
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Functionalization of P4 in the coordination sphere of coinage metal cations†
Jaap E.
Borger
a,
Martijn S.
Bakker
a,
Andreas W.
Ehlers
a,
Martin
Lutz
b,
J.
Chris Slootweg
a and
Koop
Lammertsma
*ac
aDepartment of Chemistry and Pharmaceutical Sciences Vrije Universiteit Amsterdam, De Boelelaan 1083, 1081 HV Amsterdam, The Netherlands. E-mail: K.Lammertsma@vu.nl
bCrystal and Structural Chemistry, Bijvoet Center for Biomolecular Research, Utrecht University, Padualaan 8, 3584 CH Utrecht, The Netherlands
cDepartment of Chemistry, University of Johannesburg, Auckland Park, Johannesburg, 2006 South Africa
First published on 4th January 2016
Abstract
Selective functionalization of white phosphorus is achieved by addition of ArLi to unique cationic coinage metal η2–P4 complexes. This novel approach allows controlled P–C bond formation using the bulky DmpLi (Dmp = 2,6-Mes2C6H3) and the unencumbered MesLi, giving sterically diverse doubly complexed RP4 butterfly derivatives in a single step.
Controlling direct P–C bond formation using P4 as starting material is of interest in avoiding chlorinated intermediates, such as PCl3, for the production of organophosphorus compounds. Yet this task is extremely challenging due to the highly reactive nature of the P4 tetrahedron.1 Currently, several selective methods have been developed, like the use of ambiphilic carbenes pioneered by the group of Bertrand,2 and the metal-mediated radical functionalization of P4 reported by Scheer et al. (A; R = CpR, Scheme 1)3 as well as by Cummins and co-workers (R = Dmp),4 who also demonstrated facile P-functionalization chemistry by embedding photochemically generated P2 fragments into organic frameworks (B).5 In contrast, conventional methods for the formation of P–C bonds,6 such as the use of organolithium and Grignard reagents, have been less fruitful due to the low selectivity and complex product distributions associated with their reactions with P4.7 An intriguing exception was recently described by Hill, who achieved selective activation of P4 using a β-diketiminato organomagnesium compound, producing the [nBu2P4]2− dianion C,8 which is related to the thallium tetraphosphabutadienediide [Ar2P4]2− salt D reported by Power et al.9 We showed that the reactivity of bulky ArLi reagents toward P4 can be controlled in the presence of Lewis acids (B(C6F5)3 and BPh3), giving the LA-stabilized bicyclo[1.1.0]tetraphosphabutanides [ArP4·LA]−E that can subsequently be functionalized selectively generating the neutral disubstituted bicyclic phosphanes ArP4R (type A) and the doubly coordinated tetraphosphides [ArP4·(LA)2]−F.10 Key in this approach is the irreversible formation of the transient phosphide [RP4]− that is directly trapped by the Lewis acid. Note that P4 does not form an adduct with BPh3 or even B(C6F5)3,10 and therefore requires the use of sterically encumbered FLP-type ArLi/LA combinations to avoid quenching. In this work, we present an alternative strategy by using novel cationic coinage metal based Lewis adducts of P4 as synthon that now tolerate varied bulk on the ArLi reagents, as demonstrated by the selective addition of Dmp (Dmp = 2,6-dimesitylphenyl) and mesityl lithium, resulting in the formation of unique doubly complexed RP4 butterfly cations.
