
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dynamics of formation of Ru, Os, Ir and Au metal nanocrystals on doped graphitic surfaces
Anaïs
Pitto-Barry
 ,
Peter J.
Sadler
and
Nicolas P. E.
Barry
*
,
Peter J.
Sadler
and
Nicolas P. E.
Barry
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: N.Barry@warwick.ac.uk; Tel: +44 (0)2476 524375
First published on 24th December 2015
Abstract
The fabrication of precious metal (ruthenium, osmium, gold, and iridium) nanocrystals from single atoms has been studied in real-time. The dynamics of the first stage of the metal nanocrystallisation on a doped (B,S)-graphitic surface are identified, captured, and reported.
Metal nanocrystals offer considerable potential for applications in healthcare,1–3 electronics,4 and catalysis.5 Their dimensions (1–100 nm) lead to physical and chemical properties that differ from those of bulk materials. Methods for fabricating nanocrystals are generally divided into two main approaches: top-down methods and bottom-up methods.6 The former relies on a progressive removal of material until the desired nanomaterial is obtained,7 whilst the latter uses atomic or molecular precursors and gradually assembles them until the desired structure is formed.8 Examples of physical and chemical techniques for the fabrication of Ru, Au, and Ir nanocrystals are summarised in Table 1.
| Metal | Fabrication technique | Year | Ref. |
|---|---|---|---|
| Ru | Chemical reduction | 2012, 2013 | 9–11 |
| Thermal reduction on carbon-based substrates | 2011 | 12 | |
| Chemical decomposition | 2005 | 13, 14 | |
| Microwave irradiation | 2011 | 15–17 | |
| Ir | Seed-mediated growth | 2014 | 18 |
| Atomic layer deposition | 2014 | 19 | |
| Chemical reduction | 2009, 2014 | 20–22 | |
| Thermal annealing process | 2010 | 23 | |
| Au | Thermal treatment | 2009, 2014 | 24, 25 |
| Nanopatterning: seed-mediated growth with a surfactant | 2013 | 26 | |
| Colloidal lithography and surface energy-driven dewetting process | 2013 | 27 | |
| DNA-mediated self-assembly | 2013 | 28 | |
| Light-induced rapid annealing | 2014 | 29 | |
| Chemical reduction | 1994, 2009 | 30, 31 | |
Nanocrystallisation follows a three-stage approach: nucleation, evolution of nuclei into seeds, and growth of seeds into nanocrystals.32 Nucleation is the first step of any crystallisation process, and understanding this phenomenon is of importance not only in biochemistry, e.g. nucleation of bubbles in DNA for replication and transcription, crystallisation of proteins, replication of viruses, but also in materials science, e.g. growth of metal nanocrystals and nanoparticles.33,34 Recent advances allowing time-resolved investigations of self-assembly processes through atomic resolution and single-atom sensitivity include direct imaging of the dynamics of Si6 clusters embedded in graphene pores,35 observation of the trapping of metal atoms in graphene,36 capture of atomic-level random walks of a defect in graphene,37 and direct visualisation of small clusters of gold atoms.38–41 Nonetheless, imaging the early stage of a nanocrystallisation process in real-time and in real-space is challenging and little is known about growth rates of nuclei, the minuscule clusters made of a few atoms formed in the earliest stage of nanocrystal synthesis.
We have recently developed a new technology based on transmission electron microscopy, which allows the direct observation, in real-time, of atom-by-atom fabrication of metal nanocrystals.42–44 This methodology involves electron beam irradiation of metallated micelles with an aberration-corrected high-resolution transmission electron microscope (AC-TEM). Upon irradiation, the micelles form a doped graphitic support on which individual metal atoms hop and migrate to form nanocrystals, as small as 15 Å in diameter in less than 1 h. As an initial example, we observed, quantified, and controlled the growth of osmium nanocrystals in real time. Using this methodology, we report here the fabrication of gold, ruthenium, and iridium nanocrystals, precious metal nanoparticles. We capture the aggregation of individual atoms to form nuclei. The nanocrystallisation growth rate is determined for each precious metal, providing new insights into the nucleation step of nanocrystal formation. We prepared block copolymer micelles containing 16-electron dithiocarborane complexes of Ru(II), Os(II), Ir(III) or Au(III) (Fig. 1), and irradiated them with the electron beam of an aberration-corrected TEM-STEM with a Schottky thermal field-emission source (80 keV; 1.9 pA cm−2; 7.6 × 107 electrons nm−2 s−1).
