
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly diastereoselective approach to methylenecyclopropanes via boron-homologation/allylboration sequences†
A. N.
Baumann
,
A.
Music
,
K.
Karaghiosoff
and
D.
Didier
 *
*
Department of Chemistry and Pharmacy, Ludwig-Maximillians-University Munich, Butenandtstrasse 5-13, 81377 Munich, Germany. E-mail: dorian.didier@cup.uni-muenchen.de
First published on 4th January 2016
Abstract
A simple and efficient diastereoselective synthesis of methylenecyclopropanes is described, in which boron-homologation and allylboration are merged into a one-pot process, starting from in situ generated cyclopropenyllithium species. This unprecedented methodology opens a new route to strained alkylidenecycloalkanes containing a quaternary stereocenter, in high yields and excellent diastereomeric ratios.
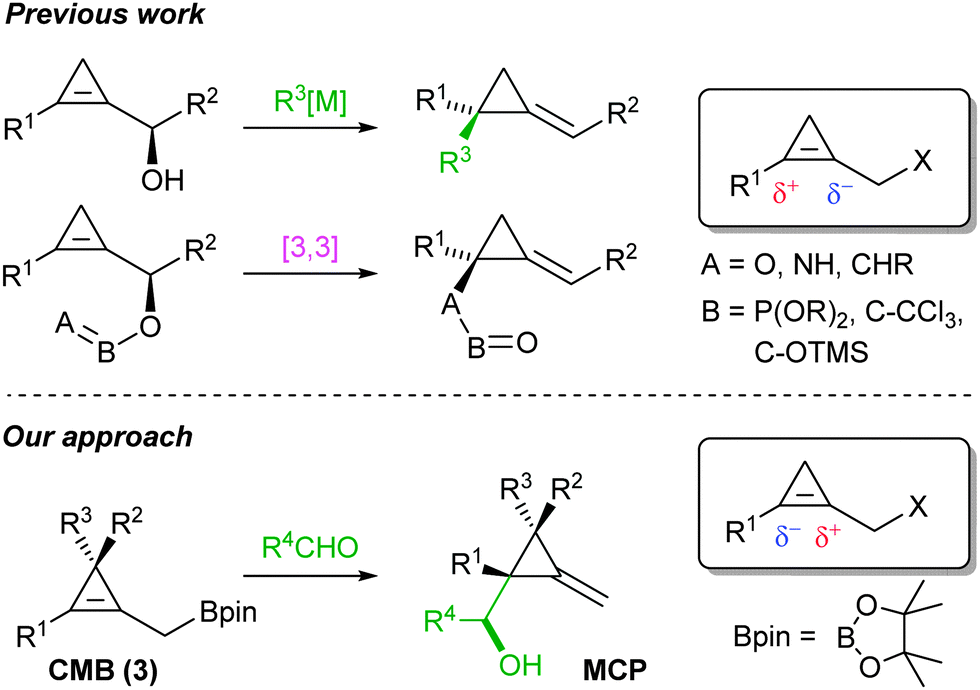
Alkylidenecyclopropanes (ACPs) possess a fascinating reactivity which continues to spark curiosity among the organic chemistry community, as they are candidates for a wide range of transformations.1 These structures have been recently employed to undergo ring expansion reactions in the presence of Lewis acids,2 or in acyclic stereocontrol through hydrometallations3 or zirconium-promoted C–C bond cleavage.4 Besides, ACPs represent valuable precursors of chiral cyclopropanes,5 architectures that can be found in a number of biologically active substrates.6
Among different diastereoselective routes for their preparation, Marek7 and Fox8 independently developed an easy and straightforward access to ACPs by using cyclopropenylcarbinol derivatives in a SN2′ reaction (Scheme 1). Cossy recently demonstrated the potential of secondary cyclopropenylcarbinol to undergo an Ireland–Claisen rearrangement, leading to ACPs by C–C bond formation.9 [3,3]-Sigmatropic rearrangements have also proven their efficiency in C–O10 and C–N11 bond forming reactions, resulting in heterosubstituted ACPs in high diastereoisomeric ratios.
 | ||
| Scheme 1 Synthetic routes towards ACPs. | ||
We hypothesized that a thoroughly designed allylic system embedded in the cyclopropyl core could allow for a nucleophilic allylation to proceed. Having recently reported the diastereoselective one-pot synthesis of methylenecyclobutanes using allylboronate derivatives,12 we envisioned that an easily prepared cyclopropenylmethyl boronic ester (CMB) would undergo the corresponding stereoselective allylboration13 reaction leading to the formation of challenging methylenecyclopropanes (MCPs) (Scheme 2).
