
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTriggering autocatalytic reaction by host–guest interactions†
Volodymyr
Sashuk
*,
Helena
Butkiewicz
,
Marcin
Fiałkowski
and
Oksana
Danylyuk
*
Institute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: vsashuk@ichf.edu.pl; odanylyuk@ichf.edu.pl
First published on 16th February 2016
Abstract
The acceleration of a sequential reaction through electrostatic alteration of substrate basicity within a supramolecular host is demonstrated. In the presence of the host, the reaction, which is autocatalytic, starts much sooner and exhibits substrate size selectivity.
Remote substrate activation is a distinctive trait of enzyme catalysis.1 Being nested in the enzyme pocket, the substrate is exposed to an electrostatic environment created by the protein residues. This entails modifying the acid–base properties (pKa) of the bound substrate, and in consequence, facilitates its hydrolysis. In some cases, the magnitude of the apparent pKa shift even reaches up to five units.2 It has long been endeavored to reproduce similar (biomimetic) action in synthetic hosts that are reminiscent of enzyme cavities.3–16 However, this has proved very challenging, as it requires a host that would meet several criteria, especially the capability of stabilizing the charged transition state. Raymond and co-workers have recently presented the first successful demonstration of electrostatic catalysis based on ion–ion interactions by using a tetrahedral metal cluster which consists of a hydrophobic cavity and negatively charged vertices.17–24 It was found that the complex alters the pKa of the encapsulated guest to the extent that it enables acid hydrolysis at basic pH.17,21 Further research, in particular by the Nau group, has revealed that a similar electrostatic action via ion–dipole interactions is also possible in cucurbit[n]uril (CB) hosts, which are a family of barrel-shaped macrocycles25–38 composed of a hydrophobic cavity and an electronegative carbonyl-fringed rim. Like the metal cluster, the CB host was shown to promote hydrolysis reactions via Coulombic stabilization of the protonated substrate.39–41 Most recently, the stabilization of positively charged species in transition state through ion–π interactions has also been suggested for resorcin[4]arene capsules.42–46 Being intrigued by these results, we pursued the studies to broaden the scope of electrostatic catalysis towards multistep chemical transformations.
Herein, we report a sequential azo coupling-type reaction promoted by electrostatic CB host–guest interactions. Azo coupling is an important chemical process used in industry for the production of azo dyes.47 Apart from the textile-dyeing industry, azo compounds are also used as pH indicators and molecular photoswitches.48,49 The coupling reaction usually occurs between an aromatic compound and diazonium salt, which has to be generated in situ due to high instability. In this respect, triazene is a robust substitute for the diazonium salt.50 We found that triazene reacts with an electron-rich arene in the presence of cucurbit[6]uril (CB6) affording an azo dye. Since the background reaction is negligible in the timescale of the catalytic event, we may talk here about triggering the reaction.
In our study, we employed berenil (S1), a commercially available triazene which is an anti-infective drug (Fig. 1a). Resorcinol (S2) served as the aromatic partner in the reaction. In a typical experiment, a stoichiometric mixture of S1, S2, and CB6 were suspended by agitation in heavy water and kept at room temperature. The experiment was carried out at near neutral pD without buffering. During the experiment, the pD of the suspension changed from 7.4 to 4.2 and the azo dye product (P3) precipitated as an orange solid. The process was monitored by 1H NMR spectroscopy following the formation of 4-aminobenzamidine (P1), which is another product of the reaction. According to the NMR data, the reaction was complete within two weeks, whereas only traces (<1 mol%) of P1 were detected in the control experiment performed without CB6. The azo dye product P3 was isolated from the CB6 precipitate by addition of calcium chloride followed by filtration and recrystallization from hydrochloric acid solution. X-ray diffraction of the isolated orange crystals, along with NMR and MS analysis, confirmed the expected structure of the azo dye (Fig. 1c). The azo product P3 was also crystallized as the inclusion complex with CB6 upon the acidification of the crude reaction mixture (Fig. 1d).