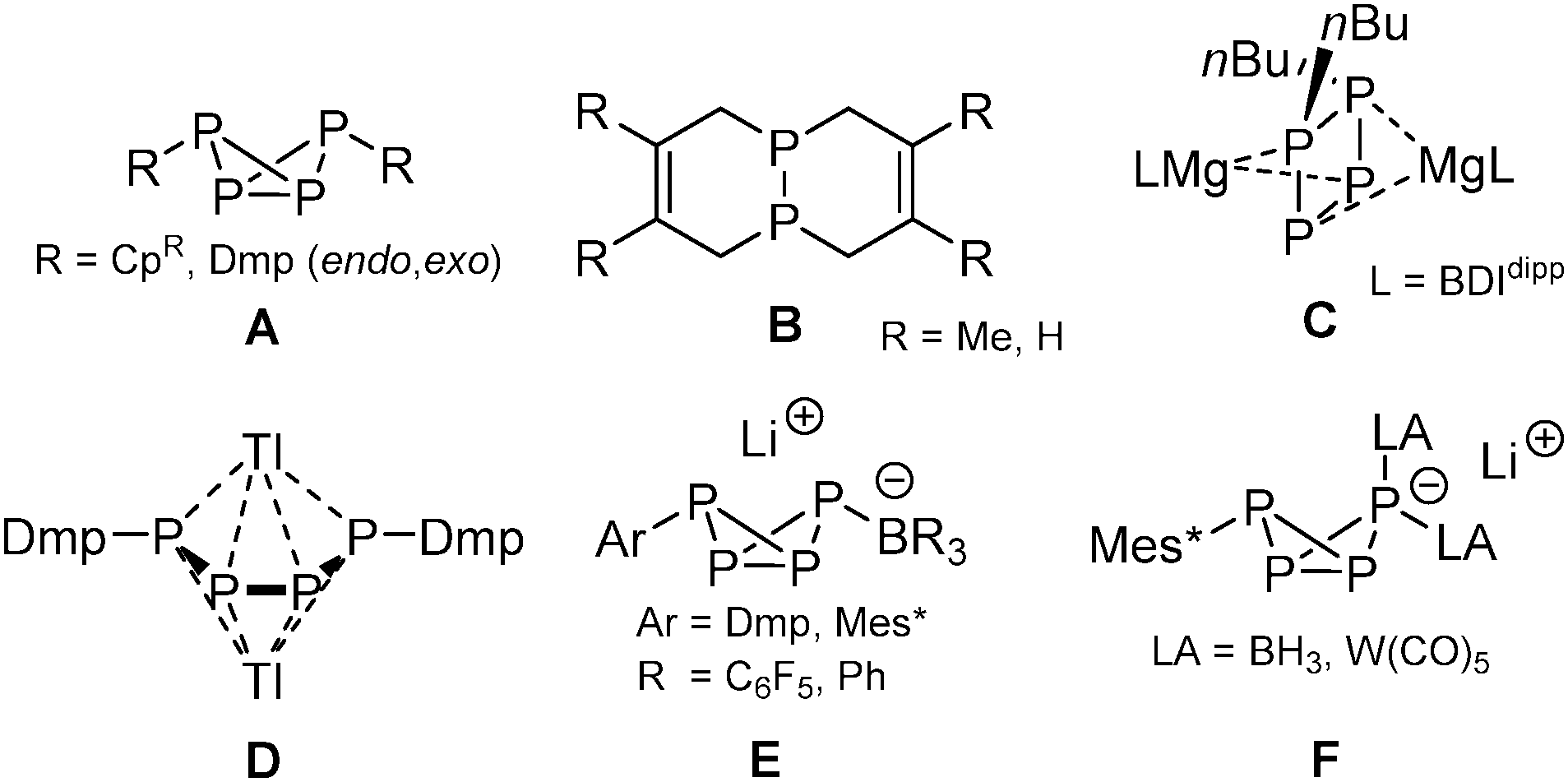 | ||
| Scheme 1 Methods allowing selective direct P–C bond formation using P4. Dmp = 2,6-dimesitylphenyl; BDIdipp = HC{C(Me)N(2,6-iPr2C6H3)}2; Mes* = 2,4,6-tBu3C6H2; CpR = CpBIG, Cp′′′, Cp*, Cp4iPr. | ||
Commercially available IPrMCl (M = Cu, Au; IPr = 1,3-bis(diisopropylphenyl)imidazol-2-ylidene) in combination with Li+ [Al(pftb)4]− (pftb = perfluoro-tert-butoxy)11,12 as chloride scavenger were found to be suitable starting materials allowing the isolation of readily available LA–P4 adducts. The complexation of P4 was achieved by dropwise addition of a solution of IPrMCl (1 equiv.; M = Cu, Au) in DCM to a suspension of white phosphorus (1.1 equiv.) and Li[Al(pftb)4] (1 equiv.) in DCM at 0 °C (Scheme 2), which resulted in a sharp downfield shifted singlet in the 31P{1H} NMR spectrum in the case of Cu(I) (−483.1 ppm), and a lower field and broadened singlet for Au(I) (−464.4 ppm), indicating both P4 tetrahedra to be coordinated dynamically to the cationic metal centers (free P4 in CD2Cl2: −522.0 ppm). The dynamics were confirmed by VT NMR spectroscopy at −90 °C,13 revealing broadening of the 31P signal for Cu–P4 complex 1a, and two broad triplets for Au–P4 analogue 1b (δ31P: −453.3 and −462.1 ppm, 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 ratio; 1JP,P = −209.8 Hz). Both novel complexes were isolated as white powders in 92% (1a) and 87% (1b) yield, respectively, and are unique examples of heteroleptic cationic P4 coinage metal complexes, complementing the homoleptic series [M(η2–P4)2]+ reported by Krossing14a–d (M = Ag, Cu) and Slattery et al.14e (M = Au), and the neutral copper complex [NacnacCu(η2–P4)] isolated by Scheer and coworkers.14f
:2 ratio; 1JP,P = −209.8 Hz). Both novel complexes were isolated as white powders in 92% (1a) and 87% (1b) yield, respectively, and are unique examples of heteroleptic cationic P4 coinage metal complexes, complementing the homoleptic series [M(η2–P4)2]+ reported by Krossing14a–d (M = Ag, Cu) and Slattery et al.14e (M = Au), and the neutral copper complex [NacnacCu(η2–P4)] isolated by Scheer and coworkers.14f
