
Tracking reaction dynamics in solution by pump–probe X-ray absorption spectroscopy and X-ray liquidography (solution scattering)
Jeongho
Kim
 *a,
Kyung Hwan
Kim
bc,
Key Young
Oang
bc,
Jae Hyuk
Lee
d,
Kiryong
Hong
e,
Hana
Cho
def,
Nils
Huse
g,
Robert W.
Schoenlein
d,
Tae Kyu
Kim
*e and
Hyotcherl
Ihee
*bc
*a,
Kyung Hwan
Kim
bc,
Key Young
Oang
bc,
Jae Hyuk
Lee
d,
Kiryong
Hong
e,
Hana
Cho
def,
Nils
Huse
g,
Robert W.
Schoenlein
d,
Tae Kyu
Kim
*e and
Hyotcherl
Ihee
*bc
aDepartment of Chemistry, Inha University, Incheon 402-751, Republic of Korea. E-mail: jkim5@inha.ac.kr
bCenter for Nanomaterials and Chemical Reactions, Institute for Basic Science, Daejeon, 305-701, Republic of Korea. E-mail: hyotcherl.ihee@kaist.ac.kr
cDepartment of Chemistry, KAIST, Daejeon 305-701, Republic of Korea
dUltrafast X-ray Science Lab, Chemical Sciences Division, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
eDepartment of Chemistry and Chemical Institute for Functional Materials, Pusan National University, Busan 609-735, Republic of Korea. E-mail: tkkim@pusan.ac.kr
fCenter for Inorganic Analysis, Division of Metrology for Quality of Life, Korea Research Institute of Standard and Science, Daejeon 305-340, Republic of Korea
gMax Planck Research Department for Structural Dynamics, University of Hamburg & Center for Free Electron Laser Science, 22607 Hamburg, Germany
First published on 19th January 2016
Abstract
Characterization of transient molecular structures formed during chemical and biological processes is essential for understanding their mechanisms and functions. Over the last decade, time-resolved X-ray liquidography (TRXL) and time-resolved X-ray absorption spectroscopy (TRXAS) have emerged as powerful techniques for molecular and electronic structural analysis of photoinduced reactions in the solution phase. Both techniques make use of a pump–probe scheme that consists of (1) an optical pump pulse to initiate a photoinduced process and (2) an X-ray probe pulse to monitor changes in the molecular structure as a function of time delay between pump and probe pulses. TRXL is sensitive to changes in the global molecular structure and therefore can be used to elucidate structural changes of reacting solute molecules as well as the collective response of solvent molecules. On the other hand, TRXAS can be used to probe changes in both local geometrical and electronic structures of specific X-ray-absorbing atoms due to the element-specific nature of core-level transitions. These techniques are complementary to each other and a combination of the two methods will enhance the capability of accurately obtaining structural changes induced by photoexcitation. Here we review the principles of TRXL and TRXAS and present recent application examples of the two methods for studying chemical and biological processes in solution. Furthermore, we briefly discuss the prospect of using X-ray free electron lasers for the two techniques, which will allow us to keep track of structural dynamics on femtosecond time scales in various solution-phase molecular reactions.
1. Introduction
Chemical and biological processes occur in a well-coordinated sequence of bond breaking and formation, resulting in the progression of the molecular structure. To better understand such dynamic processes, it is required to have experimental methods that can measure the temporal variation of chemical bonds and related molecular structures. For this purpose, researchers have developed various time-resolved optical spectroscopic tools that employ ultrashort, coherent pulses at optical frequencies from ultraviolet (UV) to infrared (IR).1–20 Time-resolved spectroscopic signals provide valuable information such as the lifetime of a specific energy state and related wave-packet motions along the potential energy surfaces.21 However, in most cases, the spectroscopic signals do not contain direct information on molecular and electronic structures in terms of atomic coordinates, bond lengths, bond angles, valence charge, and spin densities. In other words, the time-resolved spectroscopic signals at the optical frequencies are not directly related to the global molecular structures at the atomic level.To circumvent the limitations of time-resolved optical spectroscopy, there have been many efforts to develop time-resolved techniques employing structure-sensitive probes such as electron diffraction, X-ray diffraction (or scattering), and X-ray absorption. As a result, ultrafast electron diffraction (UED), time-resolved X-ray diffraction (TRXD), and time-resolved X-ray absorption spectroscopy (TRXAS) have been developed over the last decade and proven to be very effective in characterizing transient structures with the time resolution comparable to that of optical spectroscopy. Among these techniques, due to high scattering cross-section of electrons with matter, UED and ultrafast electron microscopy have been mainly applied to studying ultrafast structural dynamics of molecular systems in the gas phase, surfaces, interfaces, thin films, and membranes as well as proteins and nanostructures.22–48 In contrast, due to the high penetration depth of X-rays, TRXD is appropriate for probing transient structures in condensed media. Specifically, TRXD can be classified into two categories depending on the phase of the sample: time-resolved X-ray crystallography and time-resolved X-ray liquidography (TRXL). Time-resolved X-ray crystallography is applied to crystalline samples with long-range order and, since 1990s, has been established as a major tool for investigating structural dynamics of solid chemicals and biological systems in single crystals with atomic resolution.49–78 In contrast, TRXL, which is also called time-resolved X-ray solution scattering (TRXSS), is applied to samples in condensed media and has made great strides over the last decade as an effective method for studying structural dynamics of chemical and biological reactions in liquid and solution phases. As a result, TRXL has been used to elucidate structural dynamics of small organic, inorganic, and protein molecules in solution with ∼100 ps temporal resolution based on X-ray pulses generated from third-generation synchrotrons.79–138 Furthermore, with the recent development of X-ray free electron lasers (XFELs), both TRXL139–142 and time-resolved crystallography143 can now access ultrafast dynamics of chemical reactions and protein structural transitions on time scales down to hundreds of femtoseconds. In this review, we focus on TRXL and its applications to chemical and biological processes in solution.
In parallel with TRXL, TRXAS has emerged as a powerful tool for studying ultrafast structural/electronic dynamics because TRXAS can provide the relationship between the electronic structure and the local molecular structure. With 100 ps X-ray pulses generated from synchrotrons, TRXAS has been used to study various phenomena in transition metal complexes and interfacial dynamics of nanomaterials relevant to catalysis, solar energy conversion, and functional materials.39,144–170 Recently, the time resolution of TRXAS has been extended to femtosecond time scales with the development of X-ray pulse generation technology160,170 and XFELs.141,171–174
Here we review the basic principles and application examples of the TRXL and TRXAS along with an outlook of the future directions for the research using these techniques. In particular, we emphasize that the two time-resolved X-ray techniques are well suited for studying structural dynamics of reacting molecules in the solution phase. Since most reactions in chemistry and biology occur in the solution phase, it is important to probe the progress of the dynamic processes in their inherent environment. Considering the complementary nature of the two techniques in terms of their sensitivities to the molecular and electronic structure of the target molecules, the combination of the two techniques allows us to unambiguously elucidate the reaction dynamics in the solution phase. Because TRXL and TRXAS require X-ray pulses with high stability, high photon flux, and wide tenability (in case of TRXAS), most of the time-resolved X-ray studies of solution-phase systems have been performed at third-generation synchrotron sources. However, table-top laser plasma sources,175,176 which can achieve femtosecond temporal resolution, have also been used for time-resolved X-ray diffraction177,178 and femtosecond XAS179–182 experiments. Here we note that, in this review article, we focus on only our own research studies performed using synchrotron sources rather than present a comprehensive review of studies in the field of time-resolved X-ray methods due to the space limit of this article.