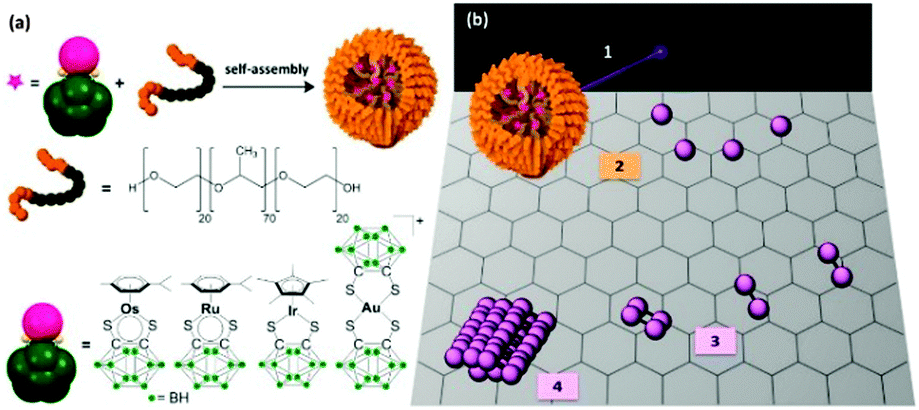 | ||
| Fig. 1 (a) Self-assembly of block copolymer micelles containing encapsulated metal (Os, Ru, Ir, or Au) carborane complexes. (b) The methodology used in this work is the following: (1) electron beam irradiation of the metallated micelles; (2) degradation and production of a graphenic surface (without attempt to indicate B and S doping) on which individual metal atoms can hop and migrate; (3) metal nucleation and formation of small molecules, clusters; (4) metal nanocrystal formation. | ||
OsMs and RuMs micelles containing [Os/Ru(p-cymene)(1,2-dicarba-closo-dodecarborane-1,2-dithiolate)] encapsulated in triblock copolymer Pluronic® P123 were synthesised following our reported procedure,44,45 while the Au-containing AuMs and Ir-containing IrMs micelles were synthesised following a similar procedure (see Materials and methods). To ensure first that the micelle degradation leads to the formation of metal nanocrystals for each metal, we irradiated the TEM grids for up to 150 min. As previously reported with OsMs, we observed structural changes within the Pluronic® film formed by the spreading of RuMs, AuMs, and IrMs within the holes of lacey carbon TEM grids upon irradiation with the high-energy electron beam. The emergence of atomic ordering within the self-supporting matrix consistent with a turbostratic graphitic structure was apparent within a few minutes. Along with these structural modifications of the self-supporting polymeric film, a rapid decomposition of the carborane-containing Ru, Au, and Ir complexes was also observed (in less than 1 min), and precious metal nanoparticles were imaged after 120 min (Fig. 2).
 | ||
| Fig. 2 AC-TEM images of Ru, Os, Ir, and Au nanocrystals (with corresponding fast Fourier transform) on a turbostratic graphitic structure obtained after 120 min electron beam irradiation. Scale bar 1 nm. | ||
Significantly, the in situ-generated precious metal nanoparticles on the self-supporting graphitic matrix are crystalline. This is illustrated by the FFT analysis (Fig. 2) of the ca. 1.5 nm-diameter Os, Ru, Au, and Ir nanocrystals depicted in Fig. 2. Such observations not only generalise this atom-by-atom fabrication methodology to a range of precious metals, but also show that various types of metal complexes can be used as precursors. The pseudo-octahedral Ru(II) complex is a half-sandwich organometallic arene complex (structurally analogous to the Os(II) precursor), whereas the half-sandwich Ir(III) complex has a cyclopentadienyl ligand, whilst the Au(III) complex has a square-planar geometry and is not an organometallic compound. Hence it is evident that the presence of an aromatic ring in the chemical structure of the precursor complex is not needed to generate the underlying graphitic support or induce formation of nanocrystals using this methodology, nor is having an octahedral complex as precursor. This work suggests that the methodology is also likely to be successful using other square-planar precious metal complexes such as Pd(II) and Pt(II) as precursors. The ability to control the formation of very small (ca. 1.5 nm diameter) Au nanocrystals is particularly significant in view of current intense interest in catalysis, sensing, and quantum plasmonics.46,47
We then imaged the early steps of nuclei aggregation for each metal (Table 2 and Fig. 3). The metal–metal distances for Au, Os, Ir, and Ru were first measured on small molecules of only a few atoms on the surface. The average Os–Os, Au–Au, Ir–Ir, and Ru–Ru distances determined for 30 different clusters per irradiation time are close to those in crystals of the respective bulk metals (Table 2). The metal–metal distances are only indicative due to the 3D nature of the nanocrystals. The slightly smaller distances observed in the nanocrystals as compared to the bulk materials might therefore be due to the stacking of several layers of metal atoms. No change in metal–metal distance was observed during nanocrystal growth, as shown by the reasonably narrow standard deviation for the average M–M distance calculated from nanoclusters/nanocrystals of various sizes (width between 0.5 and 2 nm).
