DOI:
10.1039/C5CC09991A
(Communication)
Chem. Commun., 2016,
52, 4836-4839
Ca(BH4)2–Mg2NiH4: on the pathway to a Ca(BH4)2 system with a reversible hydrogen cycle
Received
4th December 2015
, Accepted 4th March 2016
First published on 4th March 2016
Abstract
The Ca(BH4)2–Mg2NiH4 system presented here is, to the best of our knowledge, the first described Ca(BH4)2-based hydride composite that reversibly transfers boron from the Ca-based compound(s) to the reaction partner. The ternary boride MgNi2.5B2 is formed upon dehydrogenation and the formation of Ca(BH4)2 upon rehydrogenation is confirmed.
Due to their high gravimetric H2 density, metal borohydrides1 are promising candidates for solid state hydrogen storage. However, high thermal stabilities as well as poor reversibility of their decomposition products are the major obstacles to be overcome for technical applications. One of the most promising approaches to solve the reversibility issue of borohydrides is to combine them with selected materials to create reversible reactive compositions with lowered reaction enthalpy, the so-called reactive hydride composites (RHCs).2,3 The system that has attracted the most attention in recent years is LiBH4–MgH2.4 Because of its reduced desorption enthalpy compared to LiBH4, mixtures of hydrides based on Ca(BH4)2 were also investigated intensively. Unfortunately, all systems presented so far have suffered from sluggish hydrogen reaction kinetics and poor reversibility. One reason for the latter finding is the tendency of Ca(BH4)2 to form kinetically stable side products upon dehydrogenation that hinder a complete rehydrogenation. In the case of pure Ca(BH4)2, several desorption paths are proposed in the literature:| | | Ca(BH4)2 → CaH2 + 2B + 3H25–7 | (1) |
| | | Ca(BH4)2 → 2/3CaH2 + 1/3CaB6 + 10/3H25,6,8–10 | (2) |
| | | Ca(BH4)2 → 1/6CaB12H12 + 5/6CaH2 + 13/6H29–11 | (3) |
| | | Ca(BH4)2 → CaB2H6 + H2 (intermediate reaction).6,10,11 | (4) |
In fact, recent reports have indicated that the decomposition of Ca(BH4)2 may follow more than one single reaction path.6,12 In these studies, the respective decomposition products and their ratio partially depend on the applied hydrogen back-pressure as well as the dehydrogenation temperature.6,12
Also for the hydride composite Ca(BH4)2–MgH2,8,13–16 several possible decomposition pathways are reported in the literature:
| | | Ca(BH4)2 + MgH2 → CaH2 + MgB2 + 4H28 | (5) |
| | | Ca(BH4)2 + MgH2 → 2/3CaH2 + 1/3CaB6 + Mg + 13/3H28,13 | (6) |
| | | Ca(BH4)2 + MgH2 → 1/6CaB12H12 + 5/6CaH2 + Mg + 19/6H2.13 | (7) |
For this system, Bonatto-Minella
et al. demonstrated a reversibility of approximately 55% between subsequent cycles.
15 The authors rule out the possibility that the hydride composite desorbs according to
eqn (5). Instead, the formation of CaB
6 was confirmed to be the cause of the partial reversibility (
eqn (6)), which was also reported earlier by Kim
et al.8,16 Bonatto-Minella
et al. suggested that Mg promotes a better reversibility of the composite compared to pure Ca(BH
4)
2 by acting as a nucleation agent for CaB
6 due to the low
d-value mismatch {111}
CaB6/{1011}
Mg of only 0.6%.
15 Consequently, this mixture cannot be considered as a typical RHC since the overall desorption enthalpy is not lowered by the formation of reaction products between Ca(BH
4)
2 (including its decomposition products) and Mg/MgH
2. The drop in reversible capacity is attributed to the formation of CaB
12H
12 which has too high kinetic barriers to reversibly form Ca(BH
4)
2 under moderate temperature and hydrogen pressure conditions.