2. Time-resolved X-ray absorption spectroscopy
2.1 Principle of TRXAS
X-ray absorption spectroscopy measures the absorption of X-rays by specific atoms in sample molecules as a function of incident X-ray energy. The X-ray absorption spectra are characterized by absorption edges (K, L, M, …), which are unique to the type of absorbing atoms, and fine structures in the low-energy part near the edge (X-ray absorption near-edge structure: XANES)183 and in the high-energy region from tens to hundreds of eV above the edge (extended X-ray absorption fine structure: EXAFS).184 The XANES mainly originates from bound–bound transitions from the atomic core orbital to valence orbitals and thus provides information on the electronic structure of molecules. Highly oscillatory EXAFS features arise from single scattering events between the absorbing central atom and the nearest neighbouring atoms and thus gives information on the local geometric structure around the central atom. Overall, the measured TRXAS spectra probe electronic transitions from the core level to valence orbitals and thus provide a direct means of obtaining excited-state electronic structures/configurations and local geometric structures that may be inaccessible by ultrafast optical spectroscopy. Thus far, however, full structural characterization of transient species by time-resolved EXAFS in the hard X-ray range has been limited to only a few molecular systems with high quantum yield147,149,153,156,165,170 because TRXAS signals have a relatively low signal-to-noise ratio in the EXAFS range (typically one order of magnitude less than in the XANES region) and the EXAFS features may be obscured by the overlap of different absorption edges.TRXAS requires short X-ray pulses that are synchronized with pump laser pulses and monochromatic near the X-ray absorption edges of the atoms of interest in the sample. Thus far, most of the TRXAS experiments for solution-phase reactions have been conducted at third-generation synchrotrons with the time resolution of a few tens of ps, while femtosecond TRXAS experiments have been performed for several prototype molecules at XFELs. One of the beamlines where TRXAS measurements can be implemented is the ultrafast X-ray facility of Advanced Light Source (ALS) at the Lawrence Berkeley National Laboratory (LBNL).154,162,163,169,170,185–187 The beamline has a 4 kHz amplified laser system, which is synchronized with a RF clock of the synchrotron, and frequency converters and provides fs and ps X-ray pulses that are widely tunable in the energy range from soft X-rays (350–1200 eV) to hard X-rays (2–8 keV). This energy range is wide enough to cover K- and L-edges of all first-row transition metals and absorption edges of common elements in functional materials, for example, sulfur K-edge and ruthenium L-edge. For TRXAS experiment at this beamline, a special filling pattern called a “camshaft” bunch is employed from the ALS storage ring consisting of 275 electron bunches spaced 2 ns apart. Specifically, the camshaft bunch (10 mA maximum current, 70 ps temporal duration) is isolated with a 100 ns-wide gap, allowing the gated detection of X-rays produced by this single bunch. The X-ray transmission through a thin liquid film (or a liquid sheet) generated from a sapphire nozzle is recorded at twice the repetition rate of the pump laser (typically 4 kHz) using a gated detector (silicon avalanche photodiode), thereby creating interleaved transmission intensities of unperturbed (laser off) and excited (laser on) sample, Iref and I(t), respectively, where t is the time delay between the pump and the probe pulses. The difference signal (ΔT = I(t) − Iref) is normalized with respect to the Iref signal on a shot-to-shot basis to account for long-term drifts of X-ray flux. This shot-to-shot collection scheme is dictated almost entirely by the shot-noise limit of the X-ray source and thereby X-ray transmission changes as low as 10−4 can be recorded in a reasonable data acquisition time. Energy-resolved scans at a particular time delay are performed by tuning a variable line-space grating monochromator. The schematic diagram of this experimental setup is shown in Fig. 1.
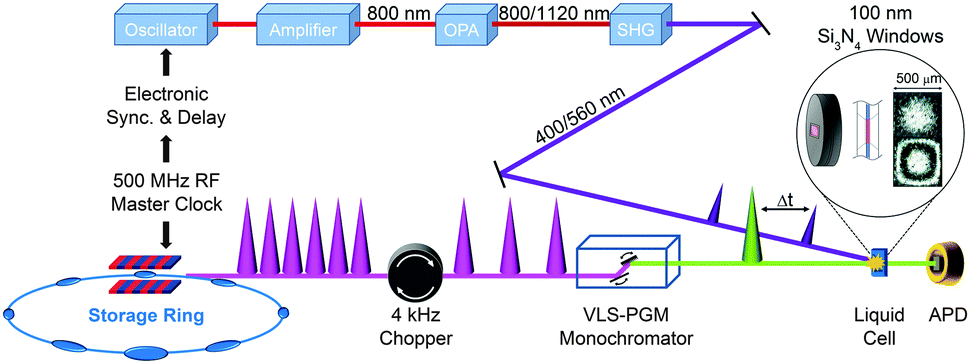 | ||
| Fig. 1 Experimental setup of TRXAS at BL6.0.2 of the Advanced Light Source. | ||
For both soft and hard X-ray ranges, we measure TRXAS signals in a direct transmission mode, which requires the sample in the form of a stable liquid sheet. For hard X-ray TRXAS experiment, naked open-jet with 100–300 μm thickness is used, as in TRXL experiment. However, due to large absorption cross section of solvents in the soft X-ray regime, TRXAS in the transmission mode requires the sample in a liquid sheet of sub-micron thickness that remains stable in vacuum environment under typical photoexcitation conditions.163,169,170 For this purpose, based on a previously reported design,188 we have developed liquid cells using silicon nitride membranes (100 nm thick) that can generate liquid sheets of less than 200 nm to 10 μm thickness. Ultimately, it is desirable to develop nanofluidic cells for flowing samples, and several promising approaches have been proposed with recent technological advances.189,190
2.2 Application of TRXAS
TRXAS can be used to determine molecular structures and associated electronic dynamics of photoexcited molecules. Over the past decade, TRXAS based on pulsed X-rays generated from synchrotrons has been applied to various molecules in chemistry, physics, biology, and materials science.148,149,151,165,191–196 In particular, our group has investigated photoinduced changes in the electronic charge distribution, the molecular structure, and the spin state of polypyridyl Fe(II) complexes.154,162,163,169,170,197 In the following sections, we present some of our own studies employing an approach of element-specific TRXAS at multiple X-ray absorption edges to better understand electronic and molecular dynamics of solvated functional materials, thereby answering many interesting scientific questions in materials science.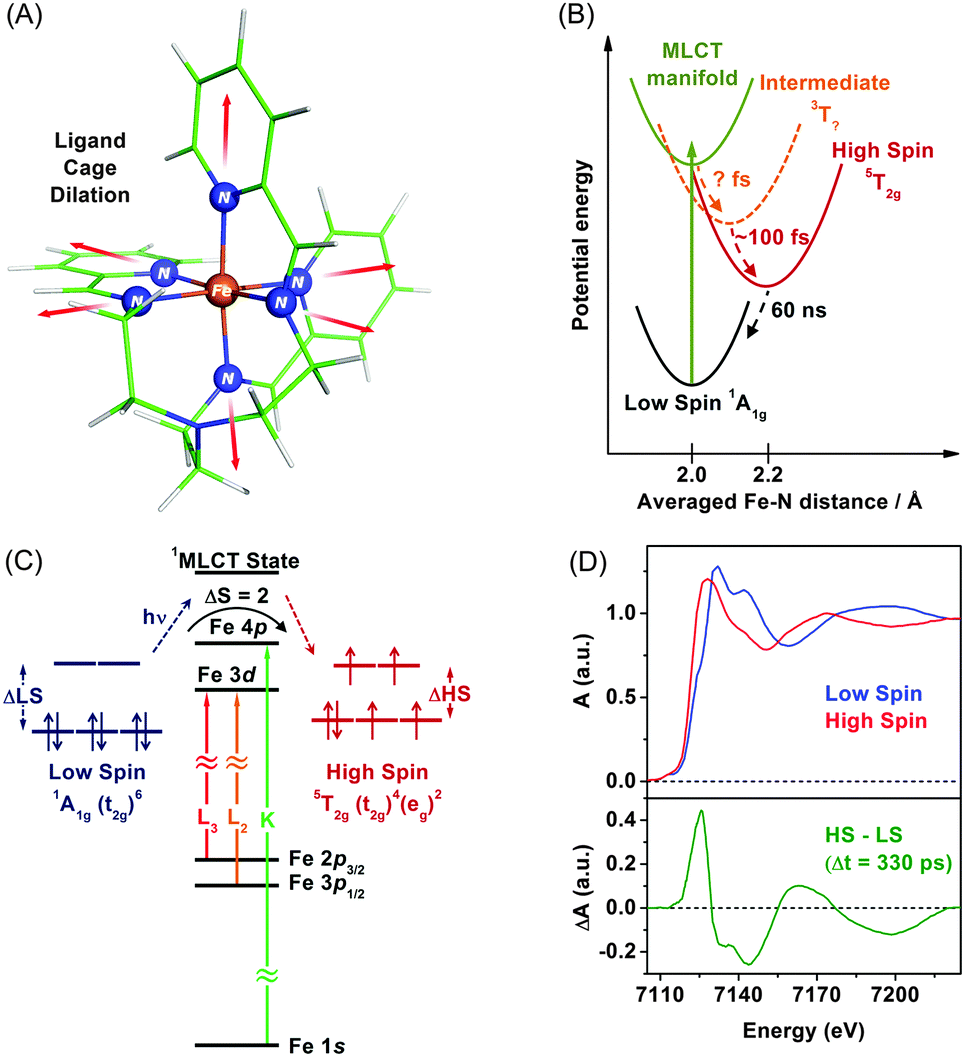 | ||
