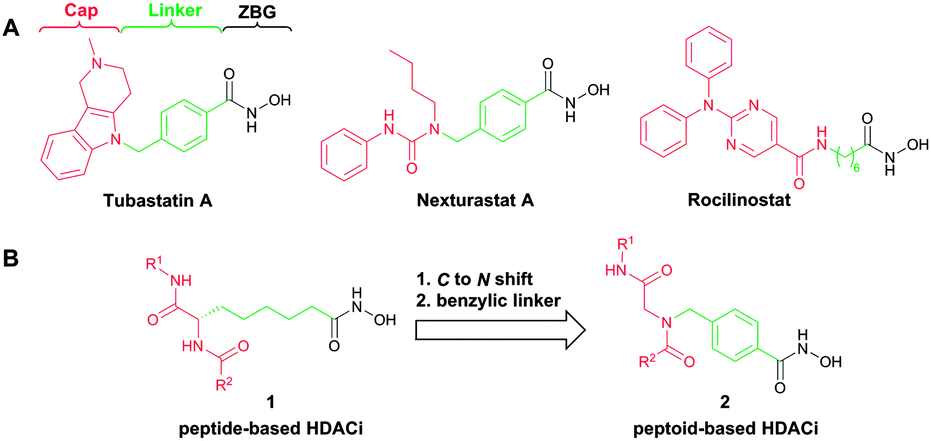 | ||
| Fig. 1 (A) Selected HDAC6-selective HDACi; (B) design of peptoid-based HDACi. | ||
All approved HDACi target multiple HDAC isoforms. Their clinical use may therefore cause serious unwanted side effects.3 Thus, the development of isoform-selective HDACi has been hypothesized to afford inhibitors with an improved safety profile.3,4 HDAC6, a class IIb enzyme, is structurally and functionally unique among the eleven human zinc-dependent HDACs.6 It is the only HDAC with two independent functional catalytic domains and a C-terminal zinc finger motif responsible for binding ubiquitinated proteins.5b Interestingly, it has been demonstrated that the second catalytic site is the major functional domain of HDAC6.7 The enzyme was initially described as tubulin deacetylase; however, it also modulates the function of other non-histone proteins implicated in regulatory processes, including cortactin, peroxiredoxins, and Hsp90.5b Due to the large number of substrates, HDAC6 is involved in numerous diseases such as autoimmune disorders, inflammation, neurodegenerative diseases, and cancer.5b Notably, the first selective HDAC6 inhibitor, rocilinostat (ACY-1215, Fig. 1A), has recently entered phase II clinical trials for the treatment of multiple myeloma and lymphoid malignancies.5a Consequently, HDAC6 has emerged as an attractive therapeutic target, and the search for novel potent and selective HDAC6 inhibitors is of high importance.
We report here on the rational design and diversity-oriented synthesis of a series of selective HDAC6 inhibitors utilizing peptoid-based cap groups. The biological evaluation of the target compounds includes whole cell HDAC and MTT assays on sensitive and chemoresistant cancer cell lines. The most active peptoid-based HDACi were investigated for their activity against selected HDAC isoforms and their enhancement of cisplatin-induced cytotoxicity. NMR spectroscopy and X-ray crystallography were employed to study the peptoid amide bond geometry. Molecular modelling, MD simulations, and docking studies allowed rationalization of the observed selectivity profile.