13 Therefore, this kinetically stable phase acts as a boron sink upon cycling.
In order to obtain complete reversibility for Ca(BH4)2-based systems, a reaction partner has to be identified that can reversibly and effectively bind boron, hence preventing the formation of competing stable phases like CaB12H12 or amorphous boron. This compound should be lightweight and preferably store hydrogen itself. Mg2NiH4,17–20 which has a hydrogen capacity of 3.6 mass%, matches these criteria. Vajo et al. showed that a mixture of LiBH4 and Mg2NiH4 forms the ternary boride MgNi2.5B2 upon desorption which can act as boron donor to form LiBH4 again.21,22 Later Afonso et al. confirmed a similar reaction mechanism for the composite NaBH4–Mg2NiH4.23
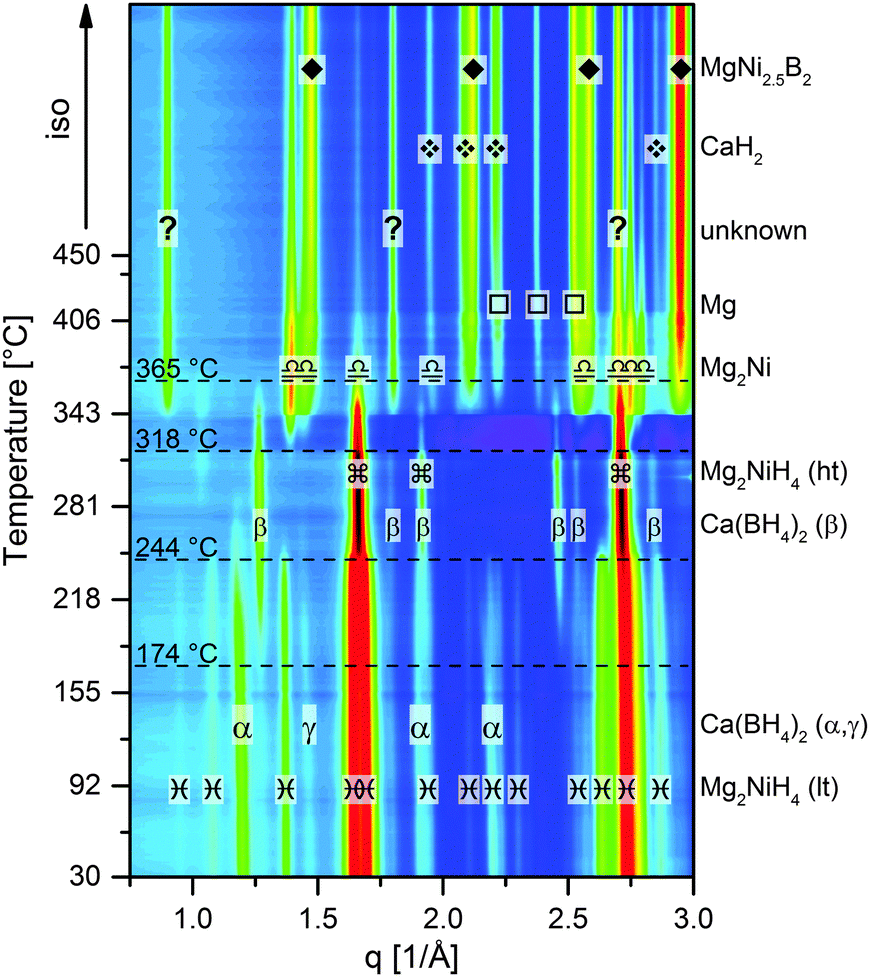
In this work, the dehydrogenation reactions of the system Ca(BH4)2–Mg2NiH4 have been investigated for the first time. Furthermore, the potential for complete rehydrogenation of the desorbed products is demonstrated. For all experiments shown in this work, an initial mixture of Ca(BH4)2 and Mg2NiH4 in a molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2.5 was prepared by 5 hours of ball milling.† To investigate the thermal dehydrogenation path, an in situ synchrotron radiation powder X-ray diffraction (SR-PXD) experiment was performed applying a temperature ramp of 5 K min−1 heating from ambient temperature to 450 °C at 1 bar H2 (Fig. 1). The room temperature diffraction pattern exhibits the diffraction peaks of the low-temperature monoclinic phase (C12/c1) of Mg2NiH4 and its high-temperature cubic phase (Fm
:2.5 was prepared by 5 hours of ball milling.† To investigate the thermal dehydrogenation path, an in situ synchrotron radiation powder X-ray diffraction (SR-PXD) experiment was performed applying a temperature ramp of 5 K min−1 heating from ambient temperature to 450 °C at 1 bar H2 (Fig. 1). The room temperature diffraction pattern exhibits the diffraction peaks of the low-temperature monoclinic phase (C12/c1) of Mg2NiH4 and its high-temperature cubic phase (Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m) (weight ratio approx. 2.3:1). Diffraction peaks of Ca(BH4)2 can be attributed to the α-(F2dd) and γ-phase (Pbca), with a weight ratio of roughly 5:1. The first event with rising temperature is the polymorphous change from α-/γ-Ca(BH4)2 to β-Ca(BH4)2 (P
m) (weight ratio approx. 2.3:1). Diffraction peaks of Ca(BH4)2 can be attributed to the α-(F2dd) and γ-phase (Pbca), with a weight ratio of roughly 5:1. The first event with rising temperature is the polymorphous change from α-/γ-Ca(BH4)2 to β-Ca(BH4)2 (P![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) ), which starts at about 174 °C. The second modification in the diffraction pattern is related to another polymorphous change: the monoclinic Mg2NiH4 converts to the cubic phase at approx. 