| Fig. 2 (A) Molecular structure of [Fe(tren(py)3)]2+. The changes of metal–ligand bonds associated with the spin transition are indicated by arrows. (B) Simplified energy levels involved in intersystem crossing after optical excitation to the metal-to-ligand charge-transfer state. (C) Schematic of orbitals associated with X-ray absorption spectroscopic measurements at multiple X-ray absorption edges. (D) (lower panel) Transient difference XAS spectrum (red) of [Fe(tren(py)3)]2+ measured at the time delay of 330 ps. (Upper panel) The steady-state XAS spectra of high-spin and low-spin Fe complexes is plotted together. | ||
Upon excitation of the metal-to-ligand charge transfer (MLCT) band, which corresponds roughly to the (t2g)6 → (t2g)5(π*)1 transition (where the π* orbital is associated with the nitrogen atom of the tren(py)3 ligand), this complex undergoes an ultrafast cascade of spin-state changes, presumably starting with singlet-to-triplet intersystem crossing in the charge-transfer manifold in less than 50 fs as suggested for [Fe(bpy)3]2+,198 and finally relaxes into the high-spin quintet 5T2g (t2g4eg2) state, which is an intermediate with the lifetime of 60 ns. Previous femtosecond optical spectroscopic studies of Fe(II) polypyridyl analogues yielded similar conclusions that the spin-state conversion with ΔS = 2 is a common feature in the ultrafast time regime following the MLCT excitation.198–202
The remarkably fast spin-state conversion in Fe(II) SCO complexes indicates that the spin-state change should be coupled with changes in the molecular structure as well as ligand-field splitting of the Fe 3d orbitals. In this regard, TRXAS is a uniquely suitable technique to examine the spin-state conversion process. For example, TRXAS measurements of [Fe(tren(py)3)]2+ and similar systems at the Fe K-edge unravelled detailed structural changes associated with the spin-state conversion.154,160 To investigate the spin-state conversion of the solvated [Fe(tren(py)3)]2+ complex, we measured TRXAS at the Fe K-edge using 100 fs optical pulses and revealed the molecular structure of the transient quintet state. Fig. 2(D) (upper panel) displays the static EXAFS spectra of the low-spin complex and its chemically-stabilized, high-spin analogue. The theoretical analysis of the static EXAFS spectra indicates the increase of Fe–N bond length by 0.23 Å in the high-spin analogue compared with the low-spin complex. Time-resolved EXAFS spectrum was obtained by taking the difference between a spectrum measured at a positive time delay (330 ps) and a reference spectrum measured at a negative time delay (−2 ns), as shown in the lower panel in Fig. 2(D). This time-resolved EXAFS spectrum at the Fe K-edge was modelled with multiple scattering calculation using the FEFF code,203 and it was found that the Fe–N bonds are elongated by 0.21 Å following the MLCT transition. According to TRXAS-EXAFS measurements on analogous Fe(II) spin-crossover complexes, the Fe–N elongation from ∼2.0 Å to ∼2.2 Å within 300 fs following the MLCT transition was commonly observed in many variants of this system despite considerable differences in the ligand structure.154,192,204
As can be seen in the Fe K-edge TRXAS measurements of Fe(II) spin-crossover complexes, we can gain insight into the molecular dynamics of 3d transition metal complexes using metal K-edge spectroscopy, which requires a relatively simple sample delivery scheme due to small absorption cross sections of solvents and air in the hard X-ray regime. While TRXAS at the metal K-edge141,144,154,191,194–196,205 has provided ample information on the local molecular structure around the metal centre, it does not give direct information on the metal valence charge and spin density because the K-edge absorption of 3d transition metals is based on the 1s → 4p transition and does not directly probe the 3d orbitals that are most relevant to chemical bonding. Although quadruple 1s → 3d transitions give such information, they are strongly broadened and thus not unambiguous probes.166,204,206 In this regard, metal L-edge spectroscopy based on the dipole-allowed 2p → 3d transitions is more informative because they directly probe unoccupied valence orbitals with high resolution owing to smaller broadening of their spectral features by almost ten times compared with those of 1s-excitations.147,207–209 Specifically, TRXAS at transition-metal L-edges directly probes changes in ligand-field splitting of metal d orbitals and spin states via selective excitation of 2p1/2 and 2p3/2 electrons to vacancies in the 3d valence orbitals of metals with spin–orbit splitting. For the Fe(II) SCO processes, fast ligand–cage dilation and spin-state changes should be coupled to changes in Fe-3d/N-2p hybridizations caused by weakening of the Fe–N bonds as well as associated changes in the ligand-field electron configurations. Therefore, to probe Fe(II) SCO processes unambiguously, it is required to measure TRXAS at multiple Fe absorption energies, for example, at both K- and L-edges.
As an effort to complement the K-edge TRXAS measurement on the SCO processes of [Fe(tren(py)3)]2+, we performed TRXAS at Fe L2,3-edges (∼700 eV) to reveal the changes of valence electron distribution following the MLCT excitation.169 In the Fe L2,3-edges the TRXAS spectrum measured at 90 ps is shown in Fig. 3(A), (positive) transient absorption and (negative) ground state bleaching (i.e. absorption loss) features are clearly observed, indicating the formation of a metastable quintet state. The temporal changes of the TRXAS signal at selected energies in Fig. 3(B) show ultrafast dynamics of the quintet formation. The spectrum of the low-spin ground state and the reconstructed spectrum of the high-spin state are shown in Fig. 3(C) together with the charge transfer multiplet (CTM) simulation results.207 The CTM method has proven very successful for simulating metal L-edge X-ray absorption spectra for various systems.