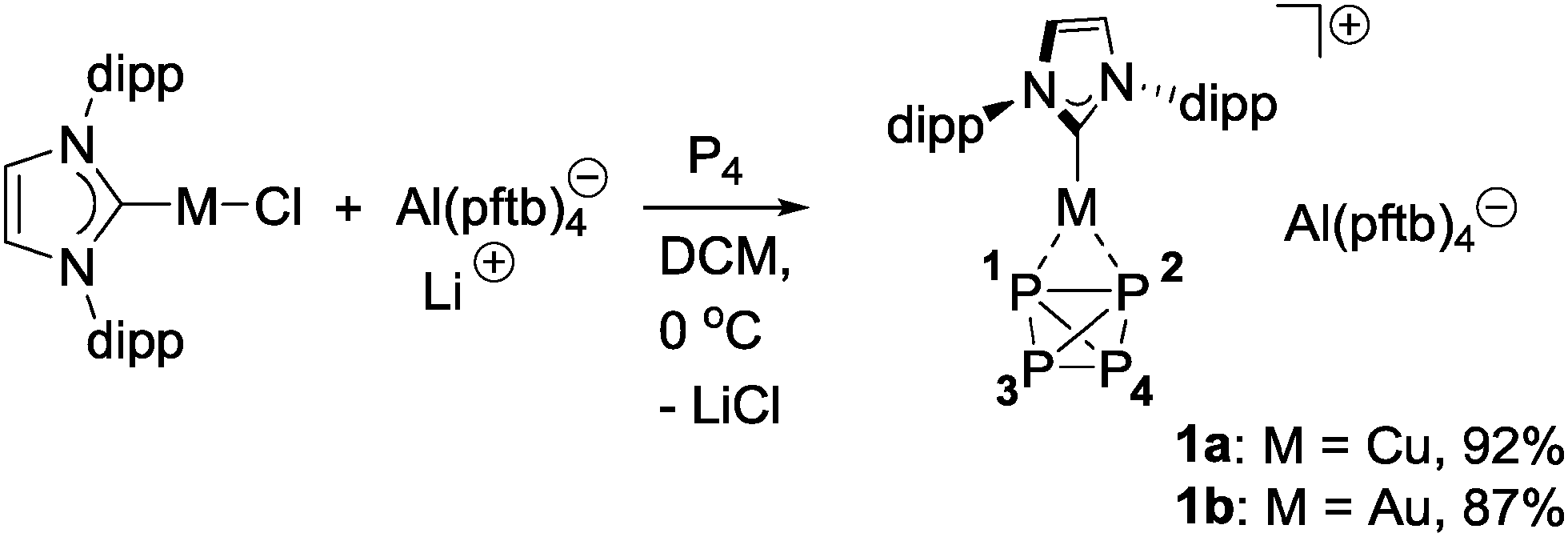 | ||
| Scheme 2 Synthesis of cationic η2–P4 complexes of copper and gold (pftb = OC(CF3)3; dipp = 2,6-diisopropylphenyl). | ||
The A2B2 spin-system of gold(I) complex 1b observed at low temperature by 31P NMR spectroscopy is indicative of η2–P4 coordination, which was confirmed by a single-crystal X-ray analysis (Fig. 1)15 that showed nearly equal Au1–P1 (2.4043(17) Å) and Au1–P2 distances (2.4286(19) Å), a distorted trigonal planar Au center with a short Au1–C1 bond (2.037(5) Å), and an acute P1–Au1–P2 angle (57.79(7)°). A comparison of the P–P bonds in “free” P4 (2.1994(3) Å, determined by gas-phase electron diffraction16) with those in 1b+ shows a contraction of the P3–P4 bond (2.148(3) Å), as well as shortened P1/P2–P3/P4 bonds (2.155(3)–2.167(4) Å), but an elongated P1–P2 bond (2.335(3) Å) due to coordination to gold, albeit less pronounced than the one found in [Au(η2–P4)2][GaCl4] (i.e. 2.410(1) Å14e).
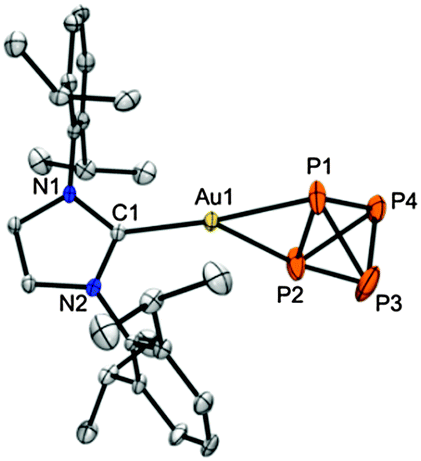 | ||
| Fig. 1 Molecular structure of 1b+ in the crystal15 (ellipsoids are set at 50% probability; [Al(OC(CF3)3)4]− counter-ion and CH2Cl2 solvent molecule omitted). Selected bond lengths [Å] and angles [°]: P1–P2 (2.335(3)), P3–P4 (2.148(3)), P1–P3/P4 (2.167(4)/2.164(3)), P2–P3/P4 (2.156(4)/2.155(3)), Au1–P1/P2 (2.4043(17)/2.4286(19)), C1–Au1 (2.037(5)); C1–Au1–P1 (156.75(14)), C1–Au1–P2 (141.92(14)), P1–Au1–P2 (57.79(7)). | ||
To analyze the bonding situation of 1 in more detail, we resorted to AIM analyses17,18 on the gas-phase optimized structures of 1a+ and 1b+,19 which revealed bond critical points (BCP) between P1 and P2 (ρ = 0.079 a.u. (ε = 1.10) in 1a+ and 0.074 a.u. (ε = 0.93) in 1b+) with only a slightly lower electron density compared to that computed for the