
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence‘Quasi-isostructural polymorphism’ in molecular crystals: inputs from interaction hierarchy and energy frameworks†
Dhananjay
Dey
a,
Sajesh P.
Thomas
b,
Mark A.
Spackman
b and
Deepak
Chopra
*a
aCrystallography and Crystal Chemistry Laboratory, Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhauri, Bhopal-462066, Madhya Pradesh, India. E-mail: dchopra@iiserb.ac.in; Fax: +91 755 6692392
bSchool of Chemistry and Biochemistry, The University of Western Australia, Crawley, WA, Australia. E-mail: mark.spackman@uwa.udu.au; Fax: +61 8 6488 7330
First published on 14th December 2015
Abstract
The polymorphs of (Z)-2-fluoro-N′-phenyl benzamidamide with multiple Z′ produce quasi-isostructural supramolecular architectures, wherein C–H⋯F interaction plays a significant role. The energy framework analysis indicates 2D structural similarities in the interaction topologies of these crystalline forms. The results point to a unique class of ‘quasi-isostructural polymorphs’ which are nearly equi-energetic crystal structures exhibiting high degrees of similarity in physical properties.
Polymorphism1 due to the presence of different numbers of symmetry-independent molecules in the asymmetric unit is of importance in the study of crystal structures and also in the area of crystal engineering.2–4 Thermodynamic and kinetic factors are very important during nucleation and crystal growth as such factors influence the final outcome of crystallization events leading to the formation of different morphologies with different molecular arrangements.5 Polymorphic structures in different crystalline environments can be compared on the basis of differences/similarities which exist in molecular conformation, packing arrangements and formation of different hydrogen bond motifs.6 Desiraju and co-workers have reported the polymorphs of pentafluorophenol and trans-1,4-bis (phenylethyl) cyclohexane-1,4-diol.7 The polymorphic forms of liquid pentafluorophenol were obtained using in situ cryocrystallization8,9 techniques under different kinetic conditions. Form I (space group: P21/c and Z = 1) was stabilized via O–H⋯O hydrogen bonded chains and form II (space group: Cc and Z′ = 3) was stabilized via O–H⋯O hydrogen bonded chains between the three symmetry-independent molecules. Nangia and co-workers further reported the existence of four polymorphs and the nineteen crystallographically independent molecular conformations of 4,4-diphenyl-2,5-cyclohexadienone10 which exhibit conformational polymorphism, conformational isomerism and concomitant polymorphism. Herein, we present a unique case of high Z′ polymorphs of (Z)-2-fluoro-N′-phenylbenzamidamide that exhibit near-isostructurality and very similar features of packing and intermolecular interactions. Although Katrusiak et al. have reported nearly isostructural high Z′ polymorphs of ethynylbenzene generated by pressure freezing.11 Examples of such polymorphs having multiple Z′ in their crystal structures and formed under ambient conditions are extremely scarce in the literature. Recently, Coles and co-workers have reported the isostructural polymorphs of 3-chloro mandelic acid obtained by a temperature dependent phase transition.12 The example reported represents a case of a change in the crystallographic symmetry with subtle changes in molecular packing and unit cell parameters. The present case reported belongs to a distinct phenomenon of “quasi-isostructural polymorphism” with significant differences in the unit cell parameters. With respect to the definition proposed by IUCr, “two crystals are said to be isostructural if they have the same crystal structure, but not necessarily the same cell dimensions nor the same chemical composition”. A detailed classification of isostructurality in molecular crystals is given in Kalman et al.13 In the current study, it is observed that the polymorphs are stable at room temperature and have been obtained from different solvents. The structural and physical characterization of these crystal forms followed by analysis of their energy frameworks points to the occurrence of this unique phenomenon of ‘quasi-isostructural polymorphism’ in molecular crystals.