244 °C. At about 318 °C, Mg2NiH4 starts to decompose to Mg2Ni and hydrogen. At slightly higher temperatures, the intensity of Ca(BH4)2 diffraction peaks starts to decrease, while the peaks of four other phases occur and intensify, namely MgNi2.5B2, Mg, CaH2 and an unknown phase. With further temperature increase, the peaks of Mg2Ni first grow until the maximum intensity is reached at roughly 365 °C, followed by a quick decline of peak intensity. The peaks of MgNi2.5B2, Mg and CaH2 grow during the remaining heating period and the following isothermal region at 450 °C. In contrast, the peaks of the unknown phase only grow until the highest intensity is attained at about 410 °C, then the peak intensities reduce much slower than those of Mg2Ni. Moreover, it must be emphasised that the diffraction peaks of Ca(BH4)2 vanish already at about 365 °C, that is when MgNi2.5B2 and the unknown phase, which potentially contains boron, are still growing. This finding strongly suggests the possibility of Mg2Ni reacting with the decomposition products of Ca(BH4)2, i.e. CaB6, CaBxHy and/or amorphous boron. Furthermore, the sequence of peak intensity changes for the different phases suggests that the unknown phase is a yet unreported Mg–Ni–B-phase. Since the reaction kinetics for the dehydrogenation are too sluggish to monitor the complete process by means of in situ SR-PXD, additional material was desorbed at a pressure of 1 bar H2 in a Sieverts apparatus and then characterised using ex situ laboratory Powder X–ray Diffraction (PXD). Fig. 2 shows the diffraction patterns‡ of seven different samples. All samples were heated to their respective maximum temperatures with 3 K min−1; subsequently the material was kept under isothermal conditions for different times. Diffractograms with the same colour represent samples with equal total desorption time, i.e. the isothermal time was altered in such a manner that the overall time of both, heating period and isotherm, was equal for desorptions of the same set. Diffractograms of samples with a total desorption time of 150 minutes are shown in black. Red and blue diffractograms indicate total desorption times of 6 and 42 hours, respectively. The sample heated up to 350 °C for 150 min shows the peaks of Mg2Ni and the unknown phase besides those of MgNi2.5B2, Mg and CaH2. However, compared to the in situ SR-PXD pattern at 350 °C the ratios of the individual phases are different: Mg2Ni almost vanished whereas the peaks of the unknown phase are much more pronounced than at any temperature in the in situ experiment. Hence, it can be deduced that the reaction path leading to this unknown phase is more dominant at lower temperatures. Nevertheless, looking at the PXD patterns of the material desorbed at 350 °C with longer isotherms, the peaks of the unknown phase first decline (6 h) and eventually vanish (42 h). Heating the composite to 450 °C results in the complete disappearance of the unknown phase already within the short isothermal period. Therefore, independent of temperature, this unknown phase clearly cannot be an end product but is an intermediate phase in this reaction. Thus, the overall desorption reaction can be summarised in the following chemical equation:
), which starts at about 174 °C. The second modification in the diffraction pattern is related to another polymorphous change: the monoclinic Mg2NiH4 converts to the cubic phase at approx. 244 °C. At about 318 °C, Mg2NiH4 starts to decompose to Mg2Ni and hydrogen. At slightly higher temperatures, the intensity of Ca(BH4)2 diffraction peaks starts to decrease, while the peaks of four other phases occur and intensify, namely MgNi2.5B2, Mg, CaH2 and an unknown phase. With further temperature increase, the peaks of Mg2Ni first grow until the maximum intensity is reached at roughly 365 °C, followed by a quick decline of peak intensity. The peaks of MgNi2.5B2, Mg and CaH2 grow during the remaining heating period and the following isothermal region at 450 °C. In contrast, the peaks of the unknown phase only grow until the highest intensity is attained at about 410 °C, then the peak intensities reduce much slower than those of Mg2Ni. Moreover, it must be emphasised that the diffraction peaks of Ca(BH4)2 vanish already at about 365 °C, that is when MgNi2.5B2 and the unknown phase, which potentially contains boron, are still growing. This finding strongly suggests the possibility of Mg2Ni reacting with the decomposition products of Ca(BH4)2, i.e. CaB6, CaBxHy and/or amorphous boron. Furthermore, the sequence of peak intensity changes for the different phases suggests that the unknown phase is a yet unreported Mg–Ni–B-phase. Since the reaction kinetics for the dehydrogenation are too sluggish to monitor the complete process by means of in situ SR-PXD, additional material was desorbed at a pressure of 1 bar H2 in a Sieverts apparatus and then characterised using ex situ laboratory Powder X–ray Diffraction (PXD). Fig. 2 shows the diffraction patterns‡ of seven different samples. All samples were heated to their respective maximum temperatures with 3 K min−1; subsequently the material was kept under isothermal conditions for different times. Diffractograms with the same colour represent samples with equal total desorption time, i.e. the isothermal time was altered in such a manner that the overall time of both, heating period and isotherm, was equal for desorptions of the same set. Diffractograms of samples with a total desorption time of 150 minutes are shown in black. Red and blue diffractograms indicate total desorption times of 6 and 42 hours, respectively. The sample heated up to 350 °C for 150 min shows the peaks of Mg2Ni and the unknown phase besides those of MgNi2.5B2, Mg and CaH2. However, compared to the in situ SR-PXD pattern at 350 °C the ratios of the individual phases are different: Mg2Ni almost vanished whereas the peaks of the unknown phase are much more pronounced than at any temperature in the in situ experiment. Hence, it can be deduced that the reaction path leading to this unknown phase is more dominant at lower temperatures. Nevertheless, looking at the PXD patterns of the material desorbed at 350 °C with longer isotherms, the peaks of the unknown phase first decline (6 h) and eventually vanish (42 h). Heating the composite to 450 °C results in the complete disappearance of the unknown phase already within the short isothermal period. Therefore, independent of temperature, this unknown phase clearly cannot be an end product but is an intermediate phase in this reaction. Thus, the overall desorption reaction can be summarised in the following chemical equation:
| | | Ca(BH4)2 + 2.5Mg2NiH4 → CaH2 + MgNi2.5B2 + 4Mg + 8H2. | (8) |
 |
| | Fig. 1
In situ SR-PXD analysis of Ca(BH4)2–Mg2NiH4, the sample was heated at 5 K min−1 from RT to 450 °C at 1 bar H2. | |
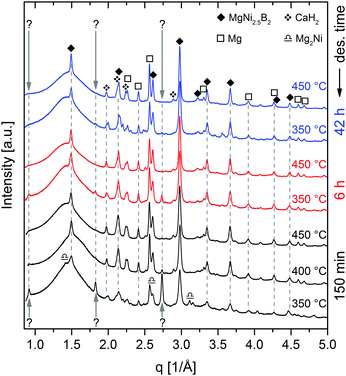 |
| | Fig. 2
Ex situ PXD pattern of Ca(BH4)2–Mg2NiH4 desorbed at 1 bar H2 and different final temperatures and times, respectively. | |
The quantity of hydrogen released in this desorption reaction amounts to 4.6 mass%.
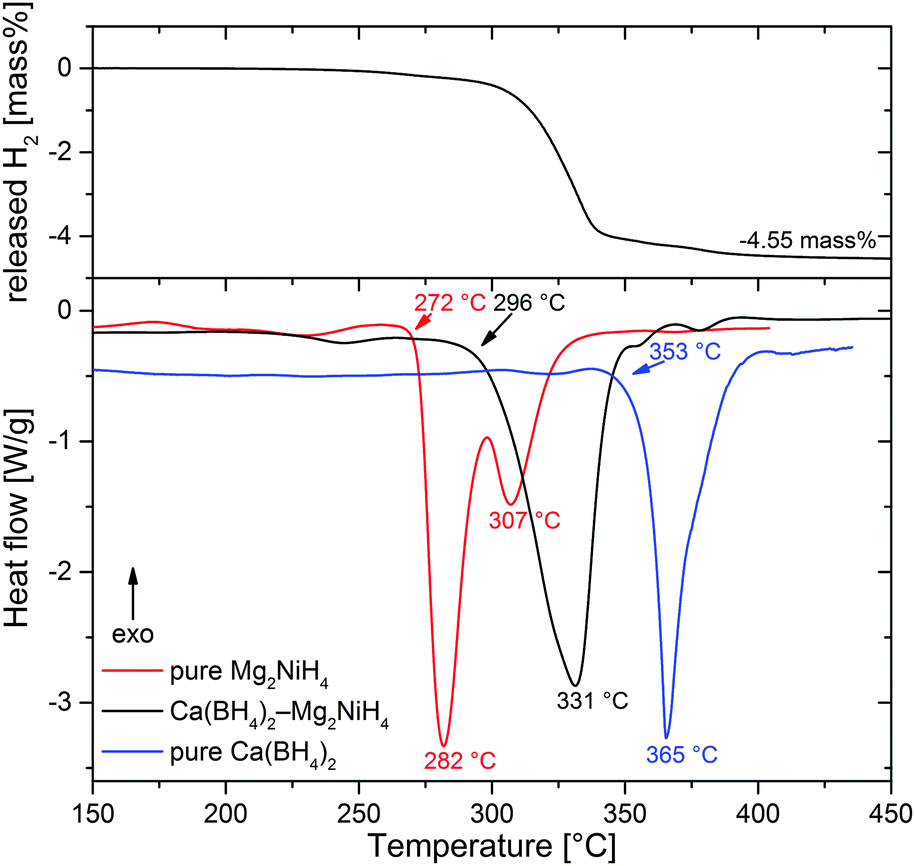
Fig. 3 presents results of Differential Scanning Calorimetry (DSC) combined with volumetric analysis for Ca(BH4)2–Mg2NiH4 in comparison to pure Mg2NiH4 and Ca(BH4)2 (all materials ball milled using the same parameters), obtained under 1 bar H2 and a heating rate of 5 K min−1. The composite shows one broad endothermic desorption peak (onset at 296 °C, maximum at 331 °C) that embraces the several sub-events. This is in agreement with the in situ SR-PXD experiment which revealed that the desorption of Mg2NiH4 triggers the described follow-up reactions and that the partial reactions overlap in time. Compared to pure Ca(BH4)2 (onset 353 °C, maximum 365 °C), desorption temperatures are shifted to lower values by about 55 K in the case of the composite. Thus, Mg2NiH4 or, more specifically, Mg2Ni clearly destabilises Ca(BH4)2. However, pure Mg2NiH4§ (onset 272 °C, first maximum 282 °C) starts to dehydrogenate at temperatures lower than those for Ca(BH4)2–Mg2NiH4. This discrepancy must be attributed to the kinetic limitations of Mg2NiH4 decomposition in the RHC with respect to pure Mg2NiH4. A possible explanation could be a reduced energy transfer to Mg2NiH4 upon ball milling due to the cushioning effect of Ca(BH4)2. As a consequence, the Mg2NiH4 within the composite would have higher activation barriers for desorption as compared to the pure Mg2NiH4. In comparison, the temperatures in the DSC experiments are roughly 20 K lower than in the in situ SR-PXD measurements. This deviation may be due to the particular experimental setup of SR-PXD experiments, where an inevitable gap between the specimen and the thermocouple may cause a temperature offset. At 4.55 mass%, the amount of released hydrogen agrees well with the theoretical capacity of the RHC of 4.6 mass%.