Recently, it has been identified via homology modelling that the catalytic domain II (CDII) of HDAC6 possesses a significantly wider channel rim in comparison with HDAC1.8a This feature has been used to develop selective HDAC6 inhibitors utilizing bulky or branched cap groups.8 Tubastatin A and nexturastat A (Fig. 1A) are important examples that occupy this unique rim region of HDAC6.8 Fairlie and co-workers previously described peptide-based HDACi utilizing 2-aminosuberic acid as key building block.9 These HDACi of type 1 (Fig. 1B) revealed remarkable anticancer activity. Notably, some compounds of this type showed a preference for HDAC6.10 However, peptides are intrinsically labile to enzymatic degradation, and the large scale synthesis of enantiomerically pure unnatural amino acids is often a challenging task. Peptoids, N-alkyl glycine derivatives, feature several advantages over peptides including proteolytic stability and increased cell permeability.11 We hypothesized that peptoid analogues 2 of the α-amino suberic acid-containing HDACi (1) having the linker attached to the nitrogen rather than to the α-carbon are promising scaffolds for the development of selective HDAC6 inhibitors. The peptoid-based cap group is intended to accommodate the significantly wider catalytic channel rim identified in human HDAC6. Furthermore, we decided to incorporate a benzyl linker, which is known to increase the selectivity for HDAC6 (Fig. 1B).8
We aimed at the development of a multicomponent approach in order to allow a rapid and diversity-oriented synthesis of the target compounds. Our retrosynthetic analysis led to an Ugi four-component reaction (U-4CR) as the key step followed by hydroxylaminolysis as a suitable synthetic pathway. Although the U-4CR has been used previously for the preparation of HDACi,12 no synthesis of peptoid-based HDACi via the U-4CR has been reported so far. Preparative conditions for the U-4CR were first optimized using intermediate 3a (R1: t-Bu; R2: 3,4-Me-Ph) as model compound (Table S1, ESI†). Different solvents, stoichiometries, concentrations, and reaction times were studied. Best results were achieved at room temperature using dry methanol as solvent in the presence of 4 Å molecular sieves. This optimized protocol was then used for the synthesis of the intermediates 3a–l. In all cases, the crude products were purified by flash column chromatography and isolated in 43–85% yield. The subsequent hydroxylaminolysis afforded the desired HDACi 2a–l in 42–69% yield (Scheme 1).
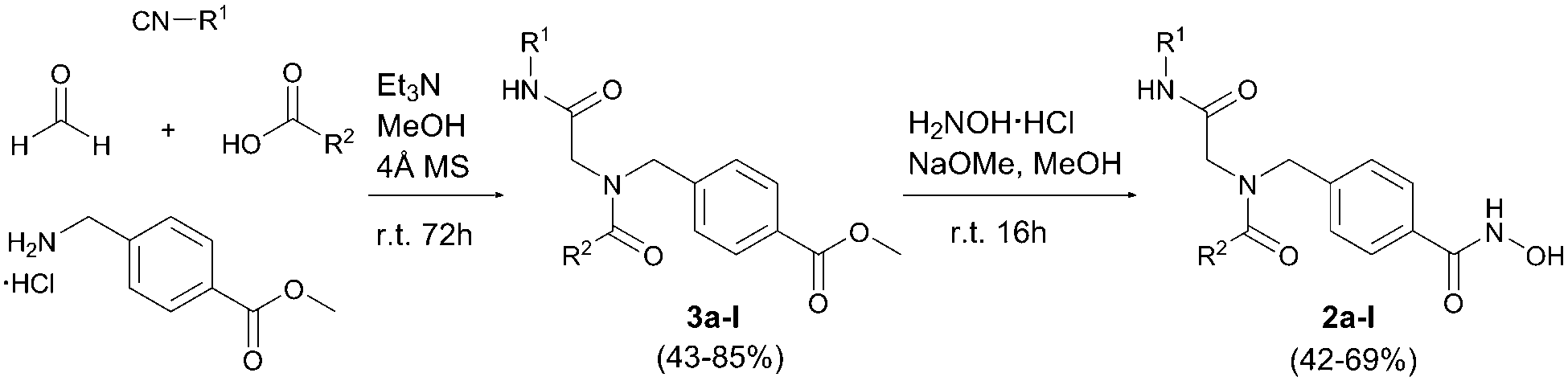 | ||
| Scheme 1 Synthesis of peptoid-based HDACi 2a–l. | ||
All synthesized peptoid-based HDACi 2a–l were first assessed in a whole cell HDAC inhibition assay and a MTT assay for cytotoxicity using the human ovarian cancer cell line A2780 and its cisplatin resistant subclone A2780 CisR. The results of both assays are summarized in Table 1. The whole cell HDAC assay provided valuable structure–activity relationships. Notably, all HDACi with a 4-dimethylamino-substituted cap group (2e, i, l) displayed remarkable activity in the whole cell HDAC assay and showed similar or better potency than the FDA-approved reference compound vorinostat. The most active compound from this series, 2i, showed IC50 values of 0.90 μM (A2780) and 0.79 μM (A2780 CisR). Interestingly, the nature of the isocyanide component (R1) used in U-4CR is important for the activity. The potency of the compounds decreased according to their R1 substituent in the order cyclohexyl > 4-tolyl > tert-butyl. All compounds (2a–l) showed cytotoxic activities against the ovarian cancer cell lines A2780/A2780 CisR. In good agreement with the data from the whole cell HDAC assay, 2e, i, l exhibited remarkable anticancer activity and displayed higher cytotoxicity than the reference compound cisplatin. Notably, 2i was the most potent compound with IC50 values of 0.34 μM (A2780) and 1.45 μM (A2780 CisR).
