
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Developing strongly luminescent platinum(IV) complexes: facile synthesis of bis-cyclometalated neutral emitters†
Fabio
Juliá
a,
Delia
Bautista
b and
Pablo
González-Herrero
 *a
*a
aDepartamento de Química Inorgánica, Facultad de Química, Universidad de Murcia, Apdo. 4021, 30071 Murcia, Spain. E-mail: pgh@um.es
bSAI, Universidad de Murcia, Apdo. 4021, 30071 Murcia, Spain
First published on 2nd December 2015
Abstract
A straightforward, one-pot procedure has been developed for the synthesis of bis-cyclometalated chloro(methyl)platinum(IV) complexes with a wide variety of heteroaromatic ligands of the 2-arylpyridine type. The new compounds exhibit phosphorescent emissions in the blue to orange colour range and represent the most efficient Pt(IV) emitters reported to date, with quantum yields up to 0.81 in fluid solutions at room temperature.
Luminescent transition-metal complexes have been exhaustively studied for their rich photochemistry and wide applicability in optical technologies.1 Thus, they are employed as electroluminescent materials,2 chemosensors,3 photocatalysts,4 probes for bioimaging,5 and photosensitizers for dye-sensitized solar cells6 and singlet-oxygen generation and photodynamic chemotherapy.7 The majority of studies concentrate on complexes of d6 [mainly Ru(II), Os(II) and Ir(III)], d8 [Pt(II), Au(III)] and d10 [Cu(I)] ions with polypyridyls or cyclometalated arylpyridine ligands.1 The quest for stable complexes that exhibit highly efficient triplet emissions, relatively long excited-state lifetimes and tunable colours has been a constant in this field, associated with the need to improve the performance and reliability of specific applications; in this regard, research on cyclometalated Pt(II)8 and Ir(III)2a,9 complexes has been particularly intense, since these systems are highly versatile and can reach very high quantum yields.
In stark contrast with the numerous photophysical studies on complexes of other d6 ions, related works on Pt(IV) complexes are scarce and appear to have been overshadowed by the strong focus on Pt(II).8f Bis-cyclometalated complexes of the type [Pt(C∧N)2(R)Cl] [C∧N = ortho-deprotonated 2-phenylpyridine (ppy) or 2-thienylpyridine (thpy); R = CH2Cl, CHCl2] were the first luminescent Pt(IV) complexes, with reported quantum yields in the range 0.05–0.15.10 Later works have described weakly emissive Pt(IV) complexes.11 Recently, we undertook the development of highly efficient Pt(IV) emitters and described a family of cationic homoleptic tris-cyclometalated complexes fac-[Pt(C∧N)3]+ that exhibit high-energy (blue) emissions with quantum yields up to 0.49 and very long lifetimes.12 We then extended the study to heteroleptic derivatives mer-[Pt(C∧N)2(C′∧N′)]+ that display lower emission energies.13 These complexes emit from essentially ligand-centred triplet excited states (3LC) with a very low but critical metal-to-ligand charge-transfer (MLCT) character, which facilitates the formation of the triplet emitting state because of the spin–orbit coupling effect induced by the metal. Here we present a series of phosphorescent bis-cyclometalated Pt(IV) complexes of the type [Pt(C∧N)2(Me)Cl] (2a–i; Scheme 1), characterized by a high stability, tunable emission energies and generally high quantum yields, which make them very attractive as triplet emitters.
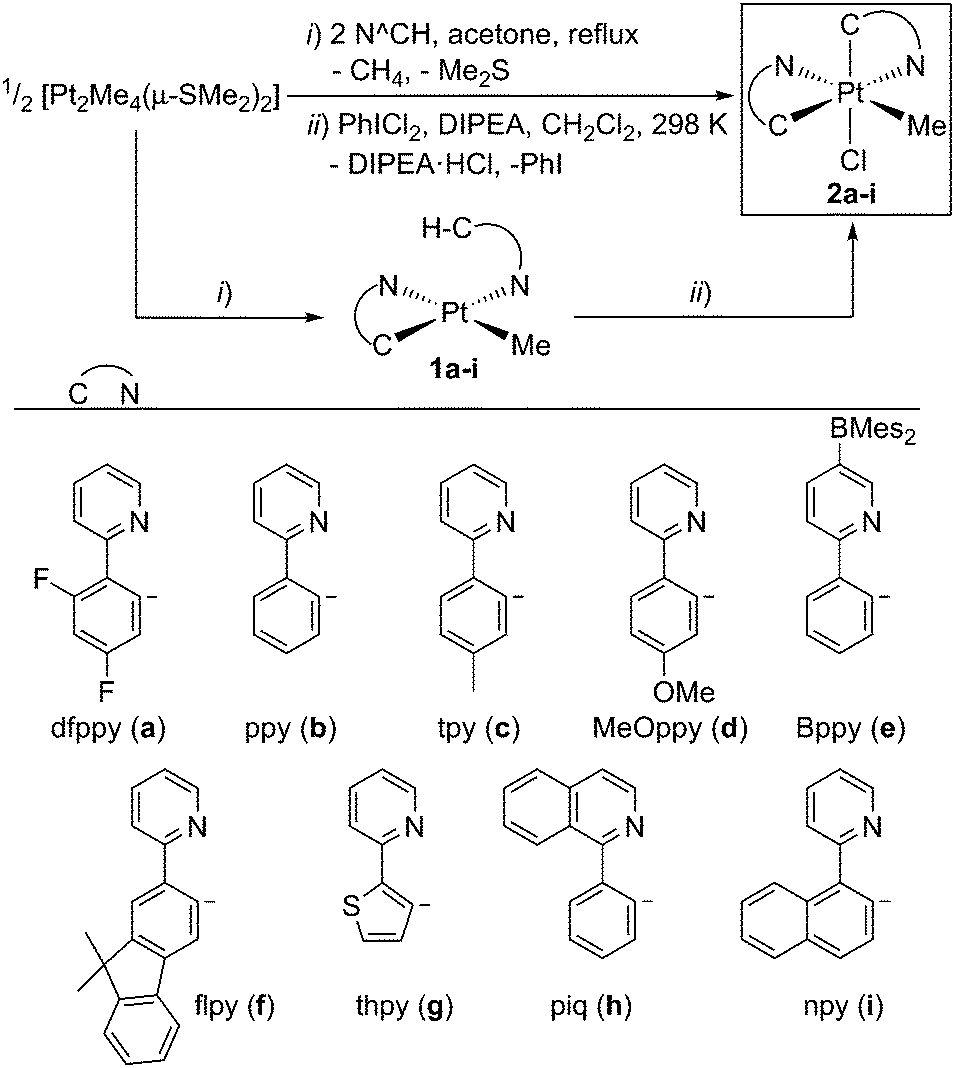 | ||
| Scheme 1 One-pot synthesis of 2a–i. DIPEA = N,N-diisopropylethylamine; Mes = mesityl. | ||
Compounds 2a–i were prepared following a one-pot, two-step procedure (Scheme 1). The first step is the reaction of [Pt2Me4(μ-SMe2)2]14 with the N∧CH compound in 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in refluxing acetone, which leads to the cyclometalation of one N∧CH molecule and the coordination of a second one through the N atom to give the monocyclometalated Pt(II) complexes 1a–i. This result is consistent with the known reactivity of the binuclear precursor, which reacts with N∧CH compounds to give cyclometalated complexes of the type [PtMe(C∧N)(SMe2)] and methane, while substitution of the labile Me2S ligand allows further derivatization.15 Treatment of the intermediate derivatives 1a–i with PhICl2 at room temperature resulted in the oxidation to Pt(IV) and subsequent metalation of the pendant aryl moiety of the coordinated N∧CH ligand to give complexes 2a–i. Similar metalations upon oxidation with PhICl2 have been reported for complexes of the type [Pt(C∧N)(N∧CH)Cl] (N∧CH trans to N).16 The metalation reaction produces HCl, which must be trapped by adding a base; otherwise, it rapidly reacts with complexes 1a–i, resulting in the substitution of the methyl ligand for a chloride (see the ESI† for details). The whole synthesis can be carried out without isolating the intermediate complexes 1a–i under atmospheric conditions and the final products can be obtained in moderate to excellent yields (57–89%). It is worth emphasising that this method allows to prepare a wide range of derivatives from an easy-to-synthesize common precursor under mild conditions, typically consuming less than 6 hours.
:2 molar ratio in refluxing acetone, which leads to the cyclometalation of one N∧CH molecule and the coordination of a second one through the N atom to give the monocyclometalated Pt(II) complexes 1a–i. This result is consistent with the known reactivity of the binuclear precursor, which reacts with N∧CH compounds to give cyclometalated complexes of the type [PtMe(C∧N)(SMe2)] and methane, while substitution of the labile Me2S ligand allows further derivatization.15 Treatment of the intermediate derivatives 1a–i with PhICl2 at room temperature resulted in the oxidation to Pt(IV) and subsequent metalation of the pendant aryl moiety of the coordinated N∧CH ligand to give complexes 2a–i. Similar metalations upon oxidation with PhICl2 have been reported for complexes of the type [Pt(C∧N)(N∧CH)Cl] (N∧CH trans to N).16 The metalation reaction produces HCl, which must be trapped by adding a base; otherwise, it rapidly reacts with complexes 1a–i, resulting in the substitution of the methyl ligand for a chloride (see the ESI† for details). The whole synthesis can be carried out without isolating the intermediate complexes 1a–i under atmospheric conditions and the final products can be obtained in moderate to excellent yields (57–89%). It is worth emphasising that this method allows to prepare a wide range of derivatives from an easy-to-synthesize common precursor under mild conditions, typically consuming less than 6 hours.
The crystal structures of 2a, 2b, 2d and 2f·CH2Cl2 were solved by X-ray crystal diffraction studies.17 The molecular structure of 2b is depicted in Fig. 1 and the others are included in the ESI† along with selected bond distances and angles. The Pt–N bond distances are rather long (range 2.121–2.156 Å) since the pyridyls are in trans to an aryl group or a methyl ligand, which exert very high trans influences. The Pt–Cl distances (2.423–2.446 Å) are similar to those found in other Pt(IV) complexes with a chloride ligand in trans to an aryl.16b Correspondingly, the Pt–C distances are very short (1.996–2.011 Å), owing to the low trans influences of the pyridyl groups or the chloride ligand.
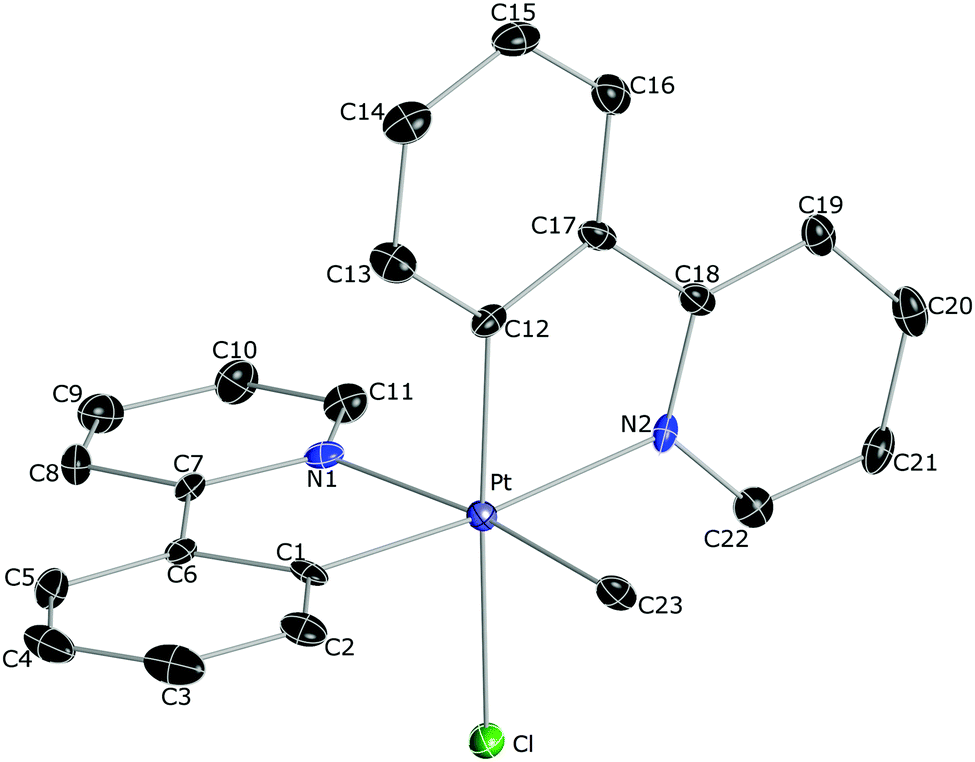 | ||
| Fig. 1 Thermal ellipsoid representation (50% probability) of the crystal structure of 2b. Hydrogen atoms are omitted for clarity. | ||
The absorption spectra of 2a–i in CH2Cl2 solution at 298 K display intense structured bands in the 250–400 nm region (Fig. 2), which are similar in energy and shape to those found for the previously reported cyclometalated Pt(IV) complexes12,13 and can be ascribed to essentially 1LC transitions within the C∧N ligands with little MLCT character. The lowest-energy band shifts from 322 to 378 nm along the sequence 2a → 2i, in line with the expected decreasing π–π* transition energies. This band has a significantly higher absorptivity for 2e and 2f (40600 or 51500 M−1 cm−1, respectively), compared to the rest of derivatives. Such intense absorptions are highly desirable for certain applications, such as bioimaging or photocatalysis.
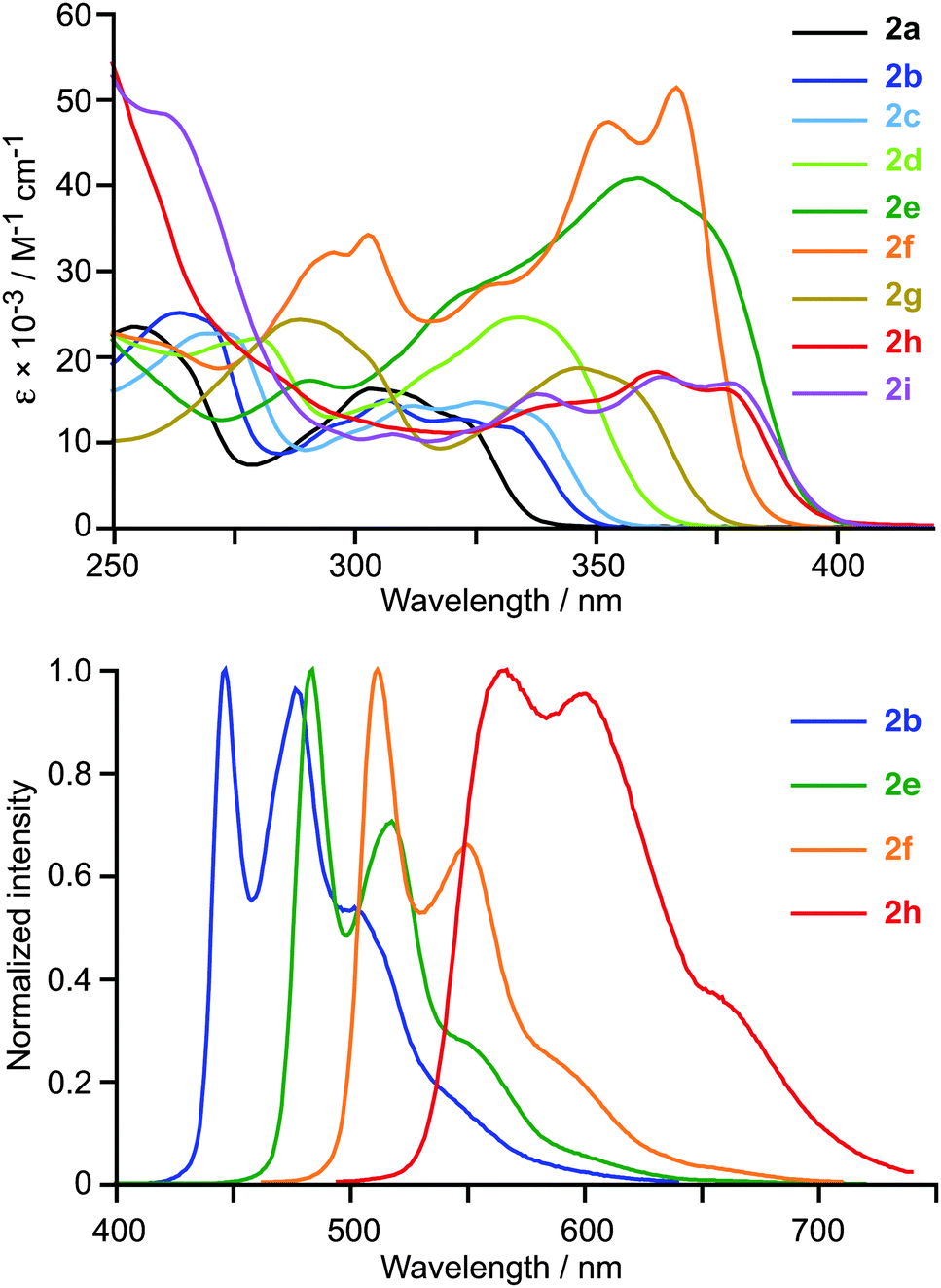 | ||
| Fig. 2 Absorption spectra of compounds 2a–i (top) and selected emission spectra (bottom) in CH2Cl2 solution at 298 K. | ||
The excitation and emission spectra of 2a–i were registered in deaerated CH2Cl2 solutions at 298 K and butyronitrile (PrCN) glasses at 77 K. The emission data are summarized in Table 1 and selected emission spectra at 298 K are shown in Fig. 2. The room-temperature emission spectra are vibrationally structured in all cases and the corresponding excitation spectra match the absorption profiles. Lifetimes are in the order of tens or hundreds of microseconds and Stokes shifts are in the range 6000–9000 cm−1. The emission energies decrease on going from 2a to 2i, in accord with the π–π* transition energies of the C∧N ligands, and the associated emission colours range from blue to orange. The vibrational structure becomes better resolved in frozen glasses at 77 K and lifetimes increase as a consequence of the partial suppression of non-radiative processes. The mentioned characteristics are indicative of an essentially 3LC emitting state, as found for the previously reported tris-cyclometalated Pt(IV) complexes.12,13 In fact, the emission spectra of 2b and fac-[Pt(ppy)3]+ are practically identical (Fig. S6, ESI†).
| Complex | 298 Ka | 77 Kb | |||||
|---|---|---|---|---|---|---|---|
| λ em /nm | Φ | τ /μs | k r × 10−3/s−1 | k nr × 10−3/s−1 | λ em /nm | τ /μs | |
| a In deaerated CH2Cl2 solution (ca. 5 × 10−6 M). b In deaerated PrCN. c Highest-energy emission peak. d Absolute quantum yield. e Emission lifetime. f Radiative rate constant, kr = Φ/τ. g Nonradiative rate constant, knr = (1 − Φ)/τ. | |||||||
| 2a | 435 | 0.57 | 154 | 3.70 | 2.79 | 430 | 267 |
| 2b | 447 | 0.60 | 99.7 | 6.02 | 4.01 | 442 | 215 |
| 2c | 453 | 0.51 | 98.0 | 5.20 | 5.00 | 447 | 258 |
| 2d | 458 | 0.28 | 74.3 | 3.77 | 9.69 | 451 | 478 |
| 2e | 484 | 0.81 | 193 | 4.20 | 0.98 | 481 | 254 |
| 2f | 512 | 0.20 | 110 | 1.82 | 7.27 | 504 | 450 |
| 2g | 514 | 0.22 | 81.8 | 2.69 | 9.53 | 508 | 289 |
| 2h | 565 | 0.05 | 17.2 | 2.91 | 55.23 | 553 | 65.6 |
| 2i | 585 | 0.04 | 14.9 | 2.68 | 64.43 | 558 | 121 |
The new compounds are very bright emitters in CH2Cl2 solutions at room temperature, showing quantum yields (Φ) higher than 0.20, with the exception of 2h and 2i. An inspection of radiative and non-radiative rate constants (kr and knr, respectively) shows that, while kr values remain within the same order of magnitude along the series, knr values are significantly higher for 2h and 2i, indicating that non-radiative processes are more effective in these cases; reasonably, this behaviour can be ascribed to the lower energy of the emitting state in these derivatives, which leads to a more significant deactivation through vibrational coupling to the ground state.18 Both kr and knr are higher in complexes 2 than in the respective tris-cyclometalated complexes fac-[Pt(C∧N)3]+ or mer-[Pt(ppy)2(C′∧N′)]+ bearing the same C∧N or C′∧N′ ligand in all the studied cases (C∧N = dfppy, ppy, tpy; C′∧N′ = thpy, piq).12,13 Notwithstanding, the new compounds exhibit higher quantum yields because the relative increase is much higher in kr than in knr. This fact suggests a significantly higher MLCT contribution to the emitting 3LC state, which is known to accelerate the radiative transition to the ground state.9c,19 In turn, the higher MLCT character is attributable to the strong electron-donating ability of the methyl ligand and the uncharged nature of complexes 2, which must necessarily correlate with a higher energy of the occupied metal d orbitals relative to the cationic tris-cyclometalated complexes and an increased contribution of these orbitals to the HOMO. The Bppy complex 2e shows an impressive quantum yield of 0.81, comparable to the best Pt(II) and Ir(III) emitters8f,19,20 and represents the most efficient Pt(IV) complex ever reported. The excellent phosphorescent efficiency of metal complexes with the Bppy ligand has been previously ascribed to the enhancement of MLCT character in the excited state, which increases kr;21 in addition, this effect combines with an important decrease in knr owing to the presence of the bulky BMes2 substituent.
The emission spectra of 2b were also registered at concentrations in the range 10−5 to 10−3 M in CH2Cl2 at 298 K to probe possible concentration effects (Fig. S8, ESI†). Although the emission profile did not change, a dramatic decrease in intensity was observed at high concentrations, indicating intermolecular quenching. A similar effect has been observed for tris-cyclometalated Pt(IV) derivatives12 and other octahedral emitters,22 most probably due to triplet–triplet annihilation.23
Additional insight into the electronic properties of the new complexes was gathered by comparing the electrochemical data of 2b, obtained using cyclic voltammetry in MeCN solution, with those of fac-[Pt(ppy)3]+13 (Table 2). The cyclic voltammogram of 2b (Fig. S9, ESI†) shows several irreversible reduction peaks that resemble those observed for fac-[Pt(ppy)3]+; the first of them corresponds a one-electron process and occurs at almost the same potential, indicating that the LUMO is based on the ppy ligand. However, while the oxidation of the tris-cyclometalated complex falls outside the solvent discharge limit, the oxidation of 2b occurs at a less positive potential and can be observed, which implies a significantly higher HOMO energy and is consistent with the above-discussed higher metal orbital involvement.
| Complex | E pa | E pc | E HOMO | E LUMO | ΔEHOMO–LUMO |
|---|---|---|---|---|---|
| a In V relative to SCE, measured in 0.1 M (Bu4N)PF6 anhydrous MeCN solution at 100 mV s−1. b In eV. c Irreversible anodic peak potential. d First irreversible cathodic peak potential. e Outside solvent window. | |||||
| 2b | 1.93 | −1.83 | −6.50 | −2.97 | 3.53 |
| fac-[Pt(ppy)3]+ | —e | −1.80 | — | −2.71 | — |
Photostability is an important issue in compounds designed for optical or photochemical applications. We have previously observed that tris-cyclometalated Pt(IV) complexes containing one thpy or piq ligand were not stable upon exposure to UV light.13 In contrast, derivatives 2g and 2h, which bear these ligands, remained unaltered upon irradiation with a 36 W UVB lamp in CD2Cl2 solution for 4 hours (Fig. S10 and S11, ESI†).
In summary, we report the one-pot method for the synthesis of luminescent bis-cyclometalated chloro(methyl)platinum(IV) complexes featuring long excited-state lifetimes and remarkably high quantum yields that can reach the level of the best Pt(II) and Ir(III) emitters. Their easier synthesis, wider tunability and higher efficiencies with respect to the previously reported Pt(IV) emitters make them an attractive alternative for diverse applications, including photocatalysis, bioimaging or chemosensing.
This work was supported by Ministerio de Economía y Competitividad, Spain (project CTQ2011-24016, with FEDER support, and FPU grant to F. J.) and Universidad de Murcia.
Notes and references
- V. W.-W. Yam and K. M.-C. Wong, Chem. Commun., 2011, 47, 11579 RSC.
- (a) M. S. Lowry and S. Bernhard, Chem. – Eur. J., 2006, 12, 7970 CrossRef CAS PubMed; (b) J. Kalinowski, V. Fattori, M. Cocchi and J. A. G. Williams, Coord. Chem. Rev., 2011, 255, 2401 CrossRef CAS; (c) W.-Y. Wong and C.-L. Ho, J. Mater. Chem., 2009, 19, 4457 RSC; (d) R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed.
- (a) B. Higgins, B. A. DeGraff and J. N. Demas, Inorg. Chem., 2005, 44, 6662 CrossRef CAS PubMed; (b) D.-L. Ma, V. P.-Y. Ma, D. S.-H. Chan, K.-H. Leung, H.-Z. He and C.-H. Leung, Coord. Chem. Rev., 2012, 256, 3087 CrossRef CAS.
- (a) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed; (b) J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC; (c) D. M. Schultz and T. P. Yoon, Science, 2014, 343, 985 CrossRef CAS PubMed.
- (a) Q. Zhao, C. Huang and F. Li, Chem. Soc. Rev., 2011, 40, 2508 RSC; (b) E. Baggaley, J. A. Weinstein and J. A. G. Williams, Coord. Chem. Rev., 2012, 256, 1762 CrossRef CAS; (c) E. Baggaley, S. W. Botchway, J. W. Haycock, H. Morris, I. V. Sazanovich, J. A. G. Williams and J. A. Weinstein, Chem. Sci., 2014, 5, 879 RSC.
- (a) M. Grätzel, Acc. Chem. Res., 2009, 42, 1788 CrossRef PubMed; (b) C. A. Bignozzi, R. Argazzi, R. Boaretto, E. Busatto, S. Carli, F. Ronconi and S. Caramori, Coord. Chem. Rev., 2013, 257, 1472 CrossRef CAS.
- (a) A. Ruggi, F. W. B. van Leeuwen and A. H. Velders, Coord. Chem. Rev., 2011, 255, 2542 CrossRef CAS; (b) P. I. Djurovich, D. Murphy, M. E. Thompson, B. Hernandez, R. Gao, P. L. Hunt and M. Selke, Dalton Trans., 2007, 3763 RSC; (c) R. Lincoln, L. Kohler, S. Monro, H. Yin, M. Stephenson, R. Zong, A. Chouai, C. Dorsey, R. Hennigar, R. P. Thummel and S. A. McFarland, J. Am. Chem. Soc., 2013, 135, 17161 CrossRef CAS PubMed.
- (a) J. Brooks, Y. Babayan, S. Lamansky, P. I. Djurovich, I. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2002, 41, 3055 CrossRef CAS PubMed; (b) A. F. Rausch, L. Murphy, J. A. G. Williams and H. Yersin, Inorg. Chem., 2012, 51, 312 CrossRef CAS PubMed; (c) S. Culham, P.-H. Lanoë, V. L. Whittle, M. C. Durrant, J. A. G. Williams and V. N. Kozhevnikov, Inorg. Chem., 2013, 52, 10992 CrossRef CAS PubMed; (d) W. A. Tarran, G. R. Freeman, L. Murphy, A. M. Benham, R. Kataky and J. A. G. Williams, Inorg. Chem., 2014, 53, 5738 CrossRef CAS PubMed; (e) A. M. Prokhorov, T. Hofbeck, R. Czerwieniec, A. F. Suleymanova, D. N. Kozhevnikov and H. Yersin, J. Am. Chem. Soc., 2014, 136, 9637 CrossRef CAS PubMed; (f) S. Huo, J. Carroll and D. A. K. Vezzu, Asian J. Org. Chem., 2015, 4, 1210 CrossRef CAS.
- (a) S. Lamansky, P. Djurovich, D. Murphy, F. Abdel-Razzaq, R. Kwong, I. Tsyba, M. Bortz, B. Mui, R. Bau and M. E. Thompson, Inorg. Chem., 2001, 40, 1704 CrossRef CAS PubMed; (b) A. B. Tamayo, B. D. Alleyne, P. I. Djurovich, S. Lamansky, I. Tsyba, N. N. Ho, R. Bau and M. E. Thompson, J. Am. Chem. Soc., 2003, 125, 7377 CrossRef CAS PubMed; (c) P.-T. Chou, Y. Chi, M.-W. Chung and C.-C. Lin, Coord. Chem. Rev., 2011, 255, 2653 CrossRef CAS; (d) Y. You and S. Y. Park, Dalton Trans., 2009, 1267 RSC; (e) Y. You and W. Nam, Chem. Soc. Rev., 2012, 41, 7061 RSC; (f) K. Beydoun, M. Zaarour, J. A. G. Williams, T. Roisnel, V. Dorcet, A. Planchat, A. Boucekkine, D. Jacquemin, H. Doucet and V. Guerchais, Inorg. Chem., 2013, 52, 12416 CrossRef CAS PubMed; (g) Y. You, S. Cho and W. Nam, Inorg. Chem., 2014, 53, 1804 CrossRef CAS PubMed; (h) P.-H. Lanoe, C. M. Tong, R. W. Harrington, M. R. Probert, W. Clegg, J. A. G. Williams and V. N. Kozhevnikov, Chem. Commun., 2014, 50, 6831 RSC; (i) B. J. Coe, M. Helliwell, S. Sánchez, M. K. Peers and N. S. Scrutton, Dalton Trans., 2015, 44, 15420 RSC.
- L. Chassot, A. Von Zelewsky, D. Sandrini, M. Maestri and V. Balzani, J. Am. Chem. Soc., 1986, 108, 6084 CrossRef CAS PubMed.
- (a) H. Kunkely and A. Vogler, Coord. Chem. Rev., 1991, 111, 15 CrossRef CAS; (b) K. P. Balashev, J. Simon and P. C. Ford, Inorg. Chem., 1991, 30, 859 CrossRef CAS; (c) D. M. Jenkins and S. Bernhard, Inorg. Chem., 2010, 49, 11297 CrossRef CAS PubMed.
- F. Juliá, D. Bautista, J. M. Fernández-Hernández and P. González-Herrero, Chem. Sci., 2014, 5, 1875 RSC.
- F. Juliá, G. Aullón, D. Bautista and P. González-Herrero, Chem. – Eur. J., 2014, 20, 17346 CrossRef PubMed.
- G. S. Hill, M. J. Irwin, C. J. Levy, L. M. Rendina, R. J. Puddephatt, R. A. Andersen and L. McLean, in Inorganic Syntheses, ed. M. Y. Darensbourg, John Wiley & Sons, Inc., 1998, p. 149 Search PubMed.
- (a) M. G. Haghighi, M. Rashidi, S. M. Nabavizadeh, S. Jamali and R. J. Puddephatt, Dalton Trans., 2010, 39, 11396 RSC; (b) S. Jamali, R. Czerwieniec, R. Kia, Z. Jamshidi and M. Zabel, Dalton Trans., 2011, 40, 9123 RSC; (c) S. Jamali, S. M. Nabavizadeh and M. Rashidi, Inorg. Chem., 2008, 47, 5441 CrossRef CAS PubMed.
- (a) J. Mamtora, S. H. Crosby, C. P. Newman, G. J. Clarkson and J. P. Rourke, Organometallics, 2008, 27, 5559 CrossRef CAS; (b) C. P. Newman, K. Casey-Green, G. J. Clarkson, G. W. V. Cave, W. Errington and J. P. Rourke, Dalton Trans., 2007, 3170 RSC.
- CCDC 1435571–1435574.
- (a) K. F. Freed and J. Jortner, J. Chem. Phys., 1970, 52, 6272 CrossRef CAS; (b) J. Englman and J. Jortner, Mol. Phys., 1970, 18, 145 CrossRef.
- H. Yersin, A. F. Rausch, R. Czerwieniec, T. Hofbeck and T. Fischer, Coord. Chem. Rev., 2011, 255, 2622 CrossRef CAS.
- Y. Chi and P.-T. Chou, Chem. Soc. Rev., 2010, 39, 638 RSC.
- (a) Z. M. Hudson, C. Sun, M. G. Helander, H. Amarne, Z.-H. Lu and S. Wang, Adv. Funct. Mater., 2010, 20, 3426 CrossRef CAS; (b) Z. M. Hudson, C. Sun, M. G. Helander, Y.-L. Chang, Z.-H. Lu and S. Wang, J. Am. Chem. Soc., 2012, 134, 13930 CrossRef CAS PubMed; (c) Y.-L. Rao and S. Wang, Inorg. Chem., 2009, 48, 7698 CrossRef CAS PubMed; (d) Z. M. Hudson and S. Wang, Dalton Trans., 2011, 40, 7805 RSC.
- C.-H. Yang, M. Mauro, F. Polo, S. Watanabe, I. Muenster, R. Fröhlich and L. De Cola, Chem. Mater., 2012, 24, 3684 CrossRef CAS.
- M. Baldo, C. Adachi and S. Forrest, Phys. Rev. B: Condens. Matter Mater. Phys., 2000, 62, 10967 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and characterization data. CCDC 1435571–1435574. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09566b |
| This journal is © The Royal Society of Chemistry 2016 |