| Metal | M–M distance (nm) | Growth rate (pm min−1) | |
|---|---|---|---|
| Nanocrystal | Bulk30,31 | ||
| Ru | 0.248 ± 0.072 | 0.2650 | 7.7 |
| Os | 0.257 ± 0.019 | 0.2705 | 17.1 |
| Ir | 0.268 ± 0.058 | 0.2715 | 5.3 |
| Au | 0.273 ± 0.049 | 0.2880 | 6.1 |
 | ||
| Fig. 3 Atom-by-atom growth of Ru (left) and Au (right) nanocrystals over time and interatomic distances within nuclei. Scale bar 1 nm. | ||
Measurements of the length of the atomic aggregates indicate a linear relationship with time (1 to 105 min) in the four cases (Ru, Os, Au, Ir – Fig. 4). These trends suggest that a degree of control over the size of the nanocrystals can be achieved by modulating the irradiation time on the TEM grids. Such reasonably linear trends allowed us to estimate the nanocrystal growth rates for these four precious metals (Table 2). Osmium nanocrystal growth, for example, is 2.2 to 3.2 × faster compared to the three other metals, while iridium nucleation is the slowest.
 | ||
| Fig. 4 Width of the clusters/crystals versus irradiation time. The error bars are the standard deviations from measurements on at least 30 clusters at each time point. | ||
The kinetic energy of the electron beam is probably the main driving force for the observed dynamic behaviour of the metal atoms observed in TEM, and was set to be the same for all the experiments reported here (1.9 pA cm−2). This may explain the narrow range of crystal growth observed for each metal. The interactions of individual Ru, Os, Au, and Ir atoms with the underlying surface may also have a direct impact on the rates of nucleation. The in situ-produced (via electron beam degradation of the metallated micelle precursors) graphitic surface which supports the growth of the metal nanocrystals is doped with boron, and sulfur atoms (10–20 boron atoms for one metal ion, and 2–4 sulfur atoms per metal ion).42–44 These hetero-atoms can act as trapping sites for the single metal atoms, slowing down their motions on the surface, with a rate-dependency related to the nature of the metal and the doping atoms. For example, the high affinity of sulfur for gold is well-known,48 and strong Au–S interactions might explain the slow rate of nanocrystal formation observed for Au compared to Os, and Ru, with Au atoms perhaps being tightly trapped on the S sites. The Au precursor is also the only complex in the series studied with two carborane ligands in its molecular structure, and therefore, the graphitic support is likely to be doped with more B and S atoms than those formed by irradiation of micelles OsMs, RuMs, and IrMs. Differences in metal–metal affinity may also have a marked effect on the rate of nucleation. The aurophilicity, or tendency of gold aggregation via gold–gold interactions, is expected to have a direct impact on the nucleation, but would be expected to facilitate the formation of Au nanocrystals. Such relativistic effects might also occur with the heavy Ir atoms, however, Ir nanocrystals have the slowest formation rate. These observations will stimulate future work combining further experiments on nanocrystal nucleation with advanced calculations and modelling.
The high-energy electron beam of high-resolution (HR) TEM is not only a powerful analytical tool but may be also utilised as an external force to modify in situ the structure and composition of various nano-scale materials, and to tailor the assembly of nanoparticles.49–55 For instance, gold nanoclusters have been reported to undergo sintering under electron irradiation, driven by surface diffusion.49 Here, we have used both capabilities of HR-TEM (analysis and modification of the structure of nanomaterials) to study the formation and growth rate of metal nanocrystals of the precious metals Ru, Os, Au, and Ir. It is possible to image the early stage dynamics of metal nanocrystal formation and control the sizes of the resulting nanocrystals on doped graphitic supports. In the present case we have generated boron/sulfur-doped supports, and as we have shown recently,43 other hetero atoms such as Se can be readily incorporated with consequent effects of the dynamics of metal migration. Understanding of the fundamental mechanisms of interactions between incident electrons and atoms/molecules might provide unique information on the chemical structure and dynamics of a wide variety of systems, including mixed metal alloys.44,56 Utilising the properties and dynamics of these small nanocrystals for applications in e.g. biotechnology and materials now presents exciting future challenges.