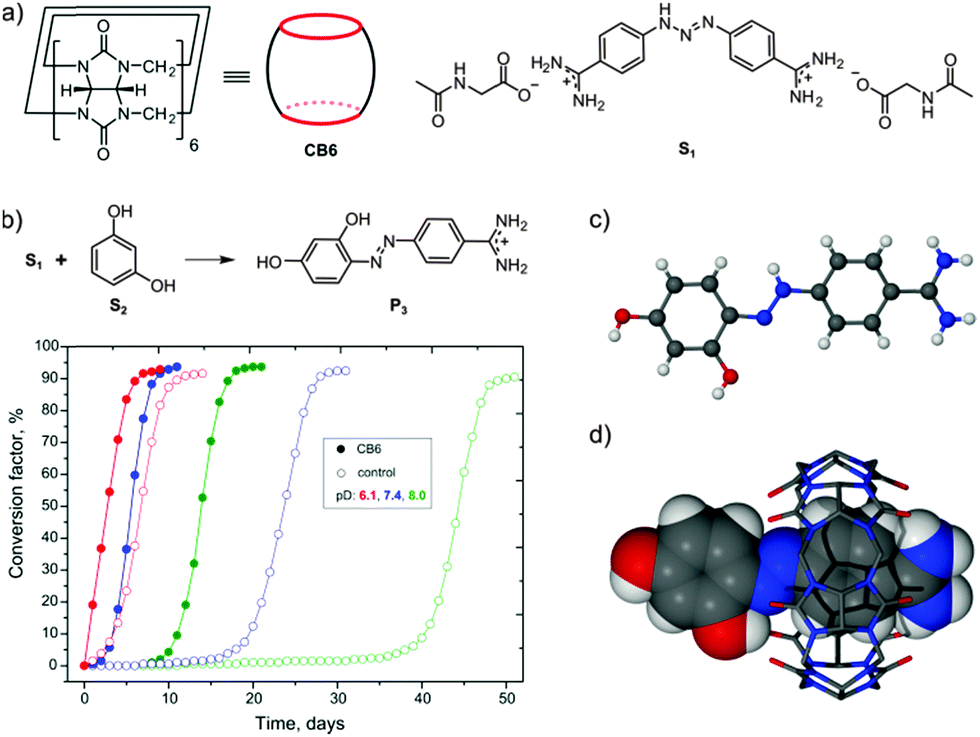 | ||
| Fig. 1 (a) Chemical structure of cucurbit[6]uril (CB6) and berenil (S1); (b) reaction scheme and plots of the time dependence of the conversion factor for azo coupling between S1 and resorcinol (S2) in the presence and absence of CB6 for different values of pD; (c) X-ray structure of azo dye product P3; (d) X-ray structure of inclusion complex between CB6 and P3. | ||
To monitor the progress of the reaction, we calculated the quantity γ = [P1]/([S2] + [P1]) × 100%. In the following, we refer to the quantity γ as the conversion factor. The time dependence of the conversion factor for the coupling reaction at different pD values is plotted in Fig. 1b. The coupling reaction is pH-sensitive, exhibiting autocatalytic behavior in nearly neutral solutions. The involvement of CB6 in the process is manifested by a shorter induction period as well as a steeper curve during the acceleration phase when compared with the control experiment. The most spectacular effect of the action of CB6 was observed in basic solution, with the lag phase being reduced by one month.