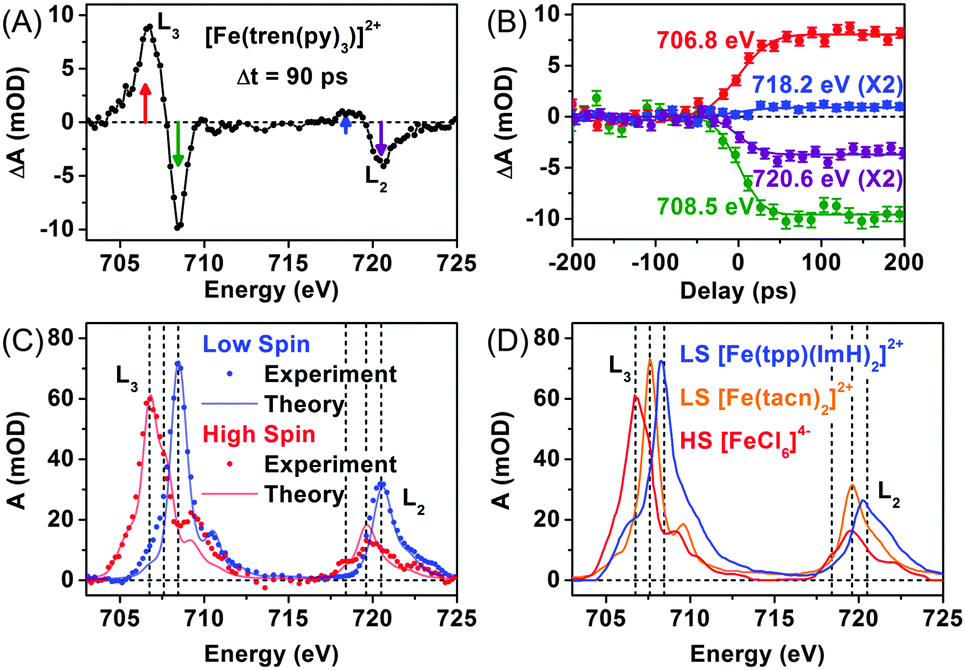 | ||
| Fig. 3 (A) TRXAS spectrum at 90 ps for [Fe(tren(py)3)]2+ SCO processes at Fe L2,3-edges. (B) Temporal changes of the TRXAS signal at selected energies indicated with colored arrows in (A). (C) L-edge spectra of the low-spin ground state (blue points) and the transient high-spin state at 90 ps delay (red points). The high-spin spectrum was reconstructed from the differential absorption spectrum shown in (A). For comparison with the experiment, theoretical spectra were obtained from charge transfer multiplet calculations (colored lines). (D) Rescaled L-edge spectra of three crystalline compounds: [Fe(tpp)(ImH)2]2+, [Fe(tacn)2]2+, and [FeCl6]4−. | ||
For comparison with the low-spin and high-spin spectra of [Fe(tren(py)3)]2+, we measured static L2,3-edges spectra of two low-spin model compounds, [Fe(tacn)2]2+ (tacn = 1,3,7-triazacyclononane) and [Fe(tpp)(ImH)2]2+ (tpp = tetraphenylporphyrin and ImH = imidazole), and a high-spin model compound, [FeCl6]4−, as shown in Fig. 3(D). The L2,3-edges spectra of both [Fe(tacn)2]2+ and [Fe(tpp)(ImH)2]2+ are of similar shape as the low-spin spectrum of [Fe(tren(py)3)]2+, but the spectrum of [Fe(tacn)2]2+ is located at lower energies than the spectra of [Fe(tpp)(ImH)2]2+ and low-spin [Fe(tren(py)3)]2+. Such a difference can be ascribed to the presence of π-conjugated ligands. Both [Fe(tacn)2]2+ and [Fe(tpp)(ImH)2]2+ have the same ligand arrangement around the iron centre as [Fe(tren(py)3)]2+, but only [Fe(tacn)2]2+ lacks the conjugated ligands that are present in [Fe(tpp)(ImH)2]2+ and [Fe(tren(py)3)]2+. The conjugated ligands tend to stabilize the valence charges of the iron centre through π-backbonding, resulting in the downshift of the unoccupied states. As a result, the L2,3-edge spectra of low-spin [Fe(tren(py)3)]2+ and [Fe(tpp)(ImH)2]2+ are shifted to higher energy than that of [Fe(tacn)2]2+ as shown in Fig. 3(C) and (D).169 The high-spin spectrum of [Fe(tren(py)3)]2+ reconstructed from the TRXAS spectrum is similar to that of the high-spin compound, [FeCl6]4−. In [FeCl6]4−, π-symmetric Cl-3p orbitals are fully occupied and therefore Cl ligands cannot serve as a π-acceptor. The resemblance of the two high-spin spectra indicates that π-backbonding in the low-spin state of [Fe(tren(py)3)]2+ is strongly inhibited due to orbital re-hybridization in the high-spin state. This interpretation is also supported by the CTM calculation,169 which supports the dilation of ligand cage as shown by the Fe K-edge measurement.154 We note that the net change of the integrated absorption for all Fe-2p transitions, which corresponds to the integrated area under the TRXAS spectrum in Fig. 3(A), is zero, indicating that the overall charge density in the Fe-3d manifold remains constant during the spin-state interconversion. This result shows that ligand 1s spectroscopy197 is sensitive to the valence charge densities of nearest-neighbour atoms. This example demonstrates that the TRXAS measurements performed at multiple absorption edges are effective for revealing the changes in both the electronic configuration and molecular geometry of metal complexes along their excited-state reaction pathways. A similar approach has been applied to intramolecular charge transfer of similar Ru(II), Ni(II), and Cu(II) complexes.144,149,151–153,161,166,167
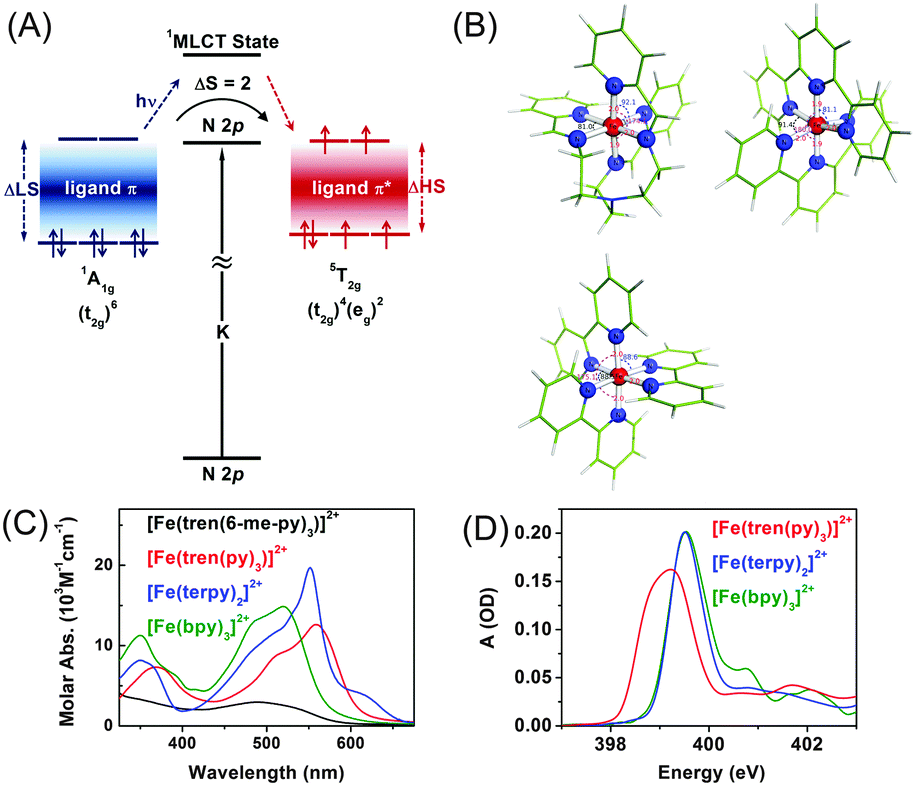 | ||
| Fig. 4 (A) Schematic of probing a Fe(II) complex in Oh-symmetry at the N K-edge (1s spectroscopy). (B) Molecular structures of [Fe(tren(py)3)]2+, [Fe(terpy)3]2+, and [Fe(bpy)3]2+. (C) Optical absorption spectra showing MLCT bands of the three Fe complexes. (D) Transmission N K-edge XAS spectra of solid [Fe(tren(py)3)]2+ (red), [Fe(terpy)3]2+ solution (blue), and [Fe(bpy)3]2+ solution (green). | ||
To understand the influence of the ligand structure on the valence charge distribution and metal–ligand interaction of Fe(II) SCO complexes, we applied ligand 1s spectroscopy to three prototypical Fe(II) polypyridyl complexes with various ligands, [Fe(tren(py)3)]2+, [Fe(terpy)2]2+ (terpy = terpyridine), and [Fe(bpy)3]2+ (bpy = bipyridine), shown in Fig. 4(B).197 Static N K-edge XAS spectra of the three complexes either in powder layers on a Si3N4 membrane or in 100 mM aqueous solution were measured as shown in Fig. 4(D). Despite different distortions, symmetries, and charge-transfer bands of the three compounds, their L-edge XAS spectra are almost identical to each other. In contrast, as shown in Fig. 4(D), their N K-edge absorption spectra are distinguishable from each other. Especially, the two compounds having pyridine-only ligands, [Fe(terpy)2]2+ and [Fe(bpy)3]2+, exhibit clearly different XAS spectra at higher energies than 400 eV compared to [Fe(tren(py)3)]2+, of which the ligand contains two distinct nitrogen species, a pyridine and an imino nitrogen. This result demonstrates the element specificity of XAS measured at the N K-edge. Thus, by performing ligand 1s TRXAS on the solvated transition metal complexes in combination with novel ab initio simulation of core-level spectra, we will be able to monitor the change in valence charge distributions of photoinduced reaction intermediates from the viewpoint of ligands, adding a new dimension to TRXAS.