| Entry | R1 | R2 | HDAC IC50 [μM] | MTT IC50 [μM] | HDAC IC50 [μM] | MTT IC50 [μM] | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| A2780 | A2780 CisR | A2780 | A2780 CisR | Cal27 | Cal27 CisR | Cal27 | Cal27 CisR | |||
| a Data from ref. 5d. | ||||||||||
| 2a | t-Bu | 3,4-Me-Ph | 5.14 | 4.97 | 3.59 | 9.39 | 2.05 | 3.22 | 14.7 | 10.7 |
| 2b | t-Bu | 3,5-Me-Ph | 5.99 | 6.16 | 4.86 | 10.7 | 2.89 | 3.83 | 25.3 | 11.7 |
| 2c | t-Bu | Ph | 7.82 | 4.82 | 32.1 | 53.6 | 7.83 | 10.6 | 37.1 | 36.7 |
| 2d | t-Bu | 1-Naphthyl | 15.0 | 15.5 | 8.33 | 31.0 | 13.9 | 18.1 | 27.7 | 34.7 |
| 2e | t-Bu | 4-Me2N-Ph | 1.23 | 1.03 | 0.94 | 1.62 | 1.08 | 0.76 | 2.92 | 3.83 |
| 2f | c-Hex | 3,5-Me-Ph | 2.72 | 2.00 | 6.05 | 19.3 | 3.31 | 2.50 | 7.50 | 12.8 |
| 2g | c-Hex | Ph | 3.88 | 3.56 | 20.0 | 32.8 | 5.45 | 3.18 | 16.9 | 14.2 |
| 2h | c-Hex | 1-Naphthyl | 6.03 | 4.47 | 31.2 | 36.5 | 5.23 | 7.51 | 34.1 | 18.0 |
| 2i | c-Hex | 4-Me2N-Ph | 0.90 | 0.79 | 0.34 | 1.45 | 0.62 | 0.48 | 2.04 | 2.70 |
| 2j | 4-Tolyl | Ph | 5.51 | 5.03 | 5.87 | 24.3 | 5.00 | 2.99 | 15.8 | 11.8 |
| 2k | 4-Tolyl | 1-Naphthyl | 8.51 | 8.94 | 6.31 | 25.4 | 3.93 | 3.76 | 19.8 | 12.3 |
| 2l | 4-Tolyl | 4-Me2N-Ph | 0.90 | 1.05 | 0.65 | 3.88 | 0.77 | 0.57 | 2.77 | 3.44 |
| Vorinostat | 0.96 | 0.97 | 2.42a | 3.12a | 0.86 | 0.73 | 2.64a | 2.08a | ||
| Cisplatin | — | — | 2.25 | 17.2 | — | — | 2.50 | 16.1 | ||
To extend the investigation of this novel class of HDACi to another pair of solid cancer cell lines, compounds 2a–l were tested for HDAC inhibitory and cytotoxic activity in the human tongue squamous cell carcinoma cell line Cal27 and its cisplatin resistant subline Cal27 CisR. We observed similar structure–activity relationships as seen against A2780/A2780 CisR. Again, 2i was the most potent HDACi (Table 1). Interestingly, the compounds showed almost equipotent cellular HDAC inhibition in the respective cancer cell pairs. However, cytotoxic activity (MTT assay) was higher in A2780 than in A2780 CisR whereas the compounds displayed about similar cytotoxicity in Cal27 and Cal27 CisR cells. Thus, resistance mechanisms induced by cisplatin may (A2780 CisR) or may not (Cal27 CisR) modulate cytotoxic effects of these novel HDACi.