Materials: the preparations of the complexes [Os/Ru(p-cym)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)], [Ir(Cp*)(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)], and [Au(1,2-dicarba-closo-dodecarborane-1,2-dithiolato)2][NBu4], OsMs and RuMs were based on previous reports.44,45 The triblock copolymer P123 [poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol)] was purchased from Sigma-Aldrich. Anhydrous tetrahydrofuran (Aldrich) was used. 18.2 MΩ cm purity water was collected from a Purelab® UHQ USF Elga system. Holey carbon grids with 200 mesh and lacey carbon grids were purchased from Quantifoil Micro Tools Gmbh and Elektron Technology UK Ltd, respectively. Synthesis of AuMs and IrMs: a tetrahydrofuran (THF) solution (1 mL) of Au or Ir complex (5 mg mL−1) was added to an aqueous solution (10 mL) of polymer P123 (5 mg mL−1) and the resultant mixture was stirred at ambient temperature for 4 h. The solution was then dialysed to remove the THF (MWCO = 1000 Da), for 48 h, and then freeze-dried. High resolution electron microscopy HR-TEM: A JEOL JEM-ARM200F HR-TEM was operated at 80 keV, 1.9 pA cm−2, with spherical aberration (Cs) tuned to approximately +1 μm and images were recorded on a Gatan SC1000 Orius CCD camera. All the images were analysed with ImageJ (Fiji) software. All the stacks were aligned with the plugin software StackReg.57 The image sequences were independently aligned using a Digital Micrograph(TM) script. Adjacent images were aligned by analysis of the normalised cross-correlation of a user-selected region in the image stack. The centre of the cross-correlation peak was found to sub-pixel accuracy using either (a) the centroid of the correlation peak or (b) fitting of parabolas to the peak in x- and y-directions, while linear interpolation was used to produce sub-pixel image shift.
We thank the Leverhulme Trust (Early Career Fellowship No. ECF-2013-414 to NPEB), the ERC (Grant No. 247450 to PJS), EPSRC (EP/F034210/1 to PJS). We also thank Dr Ana Sanchez for her help with the microscopy experiments.
Notes and references
- E.-K. Lim, T. Kim, S. Paik, S. Haam, Y.-M. Huh and K. Lee, Chem. Rev., 2014, 115, 327–394 CrossRef PubMed.
- J. Yao, M. Yang and Y. Duan, Chem. Rev., 2014, 114, 6130–6178 CrossRef CAS PubMed.
- J. A. Hubbell and A. Chilkoti, Science, 2012, 337, 303–305 CrossRef PubMed.
- D. Jariwala, V. K. Sangwan, L. J. Lauhon, T. J. Marks and M. C. Hersam, Chem. Soc. Rev., 2013, 42, 2824–2860 RSC.
- B. Cornelio, G. A. Rance, M. Laronze-Cochard, A. Fontana, J. Sapi and A. N. Khlobystov, J. Mater. Chem. A, 2013, 1, 8737–8744 CAS.
- A. Biswas, I. S. Bayer, A. S. Biris, T. Wang, E. Dervishi and F. Faupel, Adv. Colloid Interface Sci., 2012, 170, 2–27 CrossRef CAS PubMed.
- H.-D. Yu, M. D. Regulacio, E. Ye and M.-Y. Han, Chem. Soc. Rev., 2013, 42, 6006–6018 RSC.
- T. Rajagopalan, K. Venumadhav, G. Arkasubhra, C. Nripen, G. Keshab and G. Shubhra, Rep. Prog. Phys., 2013, 76, 066501 CrossRef PubMed.
- C. Koenigsmann, D. B. Semple, E. Sutter, S. E. Tobierre and S. S. Wong, ACS Appl. Mater. Interfaces, 2013, 5, 5518–5530 CAS.
- J. Watt, C. Yu, S. L. Y. Chang, S. Cheong and R. D. Tilley, J. Am. Chem. Soc., 2012, 135, 606–609 CrossRef PubMed.