3. Time-resolved X-ray liquidography
3.1 Principle of TRXL
X-rays with photon energies higher than atomic resonance scatter elastically off all atom–atom pairs of any species in solution, providing structural information over a wide range of length scales. The TRXL technique is based on this global structural sensitivity of X-ray diffraction. Here we note that the diffraction from liquid is often called diffuse scattering to distinguish it from the Bragg diffraction peaks from a well-ordered molecule system. The experimental setup of the TRXL comprises a pulsed laser system to excite the sample, a high-speed mechanical chopper that selects X-ray pulses at a reduced repetition rate, an integrating charge-coupled device (CCD) area detector, and a sample-flowing system, as shown in Fig. 5. High-flux X-rays are focused onto a circulating liquid sample either in a flowing jet or in a capillary, and scattered X-rays are measured by a two-dimensional area detector, giving X-ray scattering patterns at various time delays after laser excitation. Such experimental setup for TRXL has been initially established at the European Synchrotron Radiation Facility (ESRF)92,212 and later at other synchrotrons.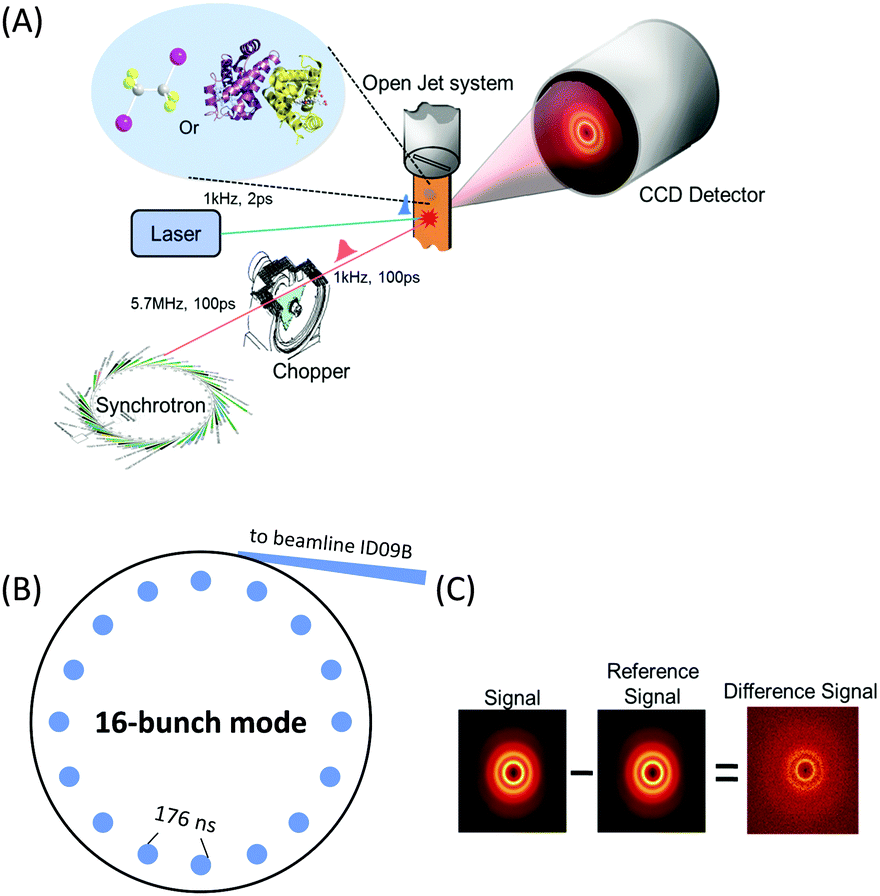 | ||
| Fig. 5 (A) Schematic of the experimental setup for the TRXL experiment. X-rays generated from a synchrotron source are directed through a beamline and selected by a high-speed mechanical X-ray chopper. The laser pulses excite the solutes in the sample and the time-delayed X-ray pulses probe the photoinduced structural changes as scattering patterns on the CCD detector. (B) Synchrotron filling (16-bunch mode) pattern at ESRF adequate for TRXL experiment. The time interval between X-ray pulses is 176 ns and a mechanical chopper can isolate a single pulse from the adjacent pulses to reach the ultimate time resolution, which is limited by the X-ray pulse length itself. (C) Procedure of extracting the difference scattering pattern. The difference scattering image at a given positive time delay is generated by subtracting the reference image at −3 ns from the scattering image at the positive delay. | ||
For the realization of TRXL experiment, many technical challenges need to be overcome. First, X-rays from synchrotrons have a repetition rate of 1–500 MHz, which is much faster than the typical repetition rate (1 kHz) of an amplified femtosecond laser. Since laser and X-ray pulses have to arrive on the sample in pairs, a high-speed mechanical chopper is used to reduce the repetition rate of X-rays. At ID09 beamline at ESRF, the mechanical chopper isolates a sub-train of pulses at 986.3 Hz, the 360th sub-harmonic of the RF clock of synchrotron. A phase shifter generates a delayed RF clock that is synchronized with the repetition rate of the laser oscillator (88.05 MHz) and the amplifier (986.3 Hz). The amplified laser pulse is in phase with the chopper. The time delay between the laser and X-ray pulse is controlled electronically by shifting the phase of the oscillator feedback loop using a delay generator with 10 ps resolution. Another experimental challenge is the extremely small difference in the scattering intensity between before and after laser excitation. Because the signal-to-noise ratio of the difference scattering depends on the X-ray flux, the full flux of a single X-ray pulse from the third-generation synchrotron should be utilized. At ESRF, polychromatic X-rays (peaked at ∼18 keV, ∼3% bandwidth) rather than monochromatic X-rays are used to increase the flux and therefore greatly shorten the data acquisition time.
Two-dimensional scattering patterns on the CCD can be circularly integrated into one-dimensional scattering curves, Sexp(q,t), as a function of the momentum transfer q = (4π/λ)sin![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) θ, where λ is the wavelength of the X-rays and 2θ is the scattering angle, and the time delay, t, between the laser and X-ray pulses. The scattering curve of the unperturbed sample (−3 ns) is subtracted from the curve measured at a positive time delay to generate the difference scattering curve, ΔSexp(q,t) and their sine-Fourier transformation, ΔSexp(r,t), gives information on the distances of all atom pairs in the sample.
θ, where λ is the wavelength of the X-rays and 2θ is the scattering angle, and the time delay, t, between the laser and X-ray pulses. The scattering curve of the unperturbed sample (−3 ns) is subtracted from the curve measured at a positive time delay to generate the difference scattering curve, ΔSexp(q,t) and their sine-Fourier transformation, ΔSexp(r,t), gives information on the distances of all atom pairs in the sample.