The plausible mechanism of the reaction is depicted in Fig. 2. In the first step, the triazene molecule S1 is encapsulated by the CB host. The encapsulation is facilitated by the template effect of the positively charged amidinium terminus, which interacts electrostatically with the electronegative CB rim. The formation of the host–guest species is clearly seen in the NMR spectrum after mixing the reactants (Fig. S1, ESI†). The proton resonances of the aromatic ring placed inside the CB cavity are shifted upfield, while the signals of the aromatic protons positioned outside the cavity move toward the downfield region. Low-temperature NMR measurements of the S1–CB6 mixture revealed that the observed shifts are attributed to the formation of two host–guest CB6 complexes (S1–CB6) comprising “frozen” tautomeric forms of the thermodynamically stable trans isomer51 of S1 (Fig. 3 and Fig. S6–S8, ESI†). The weak differentiation of the resonance structures of S1 upon the encapsulation indicates that either the NH group of the triazene moiety in both tautomers is in close proximity to the carbonyl portals of the CB rim or the hydrogen bonding is not a dominant force in the binding event, which would be consistent with previous reports.28 Hence, it is not clear which tautomer is a reaction intermediate. A possible reaction scenario involving the major tautomer is shown in Fig. 2. The enhanced electron density at the CB rim increases the basicity of the encapsulated substrate, rendering the triazene moiety susceptible to protonation and subsequent rupture with formation of 4-amidinophenyldiazonium (P2). Despite high CB6 loading, the cleavage reaction was triggered by catalytic amounts of the S1–CB6 complex (ca. 15 mol%) formed at pre-equilibrium. Importantly, the employment of sub-stoichiometric amounts of CB6 (20 mol%) did not affect the reaction rate significantly as the initial amount of the inclusion complex formed was about 5 mol% (Fig. S8, ESI†). As the reaction progresses, the vast fraction of the S1–CB6 complex is quickly consumed and not further replenished by virtue of competitive binding of P1 to CB6. In the next step, the diazonium cation P2 reacts with S2via electrophilic substitution to give azo compound pre-P3. The substitution reaction is favored by alkaline pH, though also remains feasible under acidic conditions.52 With the acidification of the reaction mixture, caused by the hydrolysis of pre-P3 to insoluble P3, the hydrolysis of S1, and consequently the overall reaction, accelerates. Although the S1–CB6 complex was not detected by NMR at this stage, the hydrolysis of S1, as judged from the kinetic slopes, does take place inside the macrocycle cavity. The plot showing the pD of the reaction mixture as a function of time is shown in Fig. S10 (ESI†). The product P3 precipitates probably due to intramolecular hydrogen bonding between the azo bond and nearby hydroxyl group.52 Of note, under strongly acidic conditions the hydrogen bond vanishes at the expense of protonation of the azo dye bond (Fig. 1c).
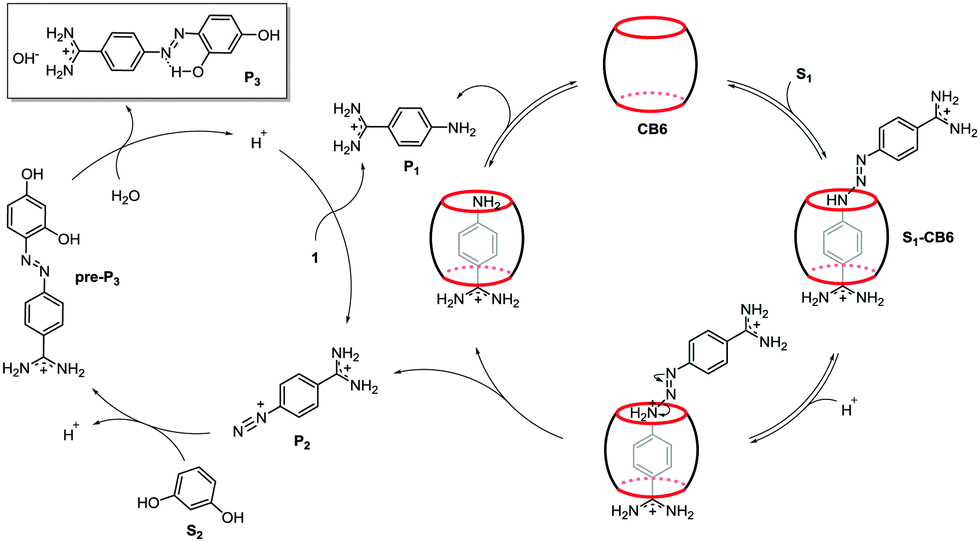 | ||
| Fig. 2 Plausible mechanism of CB6-catalyzed sequential azo coupling-type reaction. The first step of the reaction is the acid hydrolysis of S1, promoted by electrostatic action of CB6. In the next step, the generated diazonium intermediate P2 reacts with S2 to yield azo compound pre-P3. The compound pre-P3 is next transformed to final azo product P3 with loss of a proton, which acidifies the reaction mixture, accelerating the hydrolysis of S1, and consequently the overall reaction. | ||
 | ||
| Fig. 3 Partial 1H NMR spectrum of equimolar mixture of S1 and CB6 in D2O, 277 K. The tautomeric ratio was calculated by the integration of the corresponding peaks. | ||
To verify the proposed mechanism, we modeled the CB6-catalyzed sequential reaction using the following set of two rate equations:
 | (i) |
 | (ii) |