The two polymorphic forms of (Z)-2-fluoro-N′-phenylbenzamidamide were obtained by room temperature crystallization of the synthesized compound (Fig. S1, S2 and Scheme S1, ESI†) in hexane and benzene solvents respectively. Both Forms I and II crystallize in the triclinic crystal system with the P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group having two and three symmetry independent molecules (Z′) in the asymmetric unit respectively. The molecular conformations in the two forms are slightly different (see the ESI,† Tables S1, S2 and Fig. S3). In the case of Form I, two symmetry independent molecules present in the asymmetric unit are represented with red and olive colour codes for molecules A and B respectively (Fig. 1). Fig. 2 shows the overall arrangement of the molecules in the crystal packing down the (011) plane. The crystal structure consists of layers of molecules of type B interacting via the C–H⋯N dimer and the C–H⋯F dimer, respectively, along the crystallographic b axis. Two such consecutive sheets were interconnected via strong N–H⋯N H-bonds forming chains of AB molecules along the crystallographic a direction. Two nearest N–H⋯N chains (the perpendicular distance is approximately ∼4.2 Å) were connected via N–H⋯F and C–F⋯π intermolecular interactions between molecules A and C–H⋯N and C–H⋯π interactions between molecules B respectively. The next N–H⋯N sheet which comes at a perpendicular distance of ∼9.7 Å along the b direction was connected with the C–H⋯F dimer (between molecules B) and π⋯π interactions (between molecules A).
space group having two and three symmetry independent molecules (Z′) in the asymmetric unit respectively. The molecular conformations in the two forms are slightly different (see the ESI,† Tables S1, S2 and Fig. S3). In the case of Form I, two symmetry independent molecules present in the asymmetric unit are represented with red and olive colour codes for molecules A and B respectively (Fig. 1). Fig. 2 shows the overall arrangement of the molecules in the crystal packing down the (011) plane. The crystal structure consists of layers of molecules of type