- P. Lara, K. Philippot and B. Chaudret, ChemCatChem, 2013, 5, 28–45 CrossRef CAS.
- J. Liu, P. Bai and X. S. Zhao, Phys. Chem. Chem. Phys., 2011, 13, 3758–3763 RSC.
- V. Hulea, D. Brunel, A. Galarneau, K. Philippot, B. Chaudret, P. J. Kooyman and F. Fajula, Microporous Mesoporous Mater., 2005, 79, 185–194 CrossRef CAS.
- C. Amiens, B. Chaudret, D. Ciuculescu-Pradines, V. Colliere, K. Fajerwerg, P. Fau, M. Kahn, A. Maisonnat, K. Soulantica and K. Philippot, New J. Chem., 2013, 37, 3374–3401 RSC.
- D. Marquardt, C. Vollmer, R. Thomann, P. Steurer, R. Mülhaupt, E. Redel and C. Janiak, Carbon, 2011, 49, 1326–1332 CrossRef CAS.
- R. B. Nasir Baig and R. S. Varma, ACS Sustainable Chem. Eng., 2013, 1, 805–809 CAS.
- B. Zhang, X. Ni, W. Zhang, L. Shao, Q. Zhang, F. Girgsdies, C. Liang, R. Schlogl and D. S. Su, Chem. Commun., 2011, 47, 10716–10718 RSC.
- X. Xia, L. Figueroa-Cosme, J. Tao, H.-C. Peng, G. Niu, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2014, 136, 10878–10881 CrossRef CAS PubMed.
- X.-J. Liu, L. Zhu, X.-F. Li, Z.-Y. Cao, A.-D. Li and D. Wu, J. Vac. Sci. Technol., B: Nanotechnol. Microelectron.: Mater., Process., Meas., Phenom., 2014, 32, 042201 Search PubMed.
- C. Kang, L. Wang, Z. Bian, H. Guo, X. Ma, X. Qiu and L. Gao, Chem. Commun., 2014, 50, 13979–13982 RSC.
- D.-G. Lee, S. M. Kim, H. Jeong, J. Kim and I. S. Lee, ACS Nano, 2014, 8, 4510–4521 CrossRef CAS PubMed.
- J. Yang, E. Sargent, S. Kelley and J. Y. Ying, Nat. Mater., 2009, 8, 683–689 CrossRef CAS PubMed.
- T. T.-J. Wang, C.-L. Chu, I.-J. Hsieh and W.-S. Tseng, Appl. Phys. Lett., 2010, 97, 143507 CrossRef.
- Y. Qin, J. Li, Y. Kong, X. Li, Y. Tao, S. Li and Y. Wang, Nanoscale, 2014, 6, 1281–1285 RSC.
- N. Goubet, Y. Ding, M. Brust, Z. L. Wang and M.-P. Pileni, ACS Nano, 2009, 3, 3622–3628 CrossRef CAS PubMed.
- C. Zhengbo, G. Junxia, L. Jing and G. Lin, Nanotechnology, 2013, 24, 295501 CrossRef PubMed.
- Y.-K. Lin, H.-W. Ting, C.-Y. Wang, S. Gwo, L.-J. Chou, C.-J. Tsai and L.-J. Chen, Nano Lett., 2013, 13, 2723–2731 CrossRef CAS PubMed.
- S. J. Barrow, X. Wei, J. S. Baldauf, A. M. Funston and P. Mulvaney, Nat. Commun., 2012, 3, 1275 CrossRef PubMed.
- X. Chen, Y. Chen, J. Dai, M. Yan, D. Zhao, Q. Li and M. Qiu, Nanoscale, 2014, 6, 1756–1762 RSC.
- R. Ristau, R. Tiruvalam, P. Clasen, E. Gorskowski, M. Harmer, C. Kiely, I. Hussain and M. Brust, Gold Bull., 2009, 42, 133–143 CrossRef CAS.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- Y. Kim and E. W. Prohofsky, Phys. Rev. B: Condens. Matter Mater. Phys., 1987, 35, 2399–2404 CrossRef CAS.
- T. C.-J. Yang, Y. Kauffmann, L. Wu, Z. Lin, X. Jia, B. Puthen-Veettil, T. Zhang, G. Conibeer, I. Perez-Wurfl and A. Rothschild, Appl. Phys. Lett., 2014, 105, 053116 CrossRef.