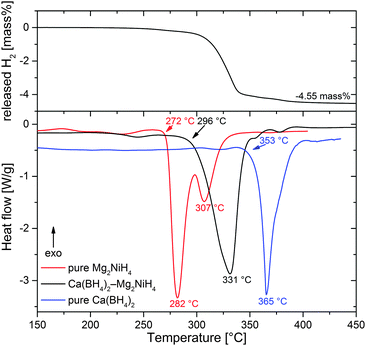 |
| | Fig. 3 Combined DSC and volumetric analyses of Ca(BH4)2–Mg2NiH4, diagrams of pure Mg2NiH4 and Ca(BH4)2 are included for reference. | |
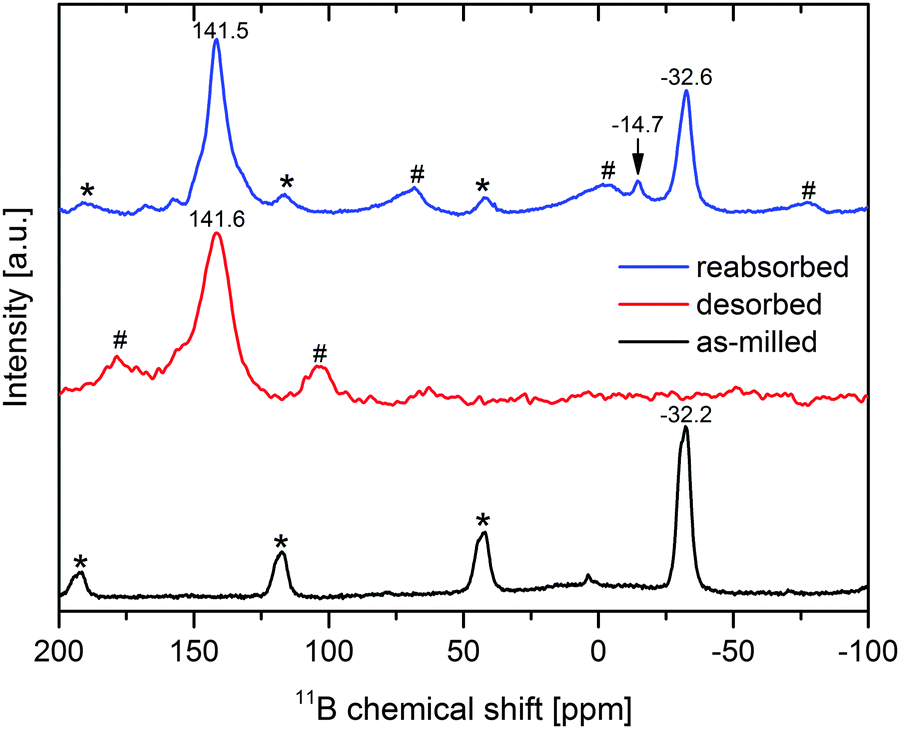
In order to investigate the possible presence of boron-containing phases that are not detectable using X-ray diffraction – CaB12H12 and B are typically nano-crystalline or amorphous7,11,13 – 11B Magic Angle Spinning Nuclear Magnetic Resonance (MAS-NMR) spectroscopy was employed. The spectra of the as-milled material and of the material desorbed for 24 h at 450 °C and dynamic H2 pressure of more than 1 bar can be found in Fig. 4 (black and red graphs). As can be seen, the initial material comprises only one boron-containing phase, that is Ca(BH4)2. More precisely, the peak at −32.2 ppm is a convolution of α- and γ-Ca(BH4)2. The desorbed sample features one peak at 141.6 ppm which is assigned to MgNi2.5B2. This result is quite remarkable since it shows that none of the typical decomposition products of Ca(BH4)2 could be detected in the desorbed state. Instead, all boron seems to be bonded in the form of MgNi2.5B2. Since the formation of the kinetically stable phases CaB12H12 and/or B is the main reason for the degradation of Ca(BH4)2-based systems, the absence of these phases may allow for full reversibility of a Ca(BH4)2-based H2 storage material. Consequently, the reabsorption potential of the Ca(BH4)2–Mg2NiH4 composite was evaluated. The decomposition products were kept in an autoclave under hydrogen with an initial pressure of 395 bar¶ and a temperature of 400 °C for 14 h. These high pressure hydrogenation conditions were chosen arbitrarily in an attempt to maximise the potential conversion of the decomposition products into the starting material. The NMR spectrum of the reabsorbed sample is shown in Fig. 4 (blue graph). There are two major resonance peaks: the first at 141.5 ppm can be assigned to MgNi2.5B2, the second, at −32.6 ppm, clearly belongs to Ca(BH4)2. Since the desorbed sample only features MgNi2.5B2 as the sole boron-containing phase, the Ca(BH4)2 formed must have received the boron from this ternary boride upon absorption. Consequently, aside from its ability to act as a boron-donor for the formation of LiBH4 and NaBH4, MgNi2.5B2 also exchanges boron reversibly with Ca(BH4)2. The 11B spectrum of the reabsorbed material also shows another small resonance peak at −14.7 ppm which cannot be assigned clearly. However, it can be assumed that it belongs to a (BxHy)z−-phase that could be an intermediate product in Ca(BH4)2 formation. The NMR spectrum indicates a converted fraction to Ca(BH4)2 of roughly one-third. However, the reabsorption did not complete under the applied conditions since there is still a high fraction of MgNi2.5B2 present in the absorbed sample. At the moment the reason for this limited reconversion is not clear yet. Since the PXD analysis of the reabsorbed sample (not presented here) only shows very small and broad Ca(BH4)2 reflexes, the Ca(BH4)2 crystallites seem to be rather small. Hence, crystallite growth appears to be restricted. Further studies will have to clarify which microstructure develops and whether the reconversion into the initial hydride is confined in some way. For instance, the reconversion may be restricted to interface areas between the desorbed reactants, and the newly formed Ca(BH4)2 may act as a diffusion barrier, leading to reduced kinetic rates and thus an incomplete rehydrogenation.
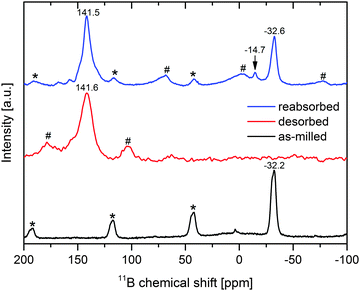 |
| | Fig. 4
11B MAS-NMR spectra of as-milled Ca(BH4)2–Mg2NiH4 (black), desorbed Ca(BH4)2–Mg2NiH4 (red) and of the reabsorbed material (blue), sidebands of Ca(BH4)2 and MgNi2.5B2 are marked with * and #, respectively. | |
In summary, the reactive hydride composite Ca(BH4)2–Mg2NiH4 features a complete boron transfer from Ca(BH4)2 to MgNi2.5B2 upon dehydrogenation. No other boron-containing phases were present among the decomposition products; this includes especially the inactive phases amorphous boron and CaB12H12. Hence, a degradation of the hydrogen storage capacity caused by these boron sinks, as observed in pure Ca(BH4)26,7,11 or Ca(BH4)2–MgH2,13–15,24 can be ruled out for this composite. Mg2NiH4 destabilises Ca(BH4)2 at a hydrogen pressure of 1 bar resulting in the lowering of desorption temperatures by about 55 K as compared to pure Ca(BH4)2. These dehydrogenation conditions might still be above the desired temperature range for technical applications. However, the absence of boron sinks must be considered as a substantial improvement of Ca(BH4)2 based RHCs. Further optimisation of the material preparation should reduce the desorption temperatures at least down to the level of Mg2NiH4. Although the system did not reabsorb entirely, it was proven that the formation of Ca(BH4)2 starting from MgNi2.5B2 as boron-donor is thermodynamically and kinetically possible. Reabsorption conditions have not been optimised yet. Hence, it is likely that rehydrogenation could also be achieved at lower temperatures and pressure. Moreover, no additives were used within this study. Therefore, further improvements of the hydrogen sorption kinetics may be feasible.
This work was supported by the Danish Council for Strategic Research via HyFillFast. Access to beam time at the synchrotron PETRA III at DESY, Hamburg, Germany is gratefully acknowledged.
Notes and references
- H.-W. Li, Y. Yan, S. Orimo, A. Züttel and C. M. Jensen, Energies, 2011, 4, 185–214 CrossRef CAS.
- M. Dornheim, S. Doppiu, G. Barkhordarian, U. Boesenberg, T. Klassen, O. Gutfleisch and R. Bormann, Scr. Mater., 2007, 56, 841–846 CrossRef CAS.
- G. Barkhordarian, T. Klassen, M. Dornheim and R. Bormann, J. Alloys Compd., 2007, 440, L18–L21 CrossRef CAS.
- U. Bösenberg, S. Doppiu, L. Mosegaard, G. Barkhordarian, N. Eigen, A. Borgschulte, T. R. Jensen, Y. Cerenius, O. Gutfleisch, T. Klassen, M. Dornheim and R. Bormann, Acta Mater., 2007, 55, 3951–3958 CrossRef.
- J. H. Kim, S. A. Jin, J. H. Shim and Y. W. Cho, J. Alloys Compd., 2008, 461, 2007–2009 CrossRef.
- Y. Yan, A. Remhof, D. Rentsch, A. Züttel, S. Giri and P. Jena, Chem. Commun., 2015, 51, 11008–11011 RSC.
- C. Bonatto Minella, S. Garroni, C. Pistidda, G. Barkhordarian, C. Rongeat, I. Lindemann, O. Gutfleisch, T. R. Jensen, Y. Cerenius, J. Christensen, M. D. Bar, T. Klassen and M. Dornheim, J. Phys. Chem. C, 2011, 115, 2497–2504 Search PubMed.