The sensitivity of TRXL to all atom pairs is a great strength, but it also poses significant challenges to the interpretation of TRXL data. To overcome this difficulty, we fit theoretical difference scattering curves, ΔStheory(q,t), to the experimental data by minimizing the difference between the experimental and theoretical curves at all time delays simultaneously. ΔStheory(q,t) is simulated using a linear combination of three major scattering contributions (solute-only, solute-solvent (cage), and solvent-only terms) based on estimated reaction dynamics and energy conservation between solutes and solvents:
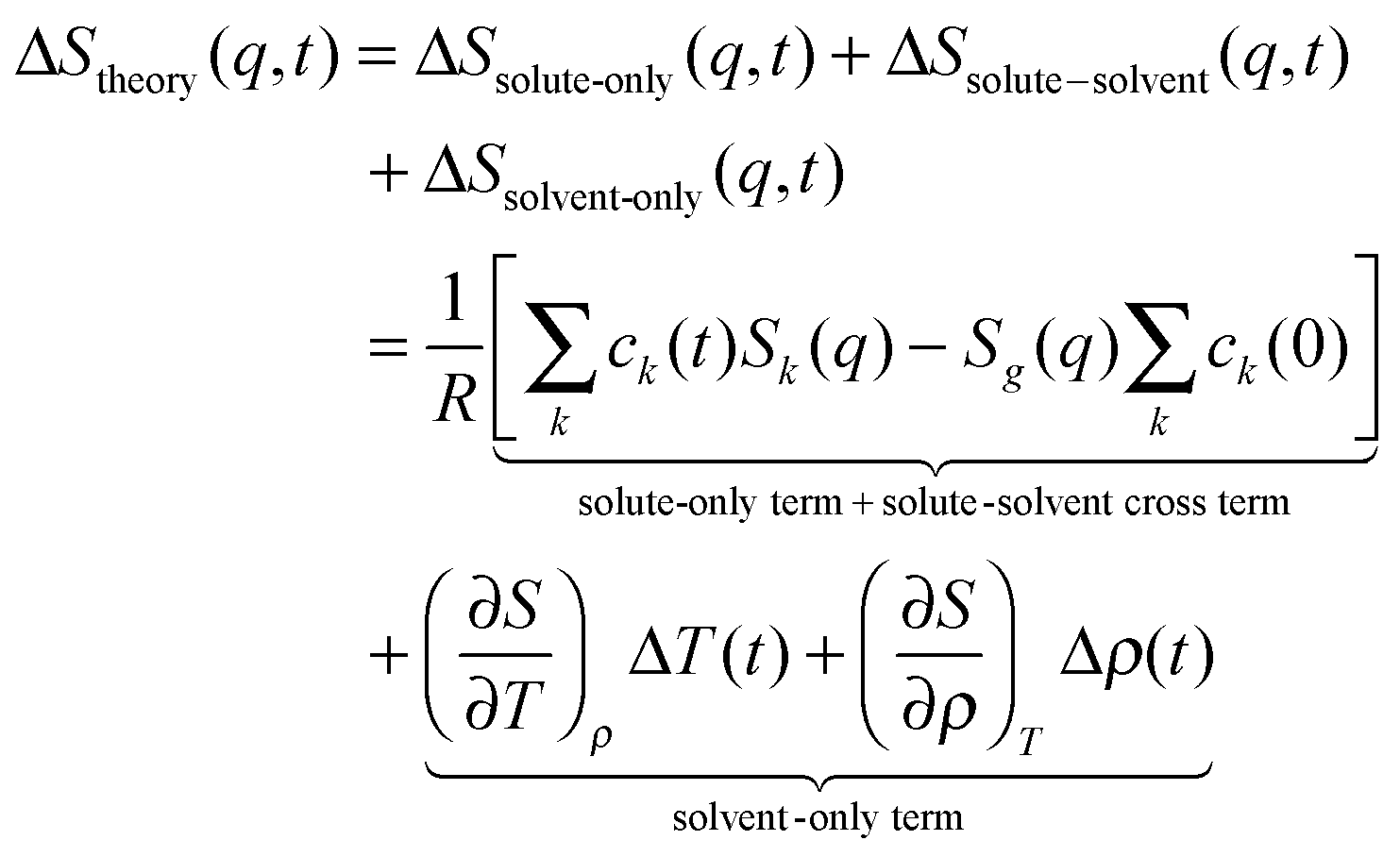 | (1) |
3.2 Application of TRXL
TRXL has been used to capture the molecular structures of transient intermediates and their spatiotemporal kinetics for various photochemical processes. The molecular systems studied by TRXL spans from small molecules79–82,84,85,87–89,92,93,98,99,105–107,110–112,114,115,128,130–133,136,140 to organometallic complexes,97,103,104,109,113,116,119,124,141 nanoparticles,83,86,90,91,94,95,100,120,125 and biological proteins96,101,102,108,117,118,121–123,126,127,129,134,135,137–139,142 in the solution phase, demonstrating wide applicability of the TRXL technique. In the following, we present some of our own studies using the TRXL technique for the investigation of structural dynamics of small molecules and proteins in solution.Later, we applied TRXL to the same reaction in cyclohexane, which has a very different polarity from methanol.128 By investigating the reaction dynamics in the two solvents, we can examine how the polarity of the solvent affects the dynamics and mechanism of the reaction. In Fig. 6, difference scattering curves measured for the photodissociation of C2H4I2 in cyclohexane are shown in the q-space and r-space. The difference scattering curves show distinct oscillatory features that are signature of structural changes of reacting molecules. The theoretical difference scattering curves, shown together in Fig. 6, were obtained by globally fitting the experimental scattering curves at various time delays.
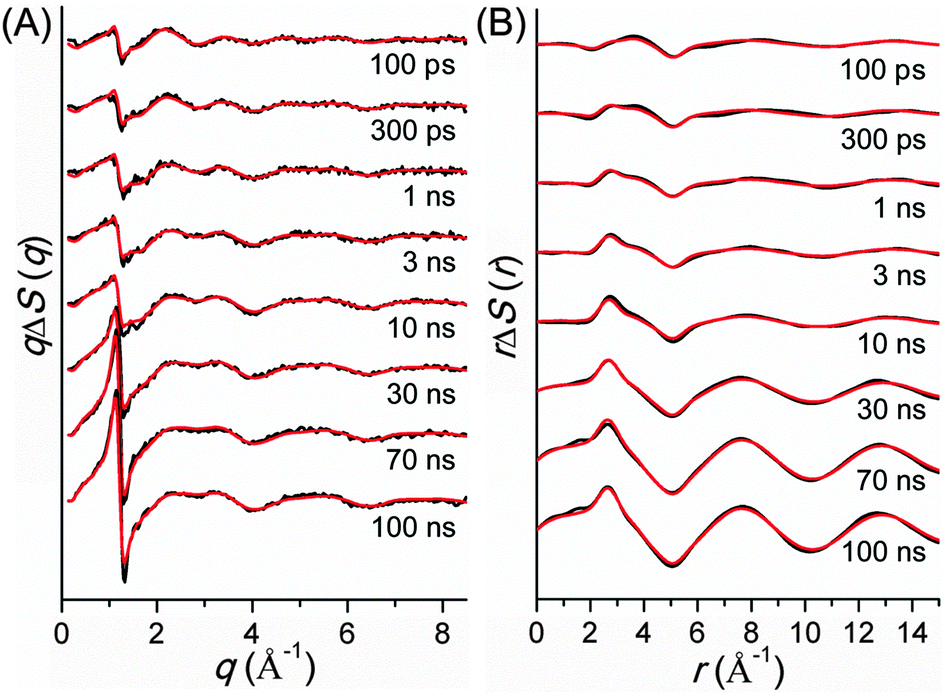 | ||
| Fig. 6 Difference scattering curves of the C2H4I2 photodissociation in cyclohexane at various time delays after photoexcitation at 267 nm. (A) Experimental (black) difference scattering curves represented as qΔS(q) in the q-space and theoretical curves (red) obtained by global fitting analysis of the experimental data. (B) Difference radial distribution functions, rΔS(r), obtained by sine-Fourier transformation of the qΔS(q) difference scattering curves shown in (A). | ||
The concentration changes of chemical species and the reaction mechanism of the photodissociation of C2H4I2 in cyclohexane and methanol are compared in Fig. 7. In both cyclohexane and methanol, on photoexcitation, one iodine atom is dissociated from the C2H4I2 molecule, generating a C2H4I˙ radical much earlier than 100 ps. However, subsequent reaction pathways starting from the C2H4I˙ radical are quite different in the two solvents. In methanol, the C2H4I radical combines with an I atom to form a C2H4I–I isomer, which then dissociates into C2H4 and I2 molecules. In contrast, in cyclohexane, the C2H4I˙ radical undergoes two competing reaction channels: (1) combines with an I atom to form a C2H4I–I isomer, or (2) dissociates into C2H4 and I.