The first equation is the hydrolysis of S1. The second reaction describes the azo coupling step. The acid specific hydrolysis rate constant, kH, in eqn (i) had the form kH = kn,c1[D+], where the superscripts n and c refer, respectively, to the neutral (control) and CB6-catalyzed reaction. The constant term in k1 was set equal to zero to account for the fact that in all experimental conditions the induction periods (the lag phases) were observed. The eqn (i) and (ii) were solved numerically and the conversion factor, γ, was calculated as a function of time for pD = 8.0, 7.4, and 6.1. The rate constants kn1, kc1, and k2 were determined by least-square fitting to the experimental values of γ (see ESI† for details). Numerical analysis of the rate equations showed the increase of both rate constants kn1 and kc1 with the decrease of pD. This tendency is in line with results reported recently for the hydrolysis of S1.53 As expected, we also observed a substantial decrease of the rate constant k2 with increasing acidity of the system. To quantify the catalytic effect of CB6, for each value of pD we calculated the acceleration factor defined as the ratio kc1/kn1. Based on the results of the numerical analysis of the rate constants, we obtained the acceleration factors 3.24, 5.47, and 5.12 for pD equal to 8.0, 7.4, and 6.1, respectively.
When S2 was substituted by phenol (S3), the coupling reaction significantly slowed down (Fig. 4a). Since the resulting azo product (P4, the X-ray structure is shown in Fig. 4b) lacks the intramolecular hydrogen bonding, the equilibrium does not shift towards its hydrolysis. As a result, the pD of the reaction mixture changed from 7.2 to 6.4, and no autocatalysis was observed. The slow reaction rate surprisingly also stems from the competitive binding of S3, which displaces encapsulated S1 from the CB cavity. It is noteworthy that this kind of competition was not observed for S2, suggesting the size selectivity of the CB6 ring. During the first day of the experiment, the content of S1–CB6 complex in the solution dropped below 0.5 mol%. The process is supposedly precipitation-driven considering the higher affinity of cationic guests toward the CB host. The precipitation of phenol–CB6 (S3–CB6) complex can be delayed by addition of calcium chloride. In this case, the process of inclusion was observed through upfield shifts of the phenolic NMR signals followed by slow crystallization of S3–CB6, whose structure was further corroborated by single crystal X-ray analysis (Fig. 4c).
 | ||
| Fig. 4 (a) Reaction scheme and plots of the time dependence of the conversion factor for azo coupling between S1 and phenol (S3) in the presence and absence of CB6; (b) X-ray structure of azo dye product P4; (c) X-ray structure of inclusion complex between S3 and CB6. | ||
To unequivocally prove the catalytic properties of CB6, the coupling reaction between S1 and S2 was conducted with a stoichiometric admixture of S3. As expected, the reaction was greatly inhibited due to competitive binding of S3 for the CB cavity, which confirms nicely the action of CB6 as a catalyst (Fig. 5). The reaction inhibition was also observed in the presence of 1,5-pentanediamine (cadaverine), which forms a strong complex with CB6 (Fig. S10, ESI†).
 | ||
| Fig. 5 (a) Plots of the time dependence of the conversion factor for CB6-catalyzed azo coupling between S1 and S2 in the presence and absence of S3; (b) schematic representation of competitive binding of S3 to CB6 in the reaction between S1 and S2 in the presence of S3. | ||
In summary, we designed a two-step azo coupling-type reaction induced by host–guest interactions. A peculiar feature of the reaction is that the first step is acid-catalyzed, whereas the next step prefers non-acidic conditions. Normally, if the reaction is performed in acidic medium, the fast hydrolysis step is followed by slow electrophilic substitution, and vice versa, the reaction is extremely sluggish in neutral and alkaline solutions. The use of the CB6 host as a catalyst facilitates the acid hydrolysis, and consequently the overall reaction at basic pH through the electrostatic enhancement of substrate basicity. Accordingly, the reaction, being autocatalytic, begins much earlier, i.e., is triggered by CB6. The presented concept is of great importance for the development of chemical transformations involving discrete reaction steps occurring at different pH values. Furthermore, this strategy, given the substrate size selectivity, may be applicable for performing acid-catalyzed reactions on selected functional groups in the presence of other functionalities sensitive to acidic conditions.
The project was supported by the Polish Ministry of Science and Higher Education (grant Iuventus Plus Nr IP2012 008272).
Notes and references
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- F. H. Westheimer, Tetrahedron, 1995, 51, 3–20 CrossRef CAS.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1734–1787 RSC.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615–621 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418–3438 CrossRef CAS PubMed.
- S. H. A. M. Leenders, R. Gramage-Doria, B. de Bruin and J. N. H. Reek, Chem. Soc. Rev., 2015, 44, 433–448 RSC.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247–262 RSC.
- C. J. Walter and J. K. M. Sanders, Angew. Chem., Int. Ed. Engl., 1995, 34, 217–219 CrossRef CAS.
- J. Kang and J. Rebek, Nature, 1997, 385, 50–52 CrossRef CAS PubMed.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251–254 CrossRef CAS PubMed.
- S. R. Shenoy, F. R. Pinacho Crisóstomo, T. Iwasawa and J. Rebek, J. Am. Chem. Soc., 2008, 130, 5658–5659 CrossRef CAS PubMed.
- P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed.
- E. Anslyn and R. Breslow, J. Am. Chem. Soc., 1989, 111, 5972–5973 CrossRef CAS.
- F. Ortega-Caballero, C. Rousseau, B. Christensen, T. E. Petersen and M. Bols, J. Am. Chem. Soc., 2005, 127, 3238–3239 CrossRef CAS PubMed.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- A. G. Salles, S. Zarra, R. M. Turner and J. R. Nitschke, J. Am. Chem. Soc., 2013, 135, 19143–19146 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, Science, 2007, 316, 85–88 CrossRef CAS PubMed.
- W. M. Hart-Cooper, K. N. Clary, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2012, 134, 17873–17876 CrossRef CAS PubMed.
- C. J. Hastings, M. P. Backlund, R. G. Bergman and K. N. Raymond, Angew. Chem., Int. Ed., 2011, 50, 10570–10573 CrossRef CAS PubMed.
- C. J. Hastings, M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2010, 132, 6938–6940 CrossRef CAS PubMed.
- M. D. Pluth, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2008, 130, 11423–11429 CrossRef CAS PubMed.
- C. Zhao, F. D. Toste, K. N. Raymond and R. G. Bergman, J. Am. Chem. Soc., 2014, 136, 14409–14412 CrossRef CAS PubMed.
- C. Zhao, Q.-F. Sun, W. M. Hart-Cooper, A. G. DiPasquale, F. D. Toste, R. G. Bergman and K. N. Raymond, J. Am. Chem. Soc., 2013, 135, 18802–18805 CrossRef CAS PubMed.
- Z. J. Wang, K. N. Clary, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Chem., 2013, 5, 100–103 CrossRef CAS PubMed.
- J. W. Lee, S. Samal, N. Selvapalam, H.-J. Kim and K. Kim, Acc. Chem. Res., 2003, 36, 621–630 CrossRef CAS PubMed.
- J. Lagona, P. Mukhopadhyay, S. Chakrabarti and L. Isaacs, Angew. Chem., Int. Ed., 2005, 44, 4844–4870 CrossRef CAS PubMed.
- B. C. Pemberton, R. Raghunathan, S. Volla and J. Sivaguru, Chem. – Eur. J., 2012, 18, 12178–12190 CrossRef CAS PubMed.
- K. I. Assaf and W. M. Nau, Chem. Soc. Rev., 2015, 44, 394–418 RSC.