- Y. Kim, D. Reed, Y.-S. Lee, J. Y. Lee, J.-H. Shim, D. Book and Y. W. Cho, J. Phys. Chem. C, 2009, 113, 5865–5871 CAS.
- V. Ozolins, E. H. Majzoub and C. Wolverton, J. Am. Chem. Soc., 2009, 131, 230–237 CrossRef CAS PubMed.
- Y. Zhang, E. Majzoub, V. Ozoliņš and C. Wolverton, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 174107 CrossRef.
- Y. Kim, S. J. Hwang, J. H. Shim, Y. S. Lee, H. N. Han and Y. W. Cho, J. Phys. Chem. C, 2012, 116, 4330–4334 CAS.
- Y. Kim, S. Hwang, Y. Lee, J. Suh, H. N. Han and Y. W. Cho, J. Phys. Chem. C, 2012, 116, 25715–25720 CAS.
- C. Bonatto Minella, S. Garroni, D. Olid, F. Teixidor, C. Pistidda, I. Lindemann, O. Gutfleisch, M. D. Bar, R. Bormann, T. Klassen and M. Dornheim, J. Phys. Chem. C, 2011, 115, 18010–18014 CAS.
- F. Karimi, P. K. Pranzas, A. Hoell, U. Vainio, E. Welter, V. S. Raghuwanshi, C. Pistidda, M. Dornheim, T. Klassen and A. Schreyer, J. Appl. Crystallogr., 2014, 47, 67–75 CrossRef CAS.
- C. B. Minella, C. Pistidda, S. Garroni, P. Nolis, M. D. Baró, O. Gutfleisch, T. Klassen, R. Bormann and M. Dornheim, J. Phys. Chem. C, 2013, 117, 3846–3852 CAS.
- Y. Kim, D. Reed, Y.-S. Lee, J.-H. Shim, H. N. Han, D. Book and Y. W. Cho, J. Alloys Compd., 2010, 492, 597–600 CrossRef CAS.
- K. Zeng, T. Klassen, W. Oelerich and R. Bormann, J. Alloys Compd., 1999, 283, 213–224 CrossRef CAS.
- Z. Gavra, M. H. Mintz, G. Kimmel and Z. Hadari, Inorg. Chem., 1979, 18, 3595–3597 CrossRef CAS.
- T. Hirata, T. Matsumoto, M. Amano and Y. Sasaki, J. Phys. F: Met. Phys., 1981, 11, 521–529 CrossRef CAS.
- M. Polanski, T. K. Nielsen, I. Kunce, M. Norek, T. Płociński, L. R. Jaroszewicz, C. Gundlach, T. R. Jensen and J. Bystrzycki, Int. J. Hydrogen Energy, 2013, 38, 4003–4010 CrossRef CAS.
- J. J. Vajo, W. Li and P. Liu, Chem. Commun., 2010, 46, 6687–6689 RSC.
- W. Li, J. J. Vajo, R. W. Cumberland, P. Liu, S. J. Hwang, C. Kim and R. C. Bowman, J. Phys. Chem. Lett., 2010, 1, 69–72 CrossRef CAS.
- G. Afonso, A. Bonakdarpour and D. P. Wilkinson, J. Phys. Chem. C, 2013, 117, 21105–21111 CAS.
- C. Bonatto Minella, S. Garroni, C. Pistidda, M. D. Baró, O. Gutfleisch, T. Klassen and M. Dornheim, J. Alloys Compd., 2015, 622, 989–994 CrossRef CAS.
- T. Klassen, U. Herr and R. S. Averback, Acta Mater., 1997, 45, 2921–2930 CrossRef CAS.
Footnotes |
| † The milling was performed in a SPEX 8000M Mill using stainless steel milling vials and stainless steel balls with a diameter of 10 mm. A ball-to-powder ratio of 10:1 was employed. |
| ‡ The background of each diffraction pattern has a broad amorphous peak between 1 and 2 Å−1 which is caused by the PMMA sample holder used to protect the material from air. |
| § Typically, Mg2NiH4 features only one single desorption peak. The presence of the double peak is a kinetic effect. A possible explanation could be a kinetic separation between nucleation and growth of Mg2Ni25 as a consequence of the rather low activation energy for desorption and a rather homogeneous particle size distribution after 5 h of ball milling. |
| ¶ Due to absorption and a small leakage the pressure dropped to approx. 180 bar at the end of the synthesis. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  c,
T.
Klassen
ad and
M.
Dornheim
a
c,
T.
Klassen
ad and
M.
Dornheim
a