naked P4 (ρ = 0.105 a.u.; ε = 0.10),13 confirming the coordinating P4 fragments to remain intact, disfavoring oxidative addition by P–P bond cleavage. Interestingly, examination of the Laplacian of the electron densities (∇2ρ) in the P1–P2 BCPs indicated a stronger P4–M+ interaction in gold complex 1b+ (0.056 a.u.) than in Cu derivative 1a+ (0.033 a.u.), which is in agreement with the observed 31P{1H} NMR shifts (−483.1 vs. −464.4 ppm for 1a and 1b, respectively). ETS-NOCV20 analyses of the M+–P4 bonds concur with these observations,18 revealing indeed a higher bonding energy for the Au complex (ΔΔE = 1.2 kcal mol−1), with the most prominent difference found for the orbital interactions, showing larger contributions for σ donation (1b+ −36.7; 1a+ −25.9 kcal mol−1) and concurrent π back-donation (1b+ −21.4; 1a+ −20.7 kcal mol−1), attributable to the influence of relativistic effects on the valence shell of Au(I).21,22
This difference in bonding energy is also reflected in the stability of 1avs.1b. Namely, dissolving 1a in toluene directly led to complete displacement of P4 at room temperature, whereas 1b is indefinitely stable under those conditions,23 rendering Au complex 1b a suitable building block for the functionalization of P4. As proof of concept, we first selected the bulky DmpLi to react with 1b, which proved successful in the synthesis of the LA-stabilized Li+ [DmpP4·B(C6F5)3]−.10a Hence, a solution of DmpLi (1 equiv.) in toluene was slowly added to a solution of 1b (1 equiv.) in toluene at −78 °C, revealing an AMX2 spin system in the 31P{1H} NMR spectrum (−105.5 (P1), −118.7 (P4) and −327.9 (P2, P3) ppm in a 1:1:2 ratio, respectively), indicative for a non-symmetrically substituted P4 butterfly.10,25 Interestingly, 1H NMR analysis revealed the presence of two NHC moieties instead of only one needed for the anticipated neutral DmpP4AuIPr 2 (Scheme 3), which suggests the formation of a doubly coordinated RP4 complex.