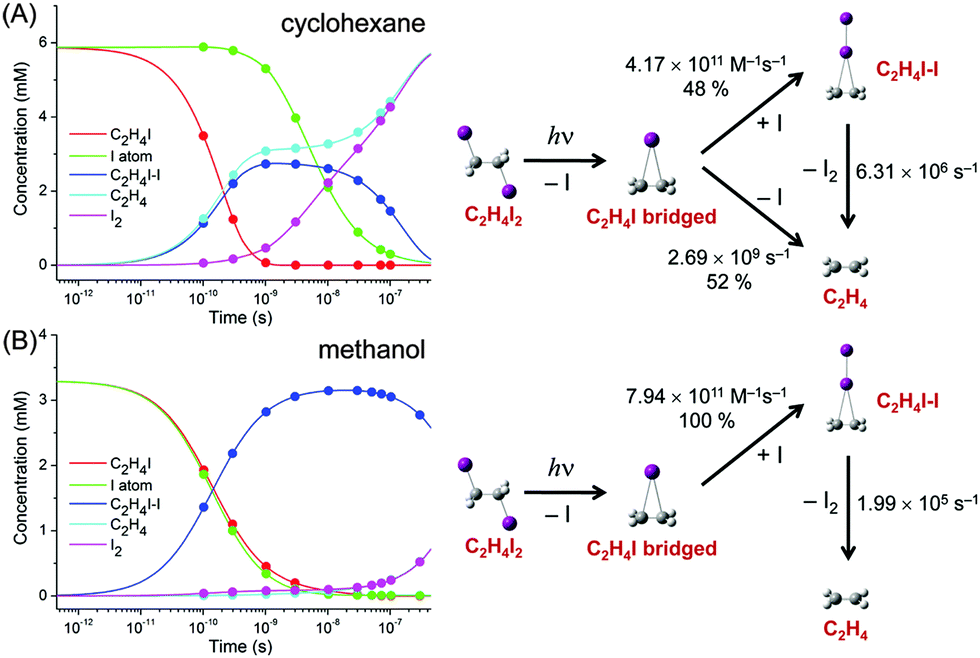 | ||
| Fig. 7 Time-dependent concentration changes of chemical species (left) and schematic of the reaction mechanism (right) of the photodissociation reaction of C2H4I2 in (A) cyclohexane and (B) methanol. C2H4I radical (red), C2H4I–I isomer (green), C2H4 (blue), I2 (cyan), and I atom (magenta). The points in the plot of concentration changes indicate the time delay points where the scattering data were measured. | ||
This difference of the reaction pathways in cyclohexane and methanol can be explained by the difference in solvent polarity. The reaction channel of isomer formation (C2H4I + I → C2H4I–I) is highly exothermic, while dissociation into C2H4 and I (C2H4I → C2H4 + I) is slightly endothermic. Since C2H4 and I are nonpolar and the C2H4I radical is polar (μ = 2.0 D), only the C2H4I˙ radical becomes more stable in methanol than in cyclohexane. As a result, the reaction channel of C2H4I → C2H4 + I is easier to occur in cyclohexane than in methanol. In contrast, the isomer formation channel, which is highly exothermic, is less likely to be influenced by the small energy shift induced by the change in solvent polarity. Therefore, the isomer formation occurs in both methanol and cyclohexane, while the dissociation into C2H4 and I is more likely to occur in cyclohexane than in methanol. The difference in the reaction pathways of C2H4I2 in methanol and cyclohexane underpins the role of the solvent in chemical reaction and also demonstrates the power of TRXL to resolve the difference in the reaction mechanism. Similarly, TRXL has also been used to investigate the spatiotemporal kinetics of photodissociation reactions of haloalkanes such as CHI3,98 CH2I2,85,107 and CBr493 in various solvents.
Time-resolved difference X-ray solution scattering curves of MbCO are shown in Fig. 8. From the analysis of scattering data using singular value decomposition (SVD) in various time ranges, we obtained four structurally distinct intermediates formed by the photolysis of the CO ligand and six kinetic components with time constants of 460 ps, 3.6 ns, 92 ns, 1.4 μs, 90 μs, and 1.2 ms. Based on the result of the SVD analysis, we performed the kinetic analysis to determine the optimum kinetic model that best fits the experimental data among a total of eighteen candidate kinetic models. The optimum model shown in Fig. 9 consists of four intermediates termed B, C, D, and S and the six kinetic components obtained from the SVD analysis were assigned to the transitions among these intermediates. Importantly, the transition from the first intermediate (B) to the second one (C) occurs biphasically with time constants of 460 ps and 3.6 ns, and the relative population ratio of these two transitions was 46% and 54%, respectively. The B-to-C transition corresponds to the movement of the photodissociated CO ligand from the primary docking site to the Xe4 site and the biphasic nature of the transition indicates that two conformational substates of B (B1 and B2) are involved in the tertiary structural relaxation associated with the transition. The biphasic transition from B to C has never been resolved with time-resolved IR spectroscopy, suggesting that the B-to-C transition from two different conformational substates can be distinguished only by their global structural changes probed by TRXL. Following the B-to-C transition, C transforms to the third intermediate (D) with the time constant of 92 ns, and D is converted to the last intermediate (S) with the time constant of 1.4 μs. These two transitions correspond to tertiary structural transitions towards the deoxy form of Mb. The C-to-D and D-to-S transitions accompany the movements of the photodissociated CO (1) from the Xe4 site (defined by G25, I28, L29, V68, and I107) to the Xe1 site (defined by L89, H93, L104, and F138) and (2) from the Xe1 site to the solvent environment, respectively. Finally, the deoxy S intermediate returns to the ground-state MbCO via bimolecular nongeminate CO recombination with the bimolecular rate constant of 230 mM−1 s−1, which can be approximated by a combination of two unimolecular time constants of 90 μs and 1.2 ms.
 | ||
| Fig. 8 Time-resolved difference scattering curves measured for a solution sample of wild-type sperm whale MbCO. Experimental curves (black) are compared with theoretical curves (red) that were generated from linear combinations of four time-independent, species-associated difference scattering curves extracted from the kinetic analysis using the kinetic model shown in Fig. 9. The time delay after photoexcitation is indicated above each difference scattering curve. | ||
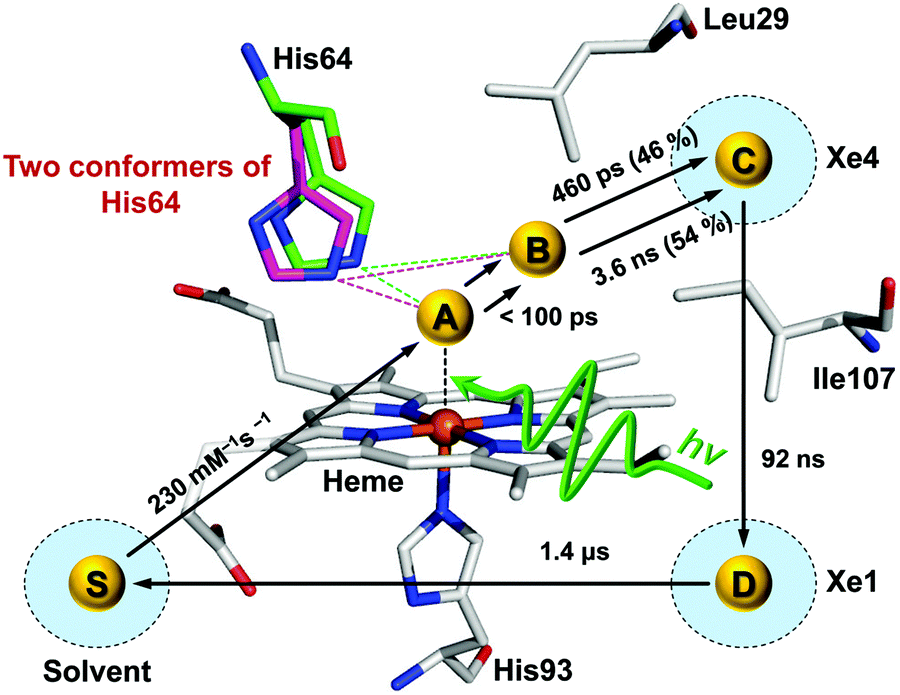 | ||
| Fig. 9 Kinetic scheme for the photoreaction of wild-type sperm whale MbCO with the movements of the CO ligand being highlighted. Yellow spheres indicate the locations of the CO for A, B, C, D, and S states. The CO ligated to the heme (A) as indicated by a black dashed line is dissociated by 532 nm laser excitation and moves from the primary docking site (B) via the Xe4 site (C) to the Xe1 site (D) and finally to the solvent environment (S). Due to the existence of the two conformational substates of B as indicated by green and magenta dashed lines, the transition from B to C exhibits biphasic kinetics. | ||
α, where α is the angle between the laser polarization and the transition dipole of a molecule. As a result, the excited molecules are aligned along the laser polarization direction and generate anisotropic X-ray scattering patterns. It has been already shown by ultrafast electron diffraction experiments that such an anisotropic pattern can help to characterize the molecular structure and dynamics more clearly.219–221 Conversely, the anisotropic scattering pattern could lead to misinterpretation of the scattering pattern. Therefore, more careful treatment is needed in the analysis of the anisotropic scattering pattern.