- A. L. Koner, C. Márquez, M. H. Dickman and W. M. Nau, Angew. Chem., Int. Ed., 2011, 50, 545–548 CrossRef CAS PubMed.
- X. Lu and E. Masson, Org. Lett., 2010, 12, 2310–2313 CrossRef CAS PubMed.
- H. Cong, Z. Tao, S. F. Xue and Q. J. Zhu, Curr. Org. Chem., 2011, 15, 86–95 CrossRef CAS.
- L. Zheng, S. Sonzini, M. Ambarwati, E. Rosta, O. A. Scherman and A. Herrmann, Angew. Chem., Int. Ed., 2015, 54, 13007–13011 CrossRef CAS PubMed.
- W. L. Mock, T. A. Irra, J. P. Wepsiec and M. Adhya, J. Org. Chem., 1989, 54, 5302–5308 CrossRef CAS.
- S. Y. Jon, Y. H. Ko, S. H. Park, H.-J. Kim and K. Kim, Chem. Commun., 2001, 1938–1939 RSC.
- B. C. Pemberton, N. Barooah, D. K. Srivatsava and J. Sivaguru, Chem. Commun., 2010, 46, 225–227 RSC.
- B. C. Pemberton, R. K. Singh, A. C. Johnson, S. Jockusch, J. P. Da Silva, A. Ugrinov, N. J. Turro, D. K. Srivastava and J. Sivaguru, Chem. Commun., 2011, 47, 6323–6325 RSC.
- C. Yang, T. Mori, Y. Origane, Y. H. Ko, N. Selvapalam, K. Kim and Y. Inoue, J. Am. Chem. Soc., 2008, 130, 8574–8575 CrossRef CAS PubMed.
- S. J. Barrow, S. Kasera, M. J. Rowland, J. del Barrio and O. A. Scherman, Chem. Rev., 2015, 115, 12320–12406 CrossRef CAS PubMed.
- N. i. Saleh, A. L. Koner and W. M. Nau, Angew. Chem., Int. Ed., 2008, 47, 5398–5401 CrossRef CAS PubMed.
- C. Klöck, R. N. Dsouza and W. M. Nau, Org. Lett., 2009, 11, 2595–2598 CrossRef PubMed.
- N. Basilio, L. García-Río, J. A. Moreira and M. Pessêgo, J. Org. Chem., 2010, 75, 848–855 CrossRef CAS PubMed.
- A. Cavarzan, A. Scarso, P. Sgarbossa, G. Strukul and J. N. H. Reek, J. Am. Chem. Soc., 2011, 133, 2848–2851 CrossRef CAS PubMed.
- G. Bianchini, G. L. Sorella, N. Canever, A. Scarso and G. Strukul, Chem. Commun., 2013, 49, 5322–5324 RSC.
- Q. Zhang and K. Tiefenbacher, J. Am. Chem. Soc., 2013, 135, 16213–16219 CrossRef CAS PubMed.
- L. Catti and K. Tiefenbacher, Chem. Commun., 2015, 51, 892–894 RSC.
- Q. Zhang and K. Tiefenbacher, Nat. Chem., 2015, 7, 197–202 CrossRef CAS PubMed.
- K. Hunger, P. Mischke and W. Rieper, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, 2000 Search PubMed.
- A. A. Beharry and G. A. Woolley, Chem. Soc. Rev., 2011, 40, 4422–4437 RSC.
- R. Klajn, Pure Appl. Chem., 2010, 82, 2247–2279 CrossRef.
- S. Bräse, Acc. Chem. Res., 2004, 37, 805–816 CrossRef PubMed.
- M. Barra and N. Chen, J. Org. Chem., 2000, 65, 5739–5744 CrossRef CAS PubMed.
- O. Macháčková, V. Štěrba and K. Valter, Collect. Czech. Chem. Commun., 1972, 37, 1851–1860 CrossRef.
- M. Campbell, R. J. Prankerd, A. S. Davie and W. N. Charman, J. Pharm. Pharmacol., 2004, 56, 1327–1332 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures, spectroscopic data, kinetic model. CCDC 1440778–1440781. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc10063a |
| This journal is © The Royal Society of Chemistry 2016 |