Based on their noteworthy activity in the whole cell HDAC assay, compounds 2e, f, i, l were selected for a detailed biological evaluation. First, they were assessed for their inhibitory activity against recombinant HDAC6. Strikingly, all four compounds were found to be potent HDAC6 inhibitors with IC50 values ranging from 1.59 nM to 11.2 nM (Table 2). In order to investigate the selectivity against other HDAC isozymes, we screened compounds 2e, f, i, l against representative examples of all other zinc dependent HDAC classes (class I: HDAC2; class IIa: HDAC4; and class IV: HDAC11). The results are presented in Table 2. Compound 2f and 2i demonstrated the highest overall selectivity for HDAC6. 2f revealed selectivity indices of 390 against HDAC2, >893 against HDAC4 and 38 against HDAC11. The most potent HDAC6 inhibitor 2i (HDAC6 IC50: 1.59 nM) exhibited selectivity indices of 126 against HDAC2, >6289 against HDAC4, and 40 against HDAC11. The HDAC6 inhibitory activity of these compounds was further validated by investigation of the acetylation status of α-tubulin in Cal27 and Cal27 CisR cells. As expected, the peptoid-based HDACi 2e, f, i, l and HDAC6-selective tubastatin A are causing α-tubulin hyperacetylation whereas class I HDAC-selective entinostat had no effect. These data confirm the inhibition of HDAC6 in a more complex cellular environment (Fig. S1, ESI†).
| Compound | HDAC2 IC50 [nM] | HDAC4 IC50 [nM] | HDAC6 IC50 [nM] | HDAC11 IC50 [nM] |
|---|---|---|---|---|
| a The inhibition of HDAC isoforms was determined by Reaction Biology Corporation (Malvern, PA, USA). | ||||
| 2e | 799 | >10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
6.52 | 227 |
| 2f | 4366 | >10000 |
11.2 | 421 |
| 2i | 200 | >10000 |
1.59 | 63.2 |
| 2l | 637 | 4377 | 2.84 | 25.0 |
| TSA | 18.9 | nd | 3.18 | 17.8 |
| TMP269 | nd | 169 | nd | nd |
The 1D NMR spectra of compounds 2a–d, f–h, j–k at room temperature and of 2e, i, l at −30 °C (Fig. S2–S4, ESI†) revealed the presence of two sets of NMR signals. The occurrence of cis/trans amide bond rotamers in peptoids is a well-known phenomenon.11 Accordingly, we assumed that the two sets of NMR signals arise from the restricted rotation around the tertiary amide bond. Indeed, variable temperature NMR studies (VT-NMR, Fig. S5 and S6, ESI†) using 2f and 2i as representative examples confirmed the presence of two rotamers. A careful inspection of the 1H NMR spectra revealed a strongly upfield-shifted position of the glycine methylene resonance signals in the case of the major conformers. This upfield shift of the major species might be attributed to shielding by the aryl ring (R2), which is only possible for the cis rotamer indicating a cis amide bond preference.13 This was further supported by single crystal X-ray crystallography. Diffraction quality crystals of 2f were obtained by vapor diffusion (see ESI†). The X-ray crystal structure of 2f confirmed the cis amide bond geometry in the solid state (Fig. S7 and S8, ESI†).