 | ||
| Scheme 3 Functionalization of P4 by reaction of ArLi with 1b, with the proposed intermediate 2 in brackets (pftb = OC(CF3)3; dipp = 2,6-diisopropylphenyl). | ||
Indeed, X-ray crystal structure determination of colorless crystals obtained by layering a DCM solution with n-pentane, displayed the non-symmetrical [DmpP4·(AuIPr)2][Al(pftb)4] 3 (Fig. 2) featuring a unique bimetallic gold fragment, with similar P4–Au1/Au2 distances (2.2924(7)/2.2860(7) Å) and a Au1–P4–Au2 angle of 128.02(3)°, which is larger than found in the triaurated cation [RP(AuPPh3)3]+ (av. 106°),24 likely due to the steric repulsion between the large NHC ligands. The P4–P2/P3 bonds (2.1919(10)/2.2077(10) Å) are slightly contracted compared to the P1–P2/P3 bonds (2.2140(10)/2.2240(11) Å), and are similar in length to the bridgehead P2–P3 bond (2.1992(11) Å). These structural parameters are akin to those reported for the cationic [Mes*2P4Cl]+ of Schulz et al.25a as well as to those of the bis-LA complexed anions [Mes*P4·(LA)2]− (LA = BH3, W(CO)5) reported by us.10b Intriguingly, the bicyclic P4 entity in 3+ is sterically highly shielded, as illustrated by a space-filling model (Fig. 2, right), reminiscent of the incorporation of white phosphorus in the self-assembled [Fe4L6]8+ container reported by Nitschke and co-workers.26
 | ||
| Fig. 2 Left: Molecular structure of 3+ in the crystal15 (ellipsoids are set at 50% probability; [Al(OC(CF3)3)4]− counter-ion and disordered solvent molecules omitted). Selected bond lengths [Å], angles and torsion angle [°]: P1–P2/P3 (2.2140(10)/2.2240(11)), P4–P2/P3 (2.1919(10)/2.2077(10)), P2–P3 (2.1992(11)), Au1–P4 (2.2924(7)), Au2–P4 (2.2860(7)), C13–P1 (1.865(3)); Au1–P4–Au2 (128.02(3)); P1–P2–P3–P4 (100.98(4)). Right: Space-filling model of 3+. | ||
The formation of 3 could be optimized by using two equivalents of 1b, which allowed its isolation in 67% yield. Bis-gold complex 3 is likely formed via neutral exo,exo-ArP4AuIPr 2 (Scheme 3) that displaces a P4 molecule from a second equivalent of gold complex 1b, which was computed to be energetically favorable by −43.1 kcal mol−1,27 and acts as a monodentate ligand (via P4) for [IPrAu]+, displaying reactivity analogous to the recent coordination of bicyclic Mes*2P4 to GaCl325b shown by Schulz et al., and of [{Cp′′′Fe(CO)2}2(μ,η1:1-P4)] toward [Cu(MeCN)]+ presented by the group of Scheer.28
Next, we assessed the reactivity of 1b toward the less encumbered nucleophile MesLi,29 which was not feasible in our original approach (E, Scheme 1)10 as combining MesLi with P4 in the presence of BPh3 exclusively produces Li+ [MesBPh3]−.13 Gratifyingly, formation of the bicyclic tetraphosphane [MesP4·(AuIPr)2][Al(pftb)4] (4) proceeded readily upon mixing MesLi and 1b (2 equiv.) in toluene at −78 °C, showing a distinct set of three 