B interacting via the C–H⋯N dimer and the C–H⋯F dimer, respectively, along the crystallographic b axis. Two such consecutive sheets were interconnected via strong N–H⋯N H-bonds forming chains of AB molecules along the crystallographic a direction. Two nearest N–H⋯N chains (the perpendicular distance is approximately ∼4.2 Å) were connected via N–H⋯F and C–F⋯π intermolecular interactions between molecules A and C–H⋯N and C–H⋯π interactions between molecules B respectively. The next N–H⋯N sheet which comes at a perpendicular distance of ∼9.7 Å along the b direction was connected with the C–H⋯F dimer (between molecules B) and π⋯π interactions (between molecules A).
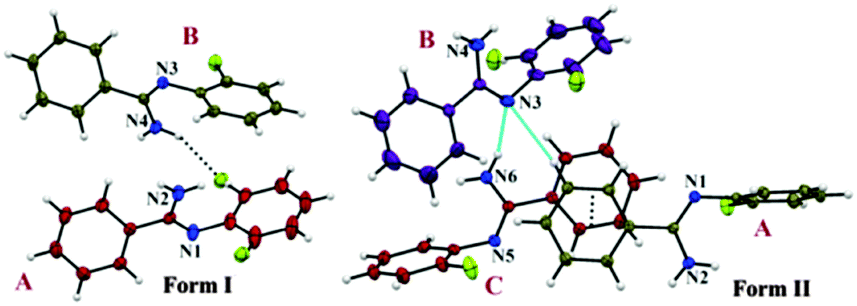 | ||
| Fig. 1 ORTEP of two polymorphic forms drawn with 50% ellipsoidal probability. Form I and II shows the asymmetric unit containing Z′ = 2 and Z′ = 3 respectively. | ||
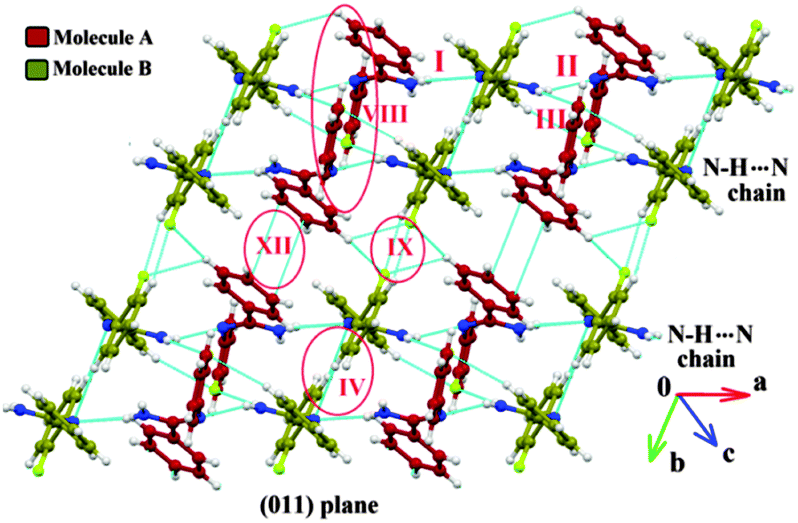 | ||
| Fig. 2 Crystal packing of Form I viewed down the (011) plane showing the alternate sheets (containing the C–H⋯N dimer and the C–H⋯F dimer) of molecules B, interconnected via strong N–H⋯N AB chains along the a direction. | ||
In the case of Form II, three symmetry independent molecules present in the asymmetric unit were denoted by different colour codes; olive (A), purple (B) and red (C) colour (Fig. 1 and 3). Fig. 3 shows the overall molecular arrangement of the three symmetry independent molecules in the crystal down the (101) plane. In this case, along the b direction two parallel N–H⋯N chains (containing molecules A, B and C) were connected through the alternate pair of sheets down the ab plane (involving molecules A and C) associated with C–H⋯F dimers and C–H⋯N dimers respectively. The distance between the nearest N–H⋯N chains is approximately ∼3.7 Å. Between the two pairs of molecular sheets (involving molecules A and C), molecules B interact with each other via weak C–F⋯π and π⋯π intermolecular interactions. Differences in the formation of different intermolecular interactions involving organic fluorine with respect to the packing between the different layers of molecules exist. This is highlighted with a red arrow in Fig. 3. In Form II, there is formation of a parallel C–H⋯F dimer whereas in Form I C–H⋯F interactions exist across the layers (shown in Fig. 2). The striking similarity in the crystal packing of these polymorphs also results in very similar thermal stability and spectral features. The thermal stabilities have been investigated via DSC and HSM measurements (Fig. 4 and Fig. S4, ESI†). The melting points of Forms I and II are 92.7 °C and 92.5 °C respectively. The corresponding crystal densities are 1.30 and 1.32 g cm−3. The FT-IR spectra also exhibit nearly identical features for these polymorphs (Fig. S5, ESI†). The experimental PXRD patterns of these polymorphs exhibit remarkable differences in the low angle region, on account of the subtle variations which exist in the crystal packing (Fig. 5 and Fig. S6–S9, ESI†).
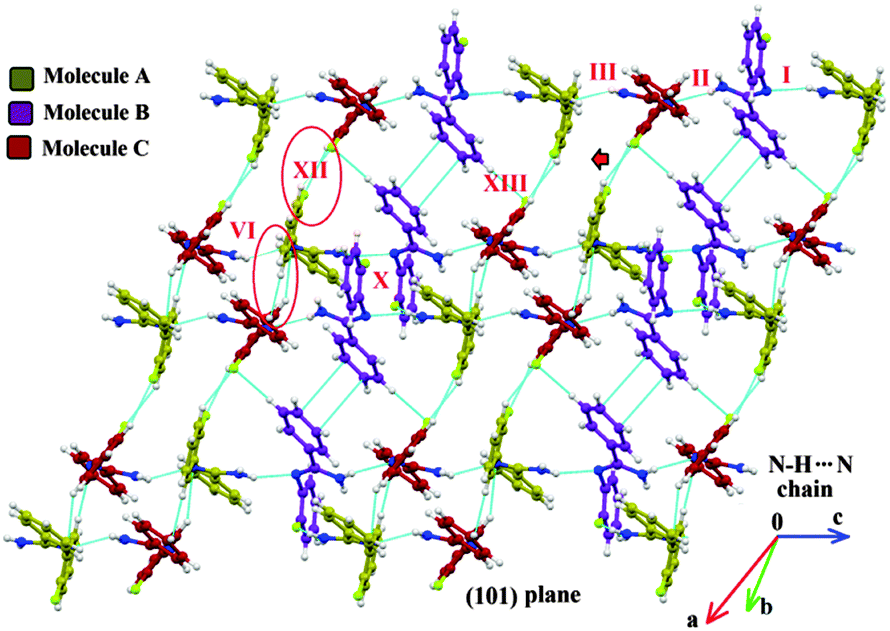 | ||
| Fig. 3 Crystal packing in Form II viewed down the (101) plane depicting the formation of different intermolecular interactions. Formation of alternate pairs of sheets (involving molecules A and B) interconnected via strong N–H⋯N A(olive)⋯B(purple)⋯C(red) chains along the crystallographic c direction. | ||
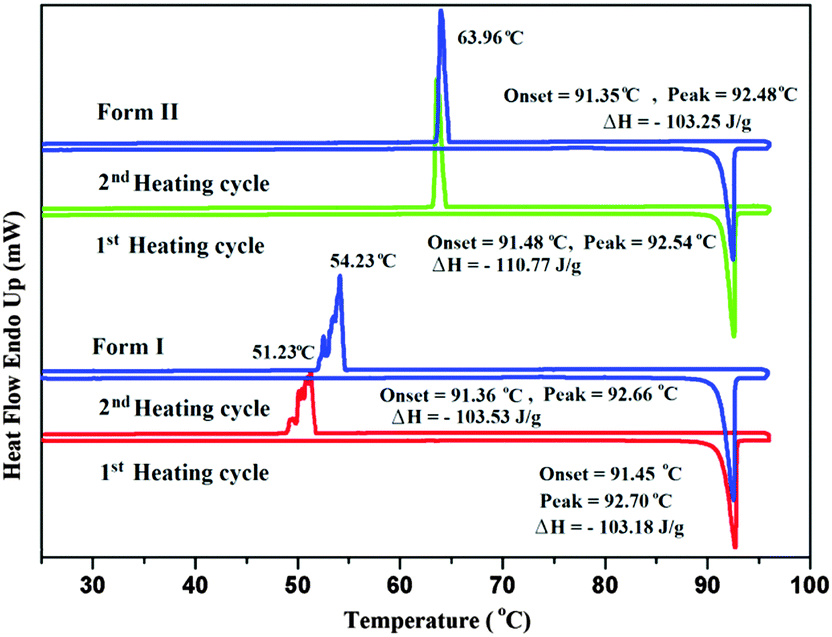 | ||
| Fig. 4 DSC traces @ 1 °C min−1 for Forms I and II. | ||
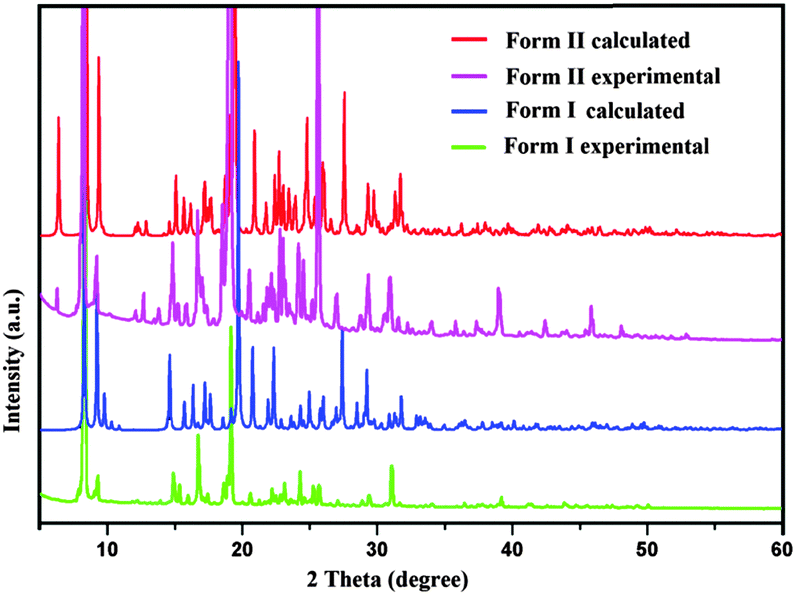 | ||
| Fig. 5 Experimental and calculated powder patterns of Forms I and II. | ||
Furthermore, the similarity/dissimilarity in these crystal structures has been quantitatively examined using Xpac analysis.14 The dissimilarity index of 10.3 obtained for these polymorphic pairs (Fig. S10, ESI†) shows that the degree of similarity in these polymorphic pairs is comparable to those found in isostructural polymorphic pairs of 6-hydroxy-4,4,5,7,8-pentamethyl-3,4-dihydrocoumarin,15 and related chemical analogues that exhibit isostructurality.16 This points to the possibility that quasi-isostructural polymorphism could be a more general phenomenon albeit underexplored in a quantitative and rigorous manner in terms of underlying intermolecular interactions.
The fact that the various physical characterizations show no explicit differences between the two polymorphs suggests that these ‘quasi-isostructural polymorphs’ might be representing two very close points in the crystal structure landscape. Hence, in order to evaluate the energetics associated with various intermolecular interactions in the crystal packing, calculations using PIXEL were performed. Tables S3 and S4 (ESI†) list the PIXEL results for the different molecular pairs (Fig. S11 and S12, ESI†) extracted from the packing in Form I and Form II respectively. A comparison of stabilization energies (PIXEL) of most important equivalent dimer motifs between Forms I and II unambiguously highlights this structural similarity (Fig. 6).
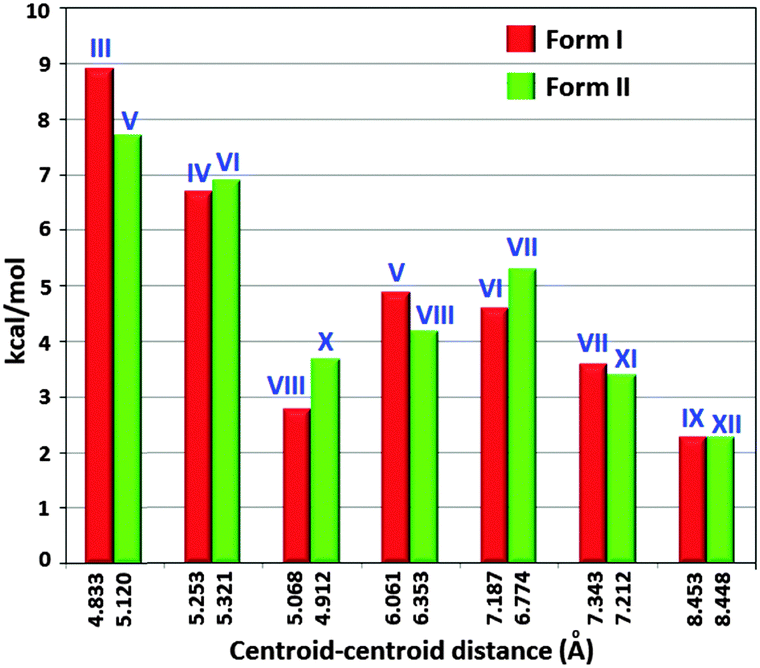 | ||
| Fig. 6 Comparison of dimeric energies between two equivalent motifs of Forms I and II. | ||
Furthermore, to visualize and differentiate the supramolecular architecture in terms of energetics for these two polymorphic forms, the calculations corresponding to the existence of energy frameworks have been performed using CrystalExplorer.17 The obtained values of interaction energies can be used to construct the three dimensional topology of interactions which are termed as energy frameworks.18 Recently, we have shown that the energy frameworks not only represent the 3D-topology of the predominant interactions but also correlate well with the observed mechanical properties of crystals such as shearing and bending properties.18 In addition, energy frameworks have also been shown to highlight the role of intermolecular interactions in liquids19 and the robustness of interactions in a drug which are reluctant to form co-crystals.20 In the present case of quasi-isostructural polymorphs, the energy frameworks were computed for a systematic comparison of the interaction topologies and their visualization. The analysis shows (Fig. S13 and Table S5; Fig. S14 and Table S6, ESI†) a remarkable similarity in the topologies corresponding to the energy frameworks in Forms I and II. The energy framework of Form I exhibits a 2D network of strongly bound molecules perpendicular to the c axis (Fig. 7 and 8). This feature is also found in Form II where a very similar 2D molecular network extends in a plane perpendicular to the b axis (Fig. 7 and 8). Similarly, the energy framework of Form I viewed down the a axis is quite similar to that of Form II viewed down the c axis, and vice versa. Interestingly, both electrostatic and dispersion energy frameworks also exhibit such a similarity between Forms I and II. These remarkable similarities observed in the energy frameworks imply a nearly isoenergetic and quasi-isostructural packing in these crystal forms. This feature can be further seen in an overlay of 2D molecular assembly (between Fig. S15 and S16, ESI†) in both forms as given in Fig. S17 (ESI†). Both the forms have similar types of molecular motifs, as shown in Scheme S2 (ESI†). From the overall molecular arrangements in the two polymorphic Forms I and II, the differences are clearly discernible from Fig. S18 (ESI†). This figure depicts the differences in crystal packing due to the presence of different symmetry independent molecules with different colour codes. In Form I, the distance (9.338 Å) between the two sheets associated with C–H⋯F and C–H⋯N dimers (involving molecules B) is slightly lower than the distance (9.744 Å) between two pairs of sheets (involving molecules A and C). The position of molecules A in Form I is invariant with respect to the position of molecules B in Form II. Furthermore, the net interaction energies of a shell of nearest neighbouring molecules around each symmetry independent molecule in both the forms were evaluated. The neighbouring-shell interaction energies for residue I and residue II in Form I were found to be −119.5 and −111 kJ mol−1. Similarly, for residues I, II, and III in Form II, the corresponding values were found to be −102.7, −115 and −118.5 kJ mol−1 respectively. This brings out the quantitative differences in neighbouring-shell interaction energy values between different molecular residues in both the polymorphs, despite the close similarity in the packing. Nevertheless, the average neighbouring-shell interaction energy values for both the polymorphs are very close (−115.3 kJ mol−1 for Form I and −112.4 kJ mol−1 for Form II).
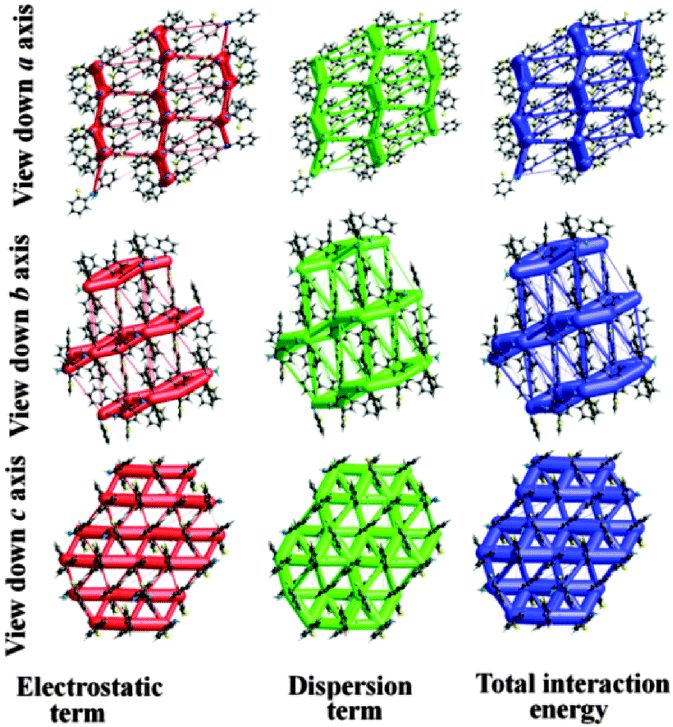 | ||
| Fig. 7 Energy frameworks corresponding to the electrostatic, dispersion and net interaction energy components in Form I. | ||
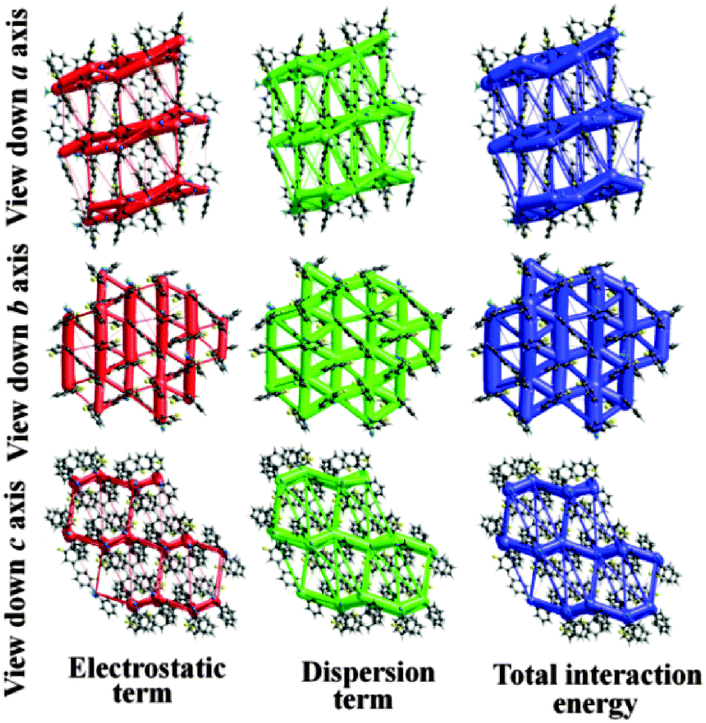 | ||
| Fig. 8 Energy frameworks corresponding to the electrostatic, dispersion and net interaction energy components in Form II. | ||
Hirshfeld surface21 associated fingerprint plots22,23 for the individual molecules present in the asymmetric unit of Forms I and II are depicted in Fig. 9. Such plots represent the underlying differences that exist between the two polymorphs. In addition such plots also highlight the differences in the crystal environment due to the existence of different symmetry-independent molecules in the asymmetric unit. For both the forms, two very sharp spikes are observed and this depicts the formation of strong hydrogen bonds (N–H⋯N) labelled with the green triangle. The middle spike is associated with the F⋯H contacts labelled with the pink triangle. Fig. S19 (ESI†) highlights the presence of two most important contacts N⋯H (6.7–8.2%) and F⋯H (7.1–17.9%) which reflect the pivotal contributions of such hydrogen bonds in the formation of polymorphs. It is noteworthy that the F⋯H (14.9%) contribution for IB was significantly greater than that for IA (8.6%), the N⋯H contribution being similar for both the symmetry-independent molecules in the case of Form I. In the case of Form II, the contribution from the F⋯H contacts was more for IIA (17.9%) in comparison to the other two molecules present in the asymmetric unit (the values being 7.1% and 10% respectively). There was a significant contribution from H⋯H contacts, in the range of 43–53% and C⋯H contacts (25–28%) which contribute to the crystal packing.
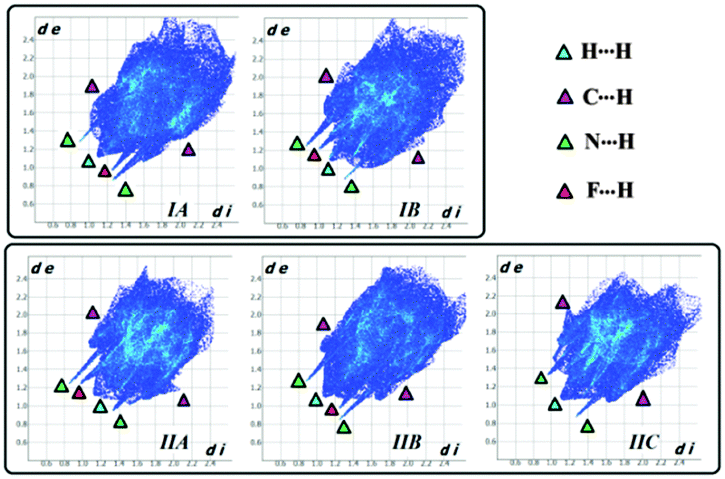 | ||
| Fig. 9 Hirshfeld surfaces associated with the fingerprint plot of the two polymorphic forms. The spikes labelled with black (H⋯H), pink (C⋯H), green (N⋯H) and red (F⋯H) triangles depict the characteristic features of the fingerprint plots. | ||
In summary, nearly isostructural and equi-energetic molecular arrangements in the two high Z′ polymorphs of (Z)-2-fluoro-N′-phenylbenzamidamide result in the occurrence of the phenomenon of ‘quasi-isostructural polymorphism’. The formation of such polymorphs under ambient conditions that exhibit remarkable similarity in thermal stabilities, densities, FT-IR spectra and powder diffraction profiles can have direct implications in the context of pharmaceutical polymorphs. Based on our analysis of the neighbouring-shell interaction energies and energy frameworks, it may be conjectured that two quasi-isostructural polymorphs of a compound might exhibit very similar physical properties such as solubility and mechanical behaviour.
D. D. thanks IISER Bhopal for the research fellowship. D. C. and D. D. thank IISER Bhopal for the research facilities and infrastructure and DST-SERB for research funding. We thank all the reviewers for their suggestions/feedback on the manuscript.
Notes and references
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, New York, 2002 Search PubMed.
- D. A. Parrish, J. R. Deschamps, R. D. Gilardi and R. J. Butcher, Cryst. Growth Des., 2008, 8, 57 CAS.
- R. Bishop and M. L. Scudder, Cryst. Growth Des., 2009, 9, 2890 CAS.
- K. M. Steed and J. W. Steed, Chem. Rev., 2015, 115, 2895 CrossRef CAS PubMed.
- G. R. Desiraju, Science, 1997, 278, 404 CrossRef CAS.
- D. Dey and D. Chopra, CrystEngComm, 2015, 17, 5288 RSC.
- D. Das, R. Banerjee, R. Mondal, J. A. K Howard, R. Boesec and G. R. Desiraju, Chem. Commun., 2006, 555 RSC.
- A. R. Choudhury, N. Winterton, A. Steiner, A. I. Cooper and K. A. Johnson, J. Am. Chem. Soc., 2005, 127, 16792 CrossRef CAS PubMed.
- D. Chopra and T. N. Guru Row, J. Indian Inst. Sci., 2007, 87, 167 CAS.
- V. S. S. Kumar, A. Addlagatta, A. Nangia, W. T. Robinson, C. K. Broder, R. Mondal, I. R. Evans, J. A. K. Howard and F. H. Allen, Angew. Chem., Int. Ed., 2002, 41, 3848 CrossRef CAS.
- K. Dziubek, M. Podsiadlo and A. Katrusiak, J. Am. Chem. Soc., 2007, 129, 12620 CrossRef CAS PubMed.
- S. J. Coles, T. L. Threlfall and G. J. Tizzard, Cryst. Growth Des., 2014, 14, 1623 CAS.
- A. Kalman, S. Parkanyi and G. Argay, Acta Crystallogr., 1993, B49, 1039 CAS.
- T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324 RSC.
- L. Fabian and A. Kalman, Acta Crystallogr., 2004, B60, 547 CAS.
- T. Gelbrich, T. L. Threlfall and M. B. Hursthouse, CrystEngComm, 2012, 14, 5454 RSC.
- M. J. Turner, S. Grabowsky, D. Jayatilaka and M. A. Spackman, J. Phys. Chem. Lett., 2014, 5, 4249 CrossRef CAS PubMed.
- M. J. Turner, S. P. Thomas, M. W. Shi, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2015, 51, 3735 RSC.
- S. P. Thomas, R. Sathishkumar and T. N. Guru Row, Chem. Commun., 2015, 51, 14225 Search PubMed.
- S. P. Thomas, D. Jayatilaka and T. N. Guru Row, Phys. Chem. Chem. Phys., 2015, 17, 25411 RSC.
- M. A. Spackman and D. Jayatilaka, CrystEngComm, 2009, 11, 19 RSC.
- M. A. Spackman and J. J. Mckinnon, CrystEngComm, 2002, 4, 378 RSC.
- J. J Mckinnon, D. Jayatilaka and M. A. Spackman, Chem. Commun., 2007, 3814 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Details of the experimental, computational methods, topological analysis, and characterization techniques: single crystal X-ray diffraction, DSC, HSM, powder X-ray diffraction, 1H-NMR and FTIR spectrum, CSD search. CCDC 982074 and 1016404. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09741j |
| This journal is © The Royal Society of Chemistry 2016 |