A docking study with HDAC2, HDAC4, and HDAC6 was performed to rationalize the selectivity profiles of 2f and 2i. 2f and 2i show the highest selectivity for binding to HDAC6 over HDAC2 and HDAC4 (Table 2) but are less selective with respect to HDAC11. Hence, docking to HDAC11 was omitted as the differences to HDAC6 are expected to be less pronounced. Due to the presence of cis/trans rotamers, it is important to consider both amide bond geometries when investigating the binding modes of 2f and 2i. Thus, both the cis- and trans-rotamers were docked into crystal structures of HDAC2 and HDAC4 and, due to the lack of a crystal structure, homology models of HDAC6 CDII using AutoDock3 in combination with DrugScore14 as successfully applied previously.5d Class IIa HDACs possess a highly conserved histidine residue in the catalytic domain that is rotated away from the binding site.15 In class I isoforms, this histidine is replaced by a tyrosine accommodating an “inward” orientation. Since the conformation of the corresponding tyrosine in HDAC6 is unknown, we generated homology models of HDAC6 CDII with Y301 (numbering based on the homology model) flipped-in and -out and subjected them to all-atom molecular dynamics (MD) simulations of 1 μs length (see ESI†). After clustering the generated protein conformations, the cluster-representative of the largest cluster was chosen for molecular docking. Comparing the docking results between the respective HDAC structures, a clear trend is apparent. Regardless of which compound and rotamer was docked, the docking energy of the ligands in HDAC6, considering the Y301 flipped-in structure, was on average 1.67 kcal mol−1 more favorable than in other HDAC isoforms (Table S2, ESI†), in qualitative agreement with the exhibited isoform selectivity (Table 2). As a reason, in the wider channel rim of HDAC6 lined with hydrophobic amino acids, the hydrophobic R1 groups of 2f and 2i become more buried than in the other isoforms (see Fig. 2B–DversusFig. 2A). In the Y301 flipped-in structure of HDAC6, hydrogen bonding between Y301 and the hydroxamic acid moiety of 2f and 2i occurs (Fig. 2B–D), leading to more favorable docking energies than in the flipped-out structure (Fig. S9 and Table S2, ESI†). 2f and 2i are not able to bind to the HDAC4 isoform, which is also reflected in the docking where only <5% of the docking poses chelated the zinc ion irrespective of the rotamer. Thus, no converged docking solution could be identified in HDAC4. Comparing the two HDACi docked to HDAC6 with Y301 flipped-in, 2i shows on average a by 1 kcal mol−1 more favorable docking energy than 2f, in semi-quantitative agreement with observed inhibition activities (Table S2, ESI†). First, the channel rim can better accommodate the two methyl groups when attached to the aniline nitrogen of 2i (Fig. 2B) than when attached in meta positions to the phenyl ring of 2f (Fig. 2D). Second, as a result, the hydrophobic groups of 2f are less buried than those of 2i, resulting in a lower energetic contribution. The trans-rotamer of 2i (Fig. 2A and B) has on average 1 kcal mol−1 more favorable docking energies than its cis-rotamer (Fig. 2C), as it can form π-stacking interactions with F139 (Fig. 2B). 2f however does not show an energy difference regarding its two rotamers, in agreement with a less buried binding pose. In summary, the predicted binding modes are in line with and provide structural explanations for the observed selectivity profiles and inhibition activities.
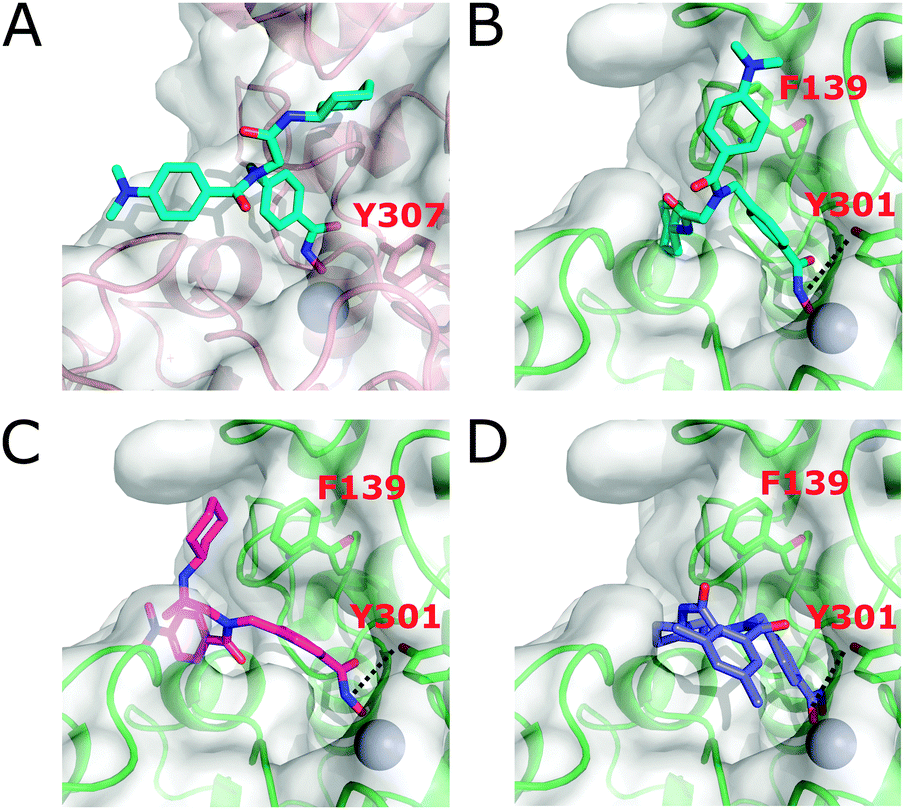 | ||
| Fig. 2 Docking poses of 2i as a trans-rotamer (A and B) and cis-rotamer (C), and 2f (D), to HDAC2 (A) and HDAC6 with Y301 flipped-in (B–D). Zinc is shown as a sphere. | ||
To further study their anticancer properties, compounds 2e, f, i, l were selected for subsequent drug combination experiments. HDACi might find broad utility as epigenetic primers resensitizing tumours to a primary agent after the tumour has developed resistance.16 Thus, we tested compounds 2e, f, i, l in combination with cisplatin using the cell lines Cal27 and the platinum-resistant subline Cal27 CisR. The cells were pretreated with the respective HDACi at a fixed concentration of 1 μM for 48 h followed by a treatment with cisplatin for 72 h. All four compounds markedly enhanced the cisplatin-induced cytotoxicity towards Cal27 and Cal27 CisR (Table 3). The strongest chemosensitizing effect was observed for 2i showing better shift factors compared with the reference compound tubastatin A. Strikingly, the combination of 2i and cisplatin led to a complete resensitization of the platinum-resistant Cal27 CisR cell line towards cisplatin (shift factor (SF): 5.57) with an IC50 of cisplatin similar to the IC50 in the native cell line Cal27. Thus, 2i possesses remarkable chemosensitizing properties leading to full reversal of cisplatin resistance in Cal27 CisR.
| Compound | Cell line | |||
|---|---|---|---|---|
| Cal27 | Cal27 CisR | |||
| IC50 | SF | IC50 | SF | |
| a Values are the mean of three experiments each performed in triplicate. The standard deviations are <10% of the mean. | ||||
| Cisplatin | 3.69 | — | 15.7 | — |
| Cisplatin + 2e | 1.28 | 2.88 | 4.09 | 3.84 |
| Cisplatin + 2f | 2.17 | 1.70 | 7.72 | 2.03 |
| Cisplatin + 2i | 0.80 | 4.61 | 2.82 | 5.57 |
| Cisplatin + 2l | 1.15 | 3.21 | 4.40 | 3.57 |
| Cisplatin + tub A | 2.96 | 1.24 | 6.16 | 2.54 |
In conclusion, we have developed a highly efficient multicomponent approach for the diversity-oriented synthesis of peptoid-based HDACi. Our data demonstrate that the peptoid-based HDACi of type 2 are a new class of potent and selective HDAC6 inhibitors with remarkable activity against a panel of cancer cells of different chemosensitivity and tissue origin. Most notably, compound 2i enhanced cisplatin-induced cytotoxicity and completely resensitized the platinum-resistant Cal27 CisR cell line towards cisplatin. Taken together, compound 2i represents a valuable lead structure for future efforts towards developing novel HDACi with optimized anticancer properties.
The Deutsche Forschungsgemeinschaft (DFG) is acknowledged for funds used to purchase the UHR-TOF maXis 4G, Bruker Daltonics, Bremen HRMS instrument used in this research. FKH acknowledges financial support from the Fonds der Chemischen Industrie (FCI).
Notes and references
- M. Biel, V. Wascholowski and A. Giannis, Angew. Chem., Int. Ed., 2005, 44, 3186 CrossRef CAS PubMed.
- J. E. Bradner, N. West, M. L. Grachan, E. F. Greenberg, S. J. Haggarty, T. Warnow and R. Mazitschek, Nat. Chem. Biol., 2010, 6, 238 CrossRef CAS.
- O. Witt, H. E. Deubzer, T. Milde and I. Oehme, Cancer Lett., 2009, 277, 8 CrossRef CAS PubMed.
- P. Bertrand, Eur. J. Med. Chem., 2010, 45, 2095 CrossRef CAS PubMed.
- (a) K. Falkenberg and R. Johnstone, Nat. Rev. Drug Discovery, 2014, 13, 673 CrossRef CAS PubMed; (b) J. H. Kalin and J. A. Bergman, J. Med. Chem., 2013, 56, 6297 CrossRef CAS PubMed; (c) F. K. Hansen, T. S. Skinner-Adams, S. Duffy, L. Marek, S. D. M. Sumanadasa, K. Kuna, J. Held, V. M. Avery, K. T. Andrews and T. Kurz, ChemMedChem, 2014, 9, 665 CrossRef CAS PubMed; (d) L. Marek, A. Hamacher, F. K. Hansen, K. Kuna, H. Gohlke, M. U. Kassack and T. Kurz, J. Med. Chem., 2013, 56, 427 CrossRef CAS; (e) R. De Vreese, T. Verhaeghe, T. Desmet and M. D'hooghe, Chem. Commun., 2013, 49, 3775 RSC.
- J.-H. Lee, A. Mahendran, Y. Yao, L. Ngo, G. Vente-Perez, M. L. Choy, N. Kim, W.-S. Ham, R. Breslow and P. A. Marks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15704 CrossRef CAS PubMed.
- H. Zou, Y. Wu, M. Navre and B. C. Sang, Biochem. Biophys. Res. Commun., 2006, 341, 45 CrossRef CAS PubMed.
- (a) K. V. Butler, J. Kalin, C. Brochier, G. Vistoli, B. L. Alan and P. Kozikowski, J. Am. Chem. Soc., 2010, 132, 10842 CrossRef CAS PubMed; (b) J. A. Bergman, K. Woan, P. Perez-Villarroel, A. Villagra, E. M. Sotomayor and A. P. Kozikowski, J. Med. Chem., 2012, 55, 9891 CrossRef CAS PubMed.
- P. Kahnberg, A. J. Lucke, M. P. Glenn, G. M. Boyle, J. D. A. Tyndall, P. G. Parsons and D. P. Fairlie, J. Med. Chem., 2006, 49, 7611 CrossRef CAS PubMed.
- P. K. Gupta, R. C. Reid, L. Liu, A. J. Lucke, S. A. Broomfield, M. R. Andrews, M. J. Sweet and D. P. Fairlie, Bioorg. Med. Chem. Lett., 2010, 20, 7067 CrossRef CAS PubMed.
- B. Yoo and K. Kirshenbaum, Curr. Opin. Chem. Biol., 2008, 12, 714 CrossRef CAS PubMed.
- A. A. Grolla, V. Podestá, M. G. Chini, S. Di Micco, A. Vallario, A. A. Genazzani, P. L. Canonico, G. Bifulco, G. C. Tron, G. Sorba and T. Pirali, J. Med. Chem., 2009, 52, 2776 CrossRef CAS PubMed.
- D. Marcovici-Mizrahi, H. E. Gottlieb, V. Marks and A. Nudelman, J. Org. Chem., 1996, 61, 8402 CrossRef CAS.
- C. A. Sotriffer, H. Gohlke and H. Klebe, J. Med. Chem., 2002, 45, 1967 CrossRef CAS PubMed.
- J. Melesina, D. Robaa, R. J. Pierce, C. Romier and W. Sippl, J. Mol. Graphics Modell., 2015, 62, 342 CrossRef CAS PubMed.
- M. Guha, Nat. Rev. Drug Discovery, 2015, 14, 225 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details (synthetic protocols, bioassays, X-ray crystallography, computational methods), characterization data of new compounds, and copies of NMR spectra. CCDC 1434499. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc10301k |
| This journal is © The Royal Society of Chemistry 2016 |