- J. Lee, W. Zhou, S. J. Pennycook, J.-C. Idrobo and S. T. Pantelides, Nat. Commun., 2013, 4, 1650 CrossRef PubMed.
- J. A. Rodríguez-Manzo, O. Cretu and F. Banhart, ACS Nano, 2010, 4, 3422–3428 CrossRef PubMed.
- J. Kotakoski, C. Mangler and J. C. Meyer, Nat. Commun., 2014, 5, 4991 CrossRef PubMed.
- S. R. Plant, L. Cao and R. E. Palmer, J. Am. Chem. Soc., 2014, 136, 7559–7562 CrossRef CAS PubMed.
- S. R. Plant, L. Cao, F. Yin, Z. W. Wang and R. E. Palmer, Nanoscale, 2014, 6, 1258–1263 RSC.
- Y. Han, D. S. He and Z. Y. Li, J. Nanopart. Res., 2013, 15, 1–7 Search PubMed.
- A. La Torre, M. del Carmen Gimenez-Lopez, M. W. Fay, C. H. Lucas, P. D. Brown and A. N. Khlobystov, Small, 2015, 11, 2756–2761 CrossRef CAS PubMed.
- A. Pitto-Barry, L. M. A. Perdigao, M. Walker, J. Lawrence, G. Costantini, P. J. Sadler and N. P. E. Barry, Dalton Trans., 2015, 44, 20308–20311 RSC.
- N. P. E. Barry, A. Pitto-Barry, J. Tran, S. E. F. Spencer, A. M. Johansen, A. M. Sanchez, A. P. Dove, R. K. O'Reilly, R. J. Deeth, R. Beanland and P. J. Sadler, Chem. Mater., 2015, 27, 5100–5105 CrossRef CAS PubMed.
- N. P. E. Barry, A. Pitto-Barry, A. M. Sanchez, A. P. Dove, R. J. Procter, J. J. Soldevila-Barreda, N. Kirby, I. Hands-Portman, C. J. Smith, R. K. O'Reilly, R. Beanland and P. J. Sadler, Nat. Commun., 2014, 5, 3851 CAS.
- N. P. E. Barry, A. Pitto-Barry, I. Romero-Canelon, J. Tran, J. J. Soldevila-Barreda, I. Hands-Portman, C. J. Smith, N. Kirby, A. P. Dove, R. K. O'Reilly and P. J. Sadler, Faraday Discuss., 2014, 175, 229–240 RSC.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS PubMed.
- E. Townsend and G. W. Bryant, Nano Lett., 2012, 12, 429–434 CrossRef CAS PubMed.
- H. Hakkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- X. Qi, Y. Huang, M. Klapper, F. Boey, W. Huang, S. D. Feyter, K. Müllen and H. Zhang, J. Phys. Chem. C, 2010, 114, 13465–13470 CAS.
- J.-U. Kim, S.-H. Cha, K. Shin, J. Y. Jho and J.-C. Lee, J. Am. Chem. Soc., 2005, 127, 9962–9963 CrossRef CAS PubMed.
- A. H. Latham and M. E. Williams, Langmuir, 2008, 24, 14195–14202 CrossRef CAS PubMed.
- A. H. Latham, M. J. Wilson, P. Schiffer and M. E. Williams, J. Am. Chem. Soc., 2006, 128, 12632–12633 CrossRef CAS PubMed.
- C. W. Huang, C. L. Hsin, C. W. Wang, F. H. Chu, C. Y. Kao, J. Y. Chen, Y. T. Huang, K. C. Lu, W. W. Wu and L. Chen, Nanoscale, 2012, 4, 4702–4706 RSC.
- J. W. Liu, J. E. Xu, Y. Ni, F. J. Fan, C. L. Zhang and S. H. Yu, ACS Nano, 2012, 6, 4500–4507 CrossRef CAS PubMed.
- Z. W. Wang and R. E. Palmer, Phys. Rev. Lett., 2012, 108, 245502 CrossRef CAS PubMed.
- T. W. Chamberlain, J. Biskupek, S. T. Skowron, P. A. Bayliss, E. Bichoutskaia, U. Kaiser and A. N. Khlobystov, Small, 2015, 11, 622–629 CrossRef CAS PubMed.
- P. Thevenaz, U. E. Ruttimann and M. Unser, IEEE Transactions on Image Processing, 1998, 7, 27–41 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |