
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing electrocatalytic hydrogen evolution by nickel salicylaldimine complexes with alkali metal cations in aqueous media†
Haiyan
Shao
abc,
Subas K.
Muduli
ad,
Phong D.
Tran‡
c and
Han Sen
Soo
*abc
aDivision of Chemistry and Biological Chemistry, School of Physical and Mathematical Sciences, Nanyang Technological University, Singapore 637371, Singapore. E-mail: hansen@ntu.edu.sg
bSingapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE), 1 Create Way, Singapore 138602, Singapore
cSolar Fuels Laboratory, Nanyang Technological University, 50 Nanyang Avenue, Singapore 639798, Singapore
dEnergy Research Institute@NTU (ERI@N), Nanyang Technological University, Research Techno Plaza, Singapore 637553, Singapore
First published on 11th January 2016
Abstract
New salicylaldimine nickel complexes, comprising only earth-abundant elements, have been developed for electrocatalytic hydrogen evolution in aqueous media. The second-sphere ether functionalities on the periphery of the complexes enhance the electrocatalytic activity in the presence of alkali metal cations. The electrocatalysts demonstrate improved performances especially in the economical and sustainable seawater reaction medium.
Global climate change has been widely attributed to the rampant consumption of fossil fuels and has stimulated recent efforts to develop alternative, more sustainable sources of fuels. An attractive solution is to develop artificial photosynthetic systems that can harvest sunlight, like a leaf, and store the solar energy in the form of chemical bonds in hydrogen (H2) and oxygen (O2) via water splitting. Energy storage is appealing since sunlight is intermittent. When alternative energy resources such as sunlight or geothermal energy can be efficiently converted into electricity, a critical challenge in creating a practical artificial photosynthetic system will be to develop commensurately efficient electrocatalysts for H2 evolution from seawater.1–4
Platinum (Pt) is a well-known electrocatalyst for the reduction of protons (H+) to H2 in commercial electrolysers, with a low overpotential (η), long durability, and high turnover frequency (TOF).3 However, Pt is a precious metal, partly due to its relative scarcity in the Earth's crust. Consequently, more earth-abundant, heterogeneous electrocatalysts have been sought and reported, including metal chalcogenides,5–7 carbides,8 phosphides,9–11 and hydroxides,12,13 in attempts to produce H2 at prices competitive with those in the production from fossil fuels.
Concurrent with the research on heterogeneous catalysts, a number of molecular complexes consisting of earth-abundant elements have been found to be exceptional H2 evolution electrocatalysts. These complexes display especially impressive TOFs in organic solvents, some of which surpass the activities of natural hydrogenase enzymes, and are supported by ligands including N2P2,14–17 macrocyclic oximes,18–20 polypyridines,21,22 thiolates,23–26 and porphyrins.27 A select few of the molecular electrocatalysts are active in aqueous media and ligated by scaffolds including PY5Me2,28 DPA-Bpy,29 and P4N2.30 Co salicylaldimine catalysts have also been used for photodriven H2 evolution in water.31 Among the most active catalysts, a common feature has been the judicious customisation of the second coordination sphere via the introduction of bioinspired H+ relays. These complexes poise the Lewis basic amine and Bronsted acidic alcohol groups in close proximity to the redox-active metal centre, similar to natural enzymatic systems. However, few studies have documented the effects of alkali metal ions in the second coordination sphere of molecular H2 evolution electrocatalysts, although copious amounts of alkali metal cations are found in seawater and biological systems.
Herein, we describe the H2 evolution activity of new nickel (Ni) complexes supported by salicylaldimine ligands with pendant, chelating ether groups that can bind Lewis acids such as alkali metal cations. The alkali metal ion-binding capabilities of salen-type ligands without pendant ether functionalities have been previously observed in molecular CO2 complexes.32 The role of alkali metal cations is important since they constitute the most practical and cheapest electrolytes, and will be especially relevant if seawater (Na+) is to become the ultimate aqueous medium for large-scale H+ reduction.
We demonstrate that our water-soluble Ni electrocatalysts, which are readily accessible in a few steps from commercially available chemicals (Scheme 1), behave as H2 evolution catalysts in both neutral and acidic aqueous solutions. Moreover, the chelating methoxyethoxy group can bind hydrated alkali metal cations to provide hydrogen-bonding stabilisation and enhanced electrocatalytic proton reduction reactivity.
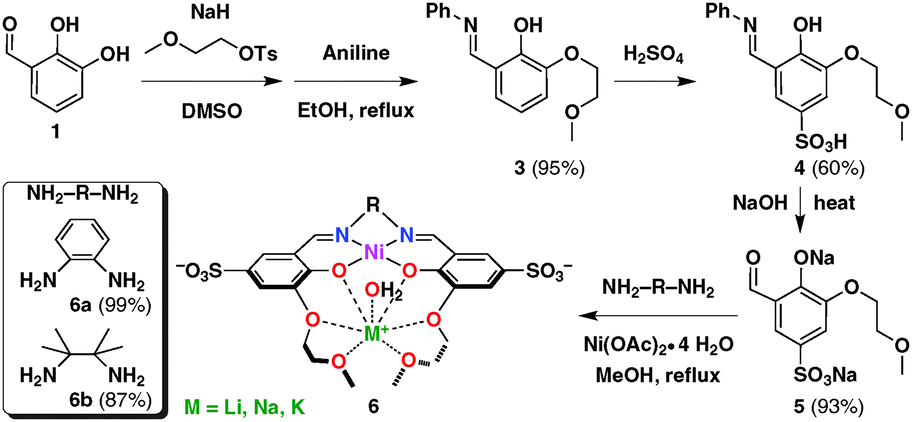 | ||
| Scheme 1 Synthesis of Ni catalysts (6) with pendant ether groups. | ||
The ligands were prepared by adaptations of previously reported methods.33–35 Compound 3 and subsequent organic molecules and Ni complexes are new and their syntheses and characterisation are included in the ESI.† The Ni complexes (6) were prepared by condensation of aldehyde 5, a diamine, and nickel acetate with high yields (Scheme 1). Two design elements were introduced to enhance H2 evolution from saline aqueous media: (i) introduction of sulfonate groups to improve water solubility and (ii) attachment of pendant, non-coordinating ether functionalities as hard Lewis bases for alkali metal cations. The salicylaldimine framework is highly modular and synthetically accessible; the amine and the aldehyde can be independently optimised and derivatised. Two distinct complexes 6 (diamine = o-phenylenediamine, 6a; diamine = 2,3-dimethylbutane-2,3-diamine, 6b) were prepared to compare their activities.
Both complexes have been characterised by NMR spectroscopy, high-resolution mass spectrometry, and elemental analyses. The deep orange 6a displays bands at 216, 312, 360, and 486 nm (ε = 2.48 × 104, 1.03 × 104, 1.19 × 104 and 3.13 × 103 M−1 cm−1, respectively, Fig. S1, ESI†), which have been used to gauge the stability of 6a under catalytic conditions (vide infra). Complex 6b functions similarly to 6a, with lower electrocatalytic activity. All subsequent studies have thus been performed with 6a. Although we have not been able to grow single crystals of 6a or 6b, we were able to prepare 6c, which has the same structure as 6a without the sulfonate groups. Unlike numerous dimeric or polymeric Ni salen complexes,336c is mononuclear (Fig. S2, ESI†).36 The salient structural parameters are listed in Tables S1–S10 (ESI†). The coordination geometry around Ni is square planar, in accordance with the d8 electronic configuration. Notably, the pendant ether arms cradle one K+ cation similar to previous reports with Na+ (other examples in the ESI†),36 potentially directing the solvent molecules in close proximity to the Ni centre. Although this only represents the solid-state structure, the UV-vis (Fig. S3, ESI†) and electrochemical experiments suggest that the alkali metal cations indeed influence the second coordination sphere around Ni in water during H2 evolution catalysis (Scheme S1, ESI†).
The cyclic voltammogram (CV, Fig. S4, ESI†) of 6a in N,N-dimethylformamide (DMF) features a quasi-reversible redox couple at −1.04 V vs. NHE assigned to a metal-based NiII/NiI redox couple. The redox wave becomes increasingly reversible at scan rates up to 1.0 V s−1, which may be due to the structural reorganisation or precipitation of the triply anionic transient species on the electrode.37,38 In the presence of acetic acid (HOAc) as a weak proton donor in DMF, the CVs show moderate current increases with little change to the onset current (Fig. S5, ESI†), indicating that 6a behaves as an electrocatalyst for H2 evolution even with weak acids. When the stronger trifluoroacetic acid (TFA) was used as the proton source, the CV displayed a dramatic increase in the catalytic current over the background (Fig. 1a), with an anodic shift of the onset current. After varying the concentration of TFA added, the CVs reveal a 2nd order dependence on the acid concentration (Fig. S6, ESI†).14 Attempts to perform a foot-of-the-wave analysis39 on H2 evolution in the presence of TFA have been complicated by significant deviations from linearity due to the anodic onset shift at higher TFA concentrations (Fig. S7, ESI†). This onset shift could be due to equilibrium protonation of the phenoxide donor at high acid concentrations (ESI†),40 which reduces the overpotential required for electrocatalytic H2 evolution by positioning protons in close proximity to the Ni centre. Instead, the foot-of-the-wave analysis using acetic acid as the proton donor demonstrates the expected linear behaviour, yielding an apparent rate constant of 17.9 M−1 s−1 in 0.10 M acetic acid at a scan rate of 100 mV s−1 (Fig. S8, ESI†).
 | ||
| Fig. 1 CVs of (a) 1.0 mM of 6a in 0.10 M n-Bu4NPF6 in DMF with different concentrations of TFA; (b) 1.0 mM 6a in 0.10 M Na2SO4 with different HOAc concentrations at scan rates of 100 mV s−1 with a glassy carbon working electrode (3 mm diameter). | ||
Interestingly, the CV of 6a in neutral water with Na2SO4 as the electrolyte exhibits a remarkable increase in the catalytic current at an onset of −1.05 V vs. NHE, corresponding to an overpotential of 0.59 V.§ Gas bubbles are also observed during cathodic scans. The overpotential of 6a is comparable to or lower than that of the other molecular electrocatalysts that contain first-row transition metals and function in neutral water with glassy carbon working electrodes.41 For instance, the overpotentials for Co complexes ligated by P4N2 ligands30 or DPA-Bpy29 are about 0.55 and 0.78 V, respectively, with glassy carbon working electrodes. When 0.10 M aqueous acetic acid is used (pH = 2.8), the catalytic onset shifts to −0.64 V, which is larger than the expected Nernstian shift of about 0.24 V due to an increase in [H+] by about four orders of magnitude (Fig. 1b). In addition, the catalytic current shows a 1st order dependence on the concentration of 6a (Fig. 2a), suggesting that H2 evolution occurs via the protonation of a mononuclear Ni intermediate, rather than bimolecular coupling of two highly anionic Ni complexes (Scheme S1, ESI†).42
 | ||
| Fig. 2 CVs of (a) different concentrations of 6a complex in 0.10 M Na2SO4, 0.10 M HOAc solution. Inset: Linear relationship between current density j and [6a] at −0.80 V; (b) 1.0 mM 6a in 0.10 M of different electrolytes in 0.10 M HOAc at scan rates of 100 mV s−1 with a glassy carbon electrode (3 mm diameter). | ||
To explore the effects of the pendant ether on 6a, CVs have been conducted in the presence of n-Bu4NCl, Li2SO4, Na2SO4, and KPF6 as the electrolytes in 0.10 M acetic acid (Fig. 2b). With n-Bu4N+ as the electrolyte, the current density increases 0.70 mA cm−2 above the background current at an overpotential of 0.84 V in the electrolyte solution with no added acids (Fig. S9d, ESI†). This indicates an appreciable, but slow H2 evolution reaction. The n-Bu4N+ cation is bulky and may hinder the access to the Ni catalytic centre by hydronium ions, similar to the proton-gated hangman iron porphyrins reported by Nocera et al.43 In contrast, with Na2SO4 as the electrolyte, the current density is 8.2 times the catalytic current with n-Bu4N+ as the electrolyte (Fig. S9, ESI†). We observe the highest electrocatalytic current with Li+ as the electrolyte (11 times compared to n-Bu4N+ as the electrolyte, Fig. S9, ESI†) among the common and affordable cations examined. This dramatic increase in catalytic current highlights the non-innocence of the electrolyte in our H2 evolution system, and emphasises the need for prudent management of second sphere effects around the catalytic centre (Scheme S1, ESI†).44–46 In 0.10 M acetic acid, the electrocatalytic current increase also followed the same trend (Fig. S10, ESI†). The most Lewis acidic Li+ corresponds to the fastest catalytic behaviour. The Li+ with the highest charge density could more effectively introduce rigidity to the ether arms and direct H+ to the Ni centre, or electrostatically reduce the overpotential via inductive effects. Since seawater contains copious amounts of Na+ ions, a judicious selection of pendant chelating groups that (size)-select for Na+ may prove fruitful in improving the catalytic performance of 6a and are being explored.
In order to probe whether a heterogeneous catalyst is the major contributor of the H2 evolution electrocatalysis, a series of standard electrochemical and spectroscopic experiments have been performed. The UV-vis, high-resolution mass, and 1H NMR spectra (Fig. S11–S13, ESI†) before and after controlled potential electrolysis (CPE) indicate that 6a remains intact in solution, although there may be some adsorption. There were no nanoparticles observed upon dynamic light scattering (DLS) measurements in the electrolyte solution after CPE for 2.5 h (Fig. S14, ESI†). The UV-vis spectra in Fig. S10 (ESI†) show that the concentration of 6a decreased to 90% and 78% after 1 h and 2.5 h, respectively.¶ We suspected that part of 6a was adsorbed on the carbon paper during the electrolysis process. To verify this conjecture, the used carbon paper electrodes in CPE measurements were rinsed thoroughly with water before being subjected to CPE again (Fig. S15, ESI†). The used electrode showed a small, but non-negligible amount of catalytic activity. X-ray photoelectron spectroscopy (XPS) experiments were conducted on 6a and on the used carbon paper electrode after CPE (Fig. S16 and S17, ESI†). The data indicate that the small amounts of deposited material on the electrode have almost identical composition to 6a, based on the XPS energies (Fig. S16 and Table S11, ESI†). We propose that some of the highly anionic 6a is deposited on the carbon paper electrode upon reduction during catalysis, and cannot be readily rinsed off. A transmission electron microscopy (TEM) image (Fig. S18, ESI†) of the solution after CPE also exhibits little evidence of nanoparticle formation. The majority of the H2 evolution electrocatalysis is still attributed to 6a in homogeneous solution instead of deposited or suspended nanoparticles.
Controlled potential electrolysis (CPE) experiments using a mercury pool electrode have been conducted to confirm the identity of the product by gas chromatography and to gauge the stability of 6a under catalytic conditions. The CPE measurements were performed in seawater obtained off the coast of Singapore (details in the ESI†) with a mercury pool working electrode and a carbon rod counter electrode, at an overpotential of 0.84 V. As shown in Fig. 3a, the amount of H2 produced increased dramatically with a TON of 6.6 mol H2 mol−1 catalyst cm−2 after 2 h, suggesting that 6a is indeed suitable as a H2 evolution catalyst with seawater as the medium. However, the Faradaic efficiency of H2 production in seawater is only around 82%, since we have a one-compartment cell and some of the O2 produced at the anode will be reduced at the cathode (Fig. S19, ESI†). To explore the effect of the cations in the electrolyte, CPE was also conducted with n-Bu4NCl, Li2SO4, Na2SO4, KPF6, and MgSO4 as the electrolytes in 0.10 M acetic acid solutions (Fig. 3b). The TON with KPF6 as the electrolyte was the highest in these experiments (Table S12, ESI†). We propose that the poor solubility of our Li and Mg complexes led to catalyst precipitation and amalgamation into the mercury pool electrode during CPE, which resulted in a discrepancy with the CV and CPE data that we collected using carbon-based electrodes. Nonetheless, these CPE measurements and the TON and TOF (Table S12, ESI†) confirm that 6a remains active for electrocatalytic H+ reduction under almost neutral, saline aqueous conditions containing Na+ or K+, even when mercury electrodes are used to suppress the activity of nanomaterials.
 | ||
| Fig. 3 The amount of H2 evolved from (a) seawater with 0.22 mM catalyst 6a (red line) and only seawater (black line); (b) 0.10 M HOAc solution with 0.25 M Li2SO4 (red), Na2SO4 (blue), and Mg2SO4 (green), and 0.5 M KPF6 (magenta) and n-Bu4NCl (black) with catalyst 6a using a mercury pool working electrode. | ||
We have prepared new mononuclear Ni complexes with salicylaldimine ligands that can be used as electrocatalysts for the production of H2 in both neutral and acidic aqueous solutions. The chelating ether groups in the structure can bind alkali metal cations to form Lewis acids and promote the H+ reduction efficiency. Future studies will focus on the modification of the Ni complexes with electron withdrawing substituents to reduce the overpotential, applying 6a in photocatalytic units for H2 evolution, and grafting the molecules on semiconductors for artificial photosynthesis.47,48
HSS is supported by a NTU start-up grant (M4081012), the Nanyang Assistant Professorship (M4081154), and an MOE Tier 1 grant (M4011154). The authors acknowledge the funding support from the Singapore-Berkeley Research Initiative for Sustainable Energy (SinBeRISE) CREATE Programme. The authors also thank Asst. Prof. Jason Xu Zhichuan from the NTU Solar Fuels Laboratory for support in electrochemistry and both Dr Li Yongxin and Dr Rakesh Ganguly for solving and refining the crystal structures in the ESI.†
Notes and references
- Z. Han and R. Eisenberg, Acc. Chem. Res., 2014, 47, 2537–2544 CrossRef CAS PubMed.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS PubMed.
- D. G. Nocera, Acc. Chem. Res., 2012, 45, 767–776 CrossRef CAS PubMed.
- P. D. Tran, L. H. Wong, J. Barber and J. S. C. Loo, Energy Environ. Sci., 2012, 5, 5902–5918 CAS.
- D. Merki, H. Vrubel, L. Rovelli, S. Fierro and X. Hu, Chem. Sci., 2012, 3, 2515–2525 RSC.
- Y. Sun, C. Liu, D. C. Grauer, J. Yano, J. R. Long, P. Yang and C. J. Chang, J. Am. Chem. Soc., 2013, 135, 17699–17702 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, M. Jaroniec and S. Z. Qiao, Angew. Chem., Int. Ed., 2015, 54, 52–65 CrossRef CAS PubMed.
- W.-F. Chen, C.-H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. Muckerman, Y. Zhu and R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 CAS.
- J. Kibsgaard and T. F. Jaramillo, Angew. Chem., Int. Ed., 2014, 53, 14433–14437 CrossRef CAS PubMed.
- E. J. Popczun, C. G. Read, C. W. Roske, N. S. Lewis and R. E. Schaak, Angew. Chem., Int. Ed., 2014, 126, 5531–5534 CrossRef.
- M. Ledendecker, S. Krick Calderón, C. Papp, H. P. Steinrück, M. Antonietti and M. Shalom, Angew. Chem., Int. Ed., 2015, 127, 12538–12542 CrossRef.
- Y. F. Xu, M. R. Gao, Y. R. Zheng, J. Jiang and S. H. Yu, Angew. Chem., Int. Ed., 2013, 52, 8546–8550 CrossRef CAS PubMed.
- Z. Zhang, R. Dua, L. Zhang, H. Zhu, H. Zhang and P. Wang, ACS Nano, 2013, 7, 1709–1717 CrossRef CAS PubMed.
- M. L. Helm, M. P. Stewart, R. M. Bullock, M. R. DuBois and D. L. DuBois, Science, 2011, 333, 863–866 CrossRef CAS PubMed.
- R. M. Bullock, A. M. Appel and M. L. Helm, Chem. Commun., 2014, 50, 3125–3143 RSC.
- G. M. Jacobsen, J. Y. Yang, B. Twamley, A. D. Wilson, R. M. Bullock, M. R. DuBois and D. L. DuBois, Energy Environ. Sci., 2008, 1, 167–174 CAS.
- M.-H. Ho, M. O'Hagan, M. Dupuis, D. L. DuBois, R. M. Bullock, W. J. Shaw and S. Raugei, Dalton Trans., 2015, 44, 10969–10979 RSC.
- N. M. Muresan, J. Willkomm, D. Mersch, Y. Vaynzof and E. Reisner, Angew. Chem., Int. Ed., 2012, 124, 12921–12925 CrossRef.
- M. J. Rose, H. B. Gray and J. R. Winkler, J. Am. Chem. Soc., 2012, 134, 8310–8313 CrossRef CAS PubMed.
- D. P. Estes, D. C. Grills and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 17362–17365 CrossRef CAS PubMed.
- S. Mandal, S. Shikano, Y. Yamada, Y.-M. Lee, W. Nam, A. Llobet and S. Fukuzumi, J. Am. Chem. Soc., 2013, 135, 15294–15297 CrossRef CAS PubMed.
- G. Connor, K. Mayer, C. Tribble and W. McNamara, Inorg. Chem., 2014, 53, 5408–5410 CrossRef CAS PubMed.
- P. Du and R. Eisenberg, Energy Environ. Sci., 2012, 5, 6012–6021 CAS.
- T. R. Simmons, G. Berggren, M. Bacchi, M. Fontecave and V. Artero, Coord. Chem. Rev., 2014, 270–271, 127–150 CrossRef CAS.
- C. S. Letko, J. A. Panetier, M. Head-Gordon and T. D. Tilley, J. Am. Chem. Soc., 2014, 136, 9364–9376 CrossRef CAS PubMed.
- L. Gan, T. L. Groy, P. Tarakeshwar, S. K. Mazinani, J. Shearer, V. Mujica and A. K. Jones, J. Am. Chem. Soc., 2015, 137, 1109–1115 CrossRef CAS PubMed.
- D. J. Graham and D. G. Nocera, Organometallics, 2014, 33, 4994–5001 CrossRef CAS.
- H. I. Karunadasa, C. J. Chang and J. R. Long, Nature, 2010, 464, 1329–1333 CrossRef CAS PubMed.
- W. M. Singh, T. Baine, S. Kudo, S. Tian, X. A. N. Ma, H. Zhou, N. J. DeYonker, T. C. Pham, J. C. Bollinger and D. L. Baker, Angew. Chem., Int. Ed., 2012, 124, 6043–6046 CrossRef.
- L. Chen, M. Wang, K. Han, P. Zhang, F. Gloaguen and L. Sun, Energy Environ. Sci., 2014, 7, 329–334 CAS.
- H. Chen, Z. Sun, S. Ye, D. Lu and P. Du, J. Mater. Chem. A, 2015, 3, 15729–15737 CAS.
- C. Floriani and G. Fachinetti, J. Chem. Soc., Chem. Commun., 1974, 615–616 RSC.
- O. F. Schall, K. Robinson, J. L. Atwood and G. W. Gokel, J. Am. Chem. Soc., 1993, 115, 5962–5969 CrossRef CAS.
- S. T. Meally, C. McDonald, P. Kealy, S. M. Taylor, E. K. Brechin and L. F. Jones, Dalton Trans., 2012, 41, 5610–5616 RSC.
- B. Choudary, T. Ramani, H. Maheswaran, L. Prashant, K. Ranganath and K. V. Kumar, Adv. Synth. Catal., 2006, 348, 493–498 CrossRef CAS.
- A. Schepartz and J. P. McDevitt, J. Am. Chem. Soc., 1989, 111, 5976–5977 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Saveant, Chem. Soc. Rev., 2013, 42, 2423–2436 RSC.
- C. Costentin, H. Dridi and J.-M. Savéant, J. Am. Chem. Soc., 2014, 136, 13727–13734 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, J. Am. Chem. Soc., 2012, 134, 11235–11242 CrossRef CAS PubMed.
- J. B. Geri and N. K. Szymczak, J. Am. Chem. Soc., 2015, 137, 12808–12814 CrossRef CAS PubMed.
- D. W. Shaffer, S. I. Johnson, A. L. Rheingold, J. W. Ziller, W. A. Goddard, R. J. Nielsen and J. Y. Yang, Inorg. Chem., 2014, 53, 13031–13041 CrossRef CAS PubMed.
- E. S. Rountree, B. D. McCarthy, T. T. Eisenhart and J. L. Dempsey, Inorg. Chem., 2014, 53, 9983–10002 CrossRef CAS PubMed.
- B. H. Solis, A. Maher, T. Honda, D. C. Powers, D. G. Nocera and S. Hammes-Schiffer, ACS Catal., 2014, 4, 4516–4526 CrossRef CAS.
- J. S. Kanady, E. Y. Tsui, M. W. Day and T. Agapie, Science, 2011, 333, 733–736 CrossRef CAS PubMed.
- S. Bang, Y.-M. Lee, S. Hong, K.-B. Cho, Y. Nishida, M. S. Seo, R. Sarangi, S. Fukuzumi and W. Nam, Nat. Chem., 2014, 6, 934–940 CrossRef CAS PubMed.
- T. D. Harris, H. S. Soo, C. J. Chang and J. R. Long, Inorg. Chim. Acta, 2011, 369, 91–96 CrossRef CAS.
- C. A. Caputo, M. A. Gross, V. W. Lau, C. Cavazza, B. V. Lotsch and E. Reisner, Angew. Chem., Int. Ed., 2014, 126, 11722–11726 CrossRef PubMed.
- S. K. Muduli, S. Wang, S. Chen, C. F. Ng, C. H. A. Huan, T. C. Sum and H. S. Soo, Beilstein J. Nanotechnol., 2014, 5, 517–523 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed synthetic procedures and characterisation, additional electrochemistry data, XPS results, TEM, and DLS data. CCDC 1435734 and 1435735. For the ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc09456a |
| ‡ Current address: Department of Advanced Materials Science and Nanotechnology, University of Science and Technology of Hanoi (Vietnam France University), 18 Hoang Quoc Viet, Cau Giay, Hanoi, Vietnam. |
| § The onset overpotential is defined as the difference between the reversible potential of the H+/H2 redox couple of neutral DI water (−0.46 V vs. NHE at pH 7.6) and the potential at which 10% of the current value at the peak potential was reached. |
| ¶ The values of the charge passed during CPE were 38 and 94 C at 1 and 2.5 h, respectively, in excess of the charge needed to reduce Ni2+ to Ni+ (0.12 and 0.26 C for 10% and 22% 6a respectively). |
| This journal is © The Royal Society of Chemistry 2016 |