31P{1H} resonances at −110.6 (P1), −119.9 (P4) and −314.5 (P2, P3) ppm (1:1:2 ratio), and an additional signal for free P4. The product could be isolated in 62% yield, and was confirmed to contain only one mesityl unit by mass spectrometry (ESI) and 1H NMR spectroscopy, and two flanking IPrAu moieties.13 In contrast to related Aryl2P4 species, which feature either bulky 2,4,6-tBu3C6H3 (Mes*)7c,10,25a,b or terphenyl4,9,10a groups, 4 is the first example of a mesityl-substituted P4 butterfly, which illustrates the merit of this novel P4-functionalization strategy in controlling direct P–C bond formation using organolithium reagents.
In summary, addition of Dmp or mesityl lithium to the coinage metal based P4–LA adduct 1b gives the unique bimetallic ArP4-butterfly cations 3 and 4. This novel approach allows for varied bulk on the organosubstituents in a single controlled step, showing facile functionalization of P4. Currently, we are defining the scope of this new methodology and are exploring the application of 1 in new P4 transformations.
Notes and references
- For reviews, see: (a) N. A. Giffin and J. D. Masuda, Coord. Chem. Rev., 2011, 255, 1342 CrossRef CAS; (b) B. M. Cossairt, N. A. Piro and C. C. Cummins, Chem. Rev., 2010, 110, 4164 CrossRef CAS PubMed; (c) M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. Rev., 2010, 110, 4178 CrossRef CAS PubMed; (d) M. Scheer, G. Balázs and A. Seitz, Chem. Rev., 2010, 110, 4236 CrossRef CAS PubMed; (e) M. Peruzzini, L. Gonsalvi and A. Romerosa, Chem. Soc. Rev., 2005, 34, 1038 RSC.
- (a) J. D. Masuda, W. W. Schoeller, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 7052 CrossRef CAS PubMed; (b) O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS PubMed; (c) C. D. Martin, C. M. Weinstein, C. E. Moore, A. L. Rheingold and G. Bertrand, Chem. Commun., 2013, 49, 4486 RSC; (d) C. L. Dorsey, B. M. Squires and T. W. Hudnall, Angew. Chem., Int. Ed., 2013, 52, 4462 CrossRef CAS PubMed.
- S. Heinl, S. Reisinger, C. Schwarzmaier, M. Bodensteiner and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 7639 CrossRef CAS PubMed.
- B. M. Cossairt and C. C. Cummins, New J. Chem., 2010, 34, 1533 RSC.
- D. Tofan and C. C. Cummins, Angew. Chem., Int. Ed., 2010, 49, 7516 CrossRef CAS PubMed.
- D. E. C. Corbridge, Phosphorus, Elsevier, Amsterdam, 2000 Search PubMed.
- (a) M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 471 CrossRef CAS; (b) M. M. Rauhut and A. M. Semsel, J. Org. Chem., 1963, 28, 473 CrossRef CAS; (c) R. Riedel, H.-D. Hausen and E. Fluck, Angew. Chem., Int. Ed., 1985, 24, 1056 CrossRef.
- M. Arrowsmith, M. S. Hill, A. L. Johnson, G. Kociok-Köhn and M. F. Mahon, Angew. Chem., Int. Ed., 2015, 54, 7882 CrossRef CAS PubMed.
- A. R. Fox, R. J. Wright, E. Rivard and P. P. Power, Angew. Chem., Int. Ed., 2005, 117, 7907 CrossRef.
- (a) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2014, 53, 12836 CrossRef CAS PubMed; (b) J. E. Borger, A. W. Ehlers, M. Lutz, J. C. Slootweg and K. Lammertsma, Angew. Chem., Int. Ed., 2016, 55, 613 CrossRef CAS.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490 CrossRef CAS.
- I. Krossing and I. Raabe, Angew. Chem., Int. Ed., 2004, 43, 2066 CrossRef CAS PubMed.
- See the ESI,† for further details.
- (a) I. Krossing, J. Am. Chem. Soc., 2001, 123, 4603 CrossRef CAS PubMed; (b) I. Krossing and L. van Wüllen, Chem. – Eur. J., 2002, 8, 700 CrossRef CAS PubMed; (c) A. Bihlmeier. M. Gonsior, I. Raabe, N. Trapp and I. Krossing, Chem. – Eur. J., 2004, 10, 5041 CrossRef PubMed; (d) G. Santiso-Quiñones, A. Reisinger, J. Slattery and I. Krossing, Chem. Commun., 2007, 5046 RSC; (e) L. C. Forfar, T. J. Clark, M. Green, S. M. Mansell, C. A. Russell, R. A. Sanguramath and J. M. Slattery, Chem. Commun., 2012, 48, 1970 RSC; (f) F. Spitzer, M. Sierka, M. Latronico, P. Mastrorilli, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 4392 CrossRef CAS PubMed.
- CCDC 1440355 (compound 1b) and 1440356 (compound 3) contain the supplementary crystallographic data for this paper.
- B. M. Cossairt, C. C. Cummins, A. R. Head, D. L. Lichtenberger, R. J. F. Berger, S. A. Hayes, N. M. Mitzel and G. Wu, J. Am. Chem. Soc., 2010, 132, 8459 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in Molecules, Claredon Press, Oxford, 1994 Search PubMed.
- The AIM and ETS-NOCV analysis was performed at ZORA-BP86-D3/TZ2P using ADF2013.01; see the ESI,† for details.
- Geometry optimizations were performed at ωB97X-D/6-31+G(2d,p) (LanL2DZ for Cu and Au); J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC ; see the ESI,† for details.
- M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS PubMed.
- This trend was also found in a computational study on homoleptic P4 complexes of group 11 cations: H.-C. Tai, I. Krossing, M. Seth and D. V. Deubel, Organometallics, 2004, 23, 2343 CrossRef CAS.
- (a) P. Pyykkö, Chem. Rev., 1988, 88, 563 CrossRef; (b) P. Pyykkö, Angew. Chem., Int. Ed., 2004, 43, 4412 CrossRef PubMed.
- Direct dissociation was observed for both complexes in Et2O, THF and MTBE.
- H. Schmidbaur, G. Weidenhiller and O. Steigelmann, Angew. Chem., Int. Ed., 1991, 30, 433 CrossRef.
- Selected examples: (a) J. Bresien, K. Faust, A. Schulz and A. Villinger, Angew. Chem., Int. Ed., 2015, 54, 6926 CrossRef CAS PubMed; (b) J. Bresien, K. Faust, C. Hering-Junghans, J. Rothe, A. Schulz and A. Villinger, Dalton Trans., 2016 10.1039/C5DT02757H; (c) D. Holschumacher, T. Bannenberg, K. Ibrom, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2010, 39, 10590 RSC; (d) M. B. Power and A. R. Barron, Angew. Chem., Int. Ed., 1991, 30, 1353 CrossRef.
- P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS PubMed.
- To reduce the computation time, all aryl substituents were replaced by phenyl groups. DFT calculations were performed at ωB97X-D/6-311+G(2d,p)//6-31G(d)(LanL2DZ for Au) using Gaussian09 (Revision D.01); see the ESI,† for details.
- C. Schwarzmaier, S. Heinl, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2015, 54, 13116 CrossRef CAS PubMed.
- Lerner et al. isolated [Li3][Mes3P4] by reaction of P4 with 3 equiv. of MesLi: A. Hübner, T. Bernert, I. Sänger, E. Alig, M. Bolte, L. Fink, M. Wagner and H.-W. Lerner, Dalton Trans., 2010, 39, 7528 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1440355 and 1440356. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc10037b |
| This journal is © The Royal Society of Chemistry 2016 |