 | ||
| Scheme 2 Access to CMBs 3 from corresponding tribromocyclopropanes 1. | ||
As CMBs (3) were identified as key intermediates in this study, their synthesis was undertaken first. Performing a double lithium-bromide permutation on a tribromocyclopropane afforded the intermediate cyclopropenyllithium species A.14 Subsequent addition of iodomethylboronic ester 2 resulted in a boron-homologation reaction15 through a 1,2-metallate rearrangement, and derivatives 3a–c were isolated in 58–70%.
With new allylic systems in hands, we investigated allylborations of aldehydes by first using the methyl derivative 3a. Interestingly, the reaction with benzaldehyde was completed within 15 min, leading to the expected methylenecyclopropane 4a in 76% yield and excellent diastereoselectivity (dr > 97![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3). Such a fast reaction time can be explained by strain release when displacing the double bond from the endo to the exo position.16 As a matter of fact, similar results were obtained for the synthesis of methylenecyclobutanes from cyclobutenylmethyl-boronic esters, with reaction times below 10 min.12 Reaction with aromatic and heteroaromatic aldehydes furnished the expected products in high yields (up to 89%) and excellent diastereoselectivities in all cases (dr > 97:3), as depicted in Scheme 3. Starting from 3a, methylcyclopropanes 4a–j were isolated with up to 89%. Slightly lower yields were obtained in cases of pyrrole and indole derivatives (4g, 58% and 4e, 52% respectively). An α,β-unsaturated aldehyde furnished the desired product 4d in good yield and excellent diastereoisomeric ratio.17
:3). Such a fast reaction time can be explained by strain release when displacing the double bond from the endo to the exo position.16 As a matter of fact, similar results were obtained for the synthesis of methylenecyclobutanes from cyclobutenylmethyl-boronic esters, with reaction times below 10 min.12 Reaction with aromatic and heteroaromatic aldehydes furnished the expected products in high yields (up to 89%) and excellent diastereoselectivities in all cases (dr > 97:3), as depicted in Scheme 3. Starting from 3a, methylcyclopropanes 4a–j were isolated with up to 89%. Slightly lower yields were obtained in cases of pyrrole and indole derivatives (4g, 58% and 4e, 52% respectively). An α,β-unsaturated aldehyde furnished the desired product 4d in good yield and excellent diastereoisomeric ratio.17
 | ||
| Scheme 3 Diastereoselective synthesis of methylenecyclopropanes 4a–p through allylboration of aldehydes from 3a–c. | ||
Changing the substituent at the vinylic position of the starting CMB to a pentyl chain (3b) did not affect the reactivity of the system nor the stereoselectivity of the allylation, and 4k was isolated in 66% yield after reaction with biphenylcarboxaldehyde. Allylboration was further performed by employing the silylated substrate 3c. With similarly high diastereomeric ratios, the introduction of nitrogen- or sulfur-containing heteroaromatic and aliphatic aldehydes resulted in building blocks of higher functionality (4l–p) in good to excellent yields (up to 85%) in only 15 min (Scheme 3).
To further expand the scope, CMB 3d bearing two methyl substituents was synthesized from the corresponding tribromocyclopropane.14 However, a notable difference of reactivity was observed when comparing to the previous systems 3a–c, and acceptable levels of starting material conversion were reached only after 16 h at room temperature. Despite comparable strained patterns, the presence of two additional substituents on 3d must play an undeniable role in lowering the reactivity of the allylic system. Such a sterically hindered moiety could partially inhibit the approach of the aldehyde, consequently increasing the reaction time (Scheme 4).
 | ||
| Scheme 4 Allylboration of aldehydes using CMB 3d. | ||
Having successfully demonstrated a new diastereoselective way of accessing MCPs, we took on the challenge of performing all the steps in a one-pot process, starting directly from tribromocyclopropanes 1. Addition of two equivalents of n-butyllithium to 1 leads to the intermediate lithium species A. Subsequent addition of 2 triggers a 1,2-metallate rearrangement, furnishing CMB B. At this point, changing the solvent of the reaction from THF to dichloromethane was detrimental for the reaction to be completed within 1 hour. THF was found to be competing with the aldehyde for coordination to the boron atom. Finally, the introduction of aldehydes allowed for the allylboration to proceed, leading to MCPs described in Scheme 5. Starting the sequence with 3a (R1 = Me, entries 1–3) furnished the expected MCPs 4a, 4i and 4j in good yields, while 3c furnished 4n (R1 = (CH2)2TMS, entry 4). In these cases, the diastereoisomeric ratios continued to be excellent and the yields were comparable to the step by step procedure. The use of 2-benzothiophene carboxaldehyde in the sequence involving 3a (entry 5) resulted in a full conversion, but a drastic drop of diastereoselectivity was observed.18
 | ||
| Scheme 5 Three-step-one-pot sequence for the diastereoselective synthesis of MCPs. | ||
With optimal conditions in hands for the one-pot formation of MCPs, we envisioned that using chiral tribromocyclopropanes (possessing an additional methyl group) could allow for the diastereocontrolled synthesis of MCPs containing three consecutive stereocenters. Aromatic and aliphatic aldehydes furnished expected “all-syn” adducts 7a–d in excellent yields and stereoselectivities (dr up to 97:3:0:0). The relative configuration of afore mentioned MCPs was attributed by analogy with 7d that could be crystallized and analysed by X-ray diffraction.19
Next, we investigated the possibility of starting from a chiral cyclopropene 8 (Scheme 6). In this specific case, the lithium species was simply generated by deprotonation of the three-membered ring in the presence of n-BuLi. The subsequent introduction of 2 to undergo a boron-homologation was then followed by the addition of benzaldehyde, after switching the solvent to dichloromethane. Through an allylboration, the homoallylic alcohol 9 was obtained as a single diastereoisomer, in 58% yield.
 | ||
| Scheme 6 Chiral cyclopropene for the one-pot synthesis of MCP. | ||
Interestingly, low temperatures were not required to observe an excellent diastereochemical outcome. We propose to explain this high diastereoselectivity by a pseudo-chair transition state involving a Zimmerman–Traxler model (Scheme 7). The chain of the aldehyde would then preferencially adopt the pseudo-equatorial position.20 When starting from chiral CMB 3e possessing a methyl group, one face of the cyclopropenyl derivative is shielded and the aldehyde approaches then from the opposite side, leading to the all-syn relative configuration.
 | ||
| Scheme 7 Proposed Zimmerman–Traxler model for allylboration reactions involving CMBs. | ||
In conclusion, we demonstrated the high potential of boron-homologation and allylboration to promote the simple synthesis of MCPs in excellent diastereoisomeric ratios. A wide variety of aldehydes was used in this unprecedented approach, showing the tolerance of the reaction towards sensitive functional groups. Ultimately, a one-pot process was elaborated, in which boron-homologation and allylboration were merged to simplify the procedure, and leading to MCPs containing up to three consecutive stereocenters.
The authors would like to thank the Chemical Industry Fund (FCI Liebig-fellowship) for financial support.
Notes and references
- (a) H. Pellissier, Tetrahedron, 2010, 66, 8341 CrossRef CAS; (b) A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed; (c) A. Brandi, S. Cicchi, F. M. Cordero and A. Goti, Chem. Rev., 2014, 114, 7317 CrossRef CAS PubMed; (d) H. Pellissier, Tetrahedron, 2014, 70, 4991 CrossRef CAS; (e) D.-H. Zhang, X.-Y. Tang and M. Shi, Acc. Chem. Res., 2014, 47, 913 CrossRef CAS PubMed.
- M. Shi, J.-M. Lu, Y. Wei and L.-X. Shao, Acc. Chem. Res., 2012, 45, 641 CrossRef CAS PubMed.
- A. Masarwa and I. Marek, Chem. – Eur. J., 2010, 16, 9712 CrossRef CAS PubMed , and references therein.
- A. Masarwa, D. Didier, T. Zabrodski, M. Schinkel, L. Ackermann and I. Marek, Nature, 2014, 505, 199 CrossRef CAS PubMed.
- (a) M. Schinkel, J. Wallbaum, S. I. Kozhushkov, I. Marek and L. Ackermann, Org. Lett., 2013, 15, 4482 CrossRef CAS PubMed; (b) S. Cui, Y. Zhang and Q. Wu, Chem. Sci., 2013, 4, 3421 RSC; (c) R. Sakae, N. Matsuda, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1228 CrossRef CAS PubMed; (d) J. C. Timmerman, B. D. Robertson and R. A. Widenhoefer, Angew. Chem., Int. Ed., 2015, 54, 2251 CrossRef CAS PubMed.
- (a) J. Salaun and M. S. Baird, Curr. Med. Chem., 1995, 2, 511 CAS; (b) W. A. Donaldson, Tetrahedron, 2001, 57, 8589 CrossRef CAS; (c) A. Reichelt and S. F. Martin, Acc. Chem. Res., 2006, 39, 433 CrossRef CAS PubMed; (d) D. Y.-K. Chen, R. H. Pouwer and J.-A. Richard, Chem. Soc. Rev., 2012, 41, 4631 RSC; (e) R. D. Taylor, M. MacCoss and A. D. G. Lawson, J. Med. Chem., 2014, 57, 5845 CrossRef CAS PubMed.
- (a) S. Simaan, A. Masarwa, P. Bertus and I. Marek, Angew. Chem., Int. Ed., 2006, 45, 3963 CrossRef CAS PubMed; (b) S. Simaan and I. Marek, Chem. Commun., 2009, 292 RSC.
- (a) Z. Yang, X. Xie and J. M. Fox, Angew. Chem., Int. Ed., 2006, 45, 3960 CrossRef CAS PubMed; (b) X. Xie, Z. Yang and J. M. Fox, J. Org. Chem., 2010, 75, 3847 CrossRef CAS PubMed.
- G. Ernouf, J.-L. Brayer, B. Folléas, J.-P. Demoute, C. Meyer and J. Cossy, Org. Lett., 2015, 17, 3786 CrossRef CAS PubMed.
- (a) I. Marek, S. Simaan and A. Masarwa, Angew. Chem., Int. Ed., 2007, 46, 7364 CrossRef CAS PubMed; (b) S. Simaan, A. Masarwa, E. Zohar, A. Stanger, P. Bertus and I. Marek, Chem. – Eur. J., 2009, 15, 8449 CrossRef CAS PubMed.
- J. K. Howard, C. Amin, B. Lainhart, J. A. Smith, J. Rimington and C. J. T. Hyland, J. Org. Chem., 2014, 79, 8462 CrossRef CAS PubMed.
- M. Eisold and D. Didier, Angew. Chem., Int. Ed., 2015, 54, 15884 CrossRef CAS PubMed.
- For recent examples of allylboration, see: (a) D. G. Hall, Pure Appl. Chem., 2008, 80, 913 CrossRef CAS; (b) M. Althaus, A. Mahmood, J. R. Suárez, S. P. Thomas and V. K. Aggarwal, J. Am. Chem. Soc., 2010, 132, 4025 CrossRef CAS PubMed; (c) M. Raducan, R. Alam and K. J. Szabó, Angew. Chem., Int. Ed., 2012, 124, 13227 CrossRef; (d) J. L.-Y. Chen, H. K. Scott, M. J. Hesse, C. L. Willis and V. K. Aggarwal, J. Am. Chem. Soc., 2013, 135, 5316 CrossRef CAS PubMed; (e) J. L.-Y. Chen and V. K. Aggarwal, Angew. Chem., Int. Ed., 2014, 53, 10992 CrossRef CAS PubMed.
- (a) M. C. Pirrung, A. B. Bleeker, Y. Inoue, F. I. Rodríguez, N. Sagawara, T. Wada, Y. Zou and B. M. Binder, Chem. Biol., 2008, 15, 313 CrossRef CAS PubMed; (b) K. F. S. Alnes and L. K. Sydnes, Monatsh. Chem., 2006, 137, 483 CrossRef CAS.
- (a) D. S. Matteson and R. H. W. Mah, J. Am. Chem. Soc., 1963, 85, 2599 CrossRef CAS; (b) D. S. Matteson and R. Ray, J. Am. Chem. Soc., 1980, 102, 7590 CrossRef CAS.
- K. B. Wildberg, Angew. Chem., Int. Ed., 1986, 98, 322 Search PubMed.
- When R1 = Ph, a drastic drop of diastereoselectivity was observed (dr = 55:45).
- Similar results were obtained employing 3a in a step-by-step procedure with 2-benzothiophene carboxaldehyde.
- CCDC 1439072 (7d).
- R. W. Hoffmann, G. Niel and A. Schlapbach, Pure Appl. Chem., 1990, 62, 1993 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed procedures and analytical data. CCDC 1439072. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09904h |
| This journal is © The Royal Society of Chemistry 2016 |