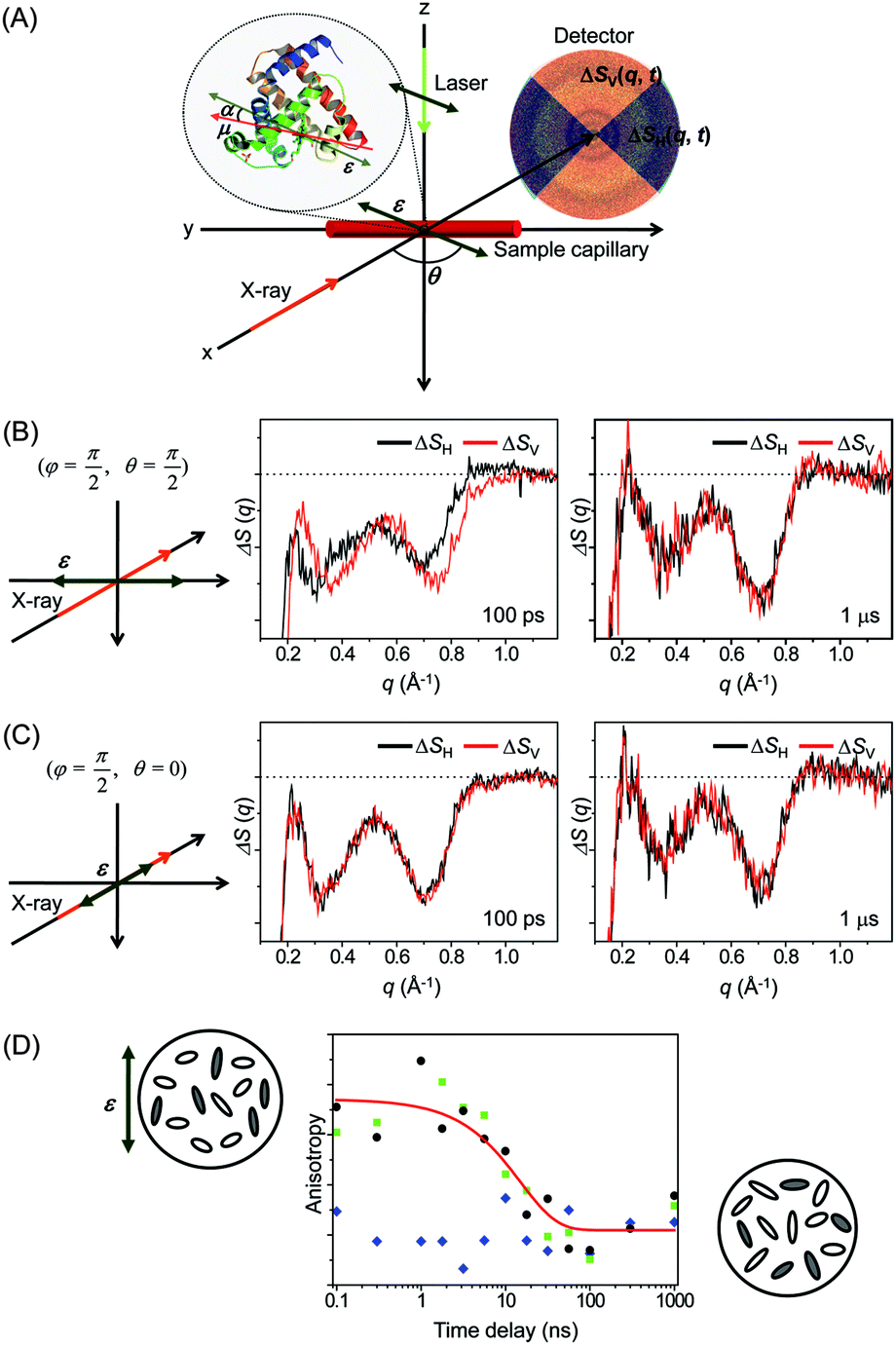 | ||
| Fig. 10 (A) Concept of X-ray scattering from selectively aligned protein molecules. With respect to the X-ray propagation direction (x axis), the polarization (ε) of the laser pulse can have any orientation defined by θ and φ in spherical coordinates, where 0 ≤ θ ≤ π/2 and 0 ≤ φ ≤ π/2. In our experiment, the laser light with linear polarization was sent along the z direction (setting φ = π/2) and only the θ value was varied between 0 and π/2. The orientation of the transition dipole (μ) of a molecule is described by the angle, α, with respect to the laser polarization (ε). We used linearly polarized light with θ = 0 (i.e. parallel to the X-ray propagation) and θ = π/2 (i.e. perpendicular to the X-ray propagation) as the laser excitation source. (B and C) 1D scattering curves, ΔSH(q,t) and ΔSV(q,t), obtained from horizontal (ΔSH) and vertical (ΔSV) cuts, respectively, of 2D scattering patterns of Mb measured at 100 ps and 1 μs time delays. The laser polarization direction and the X-ray propagation direction were adjusted to be (B) perpendicular (θ = π/2) and (C) parallel (θ = 0) to each other. (D) Time evolution of transient anisotropy obtained from the anisotropic X-ray scattering curves. Both the difference between the parallel and perpendicular cases (black circles) and the difference between the vertical and horizontal cross sections in the perpendicular case alone (green squares) provide the temporal change of anisotropy, which reflects the dynamics of rotational diffusion of the protein molecules in solution. In contrast, the difference between the same cross sections in the parallel case (blue diamonds) does not show any change in anisotropy over time. The red curve is a fit to the observed time-dependent anisotropy with the decay time constant of 15 ± 6 ns. | ||
As an effort to take advantage of anisotropic scattering patterns measured by TRXL, we investigated structural transition of the Mb protein by applying TRXL with linearly polarized pump laser pulses.122 Since Mb has a much larger size than small molecules, rotational diffusion of excited Mb molecules takes ∼10 ns. Therefore, even with 100 ps time resolution of TRXL, the effect of linearly polarized excitation on the X-ray scattering pattern can be observed. In our experiment, the excited Mb molecules are photoselectively aligned by photoinduced dissociation of a CO ligand from the metal ion of the heme group in the carboxymyoglobin (MbCO) using a linearly polarized picosecond laser pulse.
To examine the laser polarization effect on the X-ray scattering pattern, we checked the 1D curves obtained from the horizontal (ΔSH(q,t)) and vertical (ΔSV(q,t)) cuts of the 2D scattering image. When the laser polarization is perpendicular to the direction of X-ray propagation, the horizontal and vertical cross-sections are clearly different from each other at 100 ps time delay as shown in Fig. 10(B). As time goes on, the difference between the horizontal and vertical cuts, i.e. anisotropy, of the anisotropic scattering pattern becomes smaller and completely vanishes in the data at 1 μs time delay. In contrast, in Fig. 10(C), when the laser polarization is parallel to the X-ray propagation direction, no distinct difference between the horizontal and vertical cuts is seen, even at 100 ps.
To follow the time evolution of the anisotropic X-ray scattering patterns, at each time delay, we took the difference between the two scattering curves from the perpendicular and parallel cases and integrated it along the q axis to define a measure of polarization anisotropy contained in the X-ray scattering patterns. The time evolution of the anisotropy is plotted in Fig. 10(D). Also, the perpendicular case alone were used to extract the anisotropy by taking the difference between the 1D curves from the vertical and horizontal cross sections. In both cases, the observed anisotropy decay of aligned Mb molecules are fit by an exponential of ∼15 ns time constant, which agrees well with the rotational diffusion time measured by NMR (10–20 ns)222,223 and the value predicted by the Stokes–Einstein equation (11 ns). Thus, the transient anisotropy measured by picosecond X-ray solution scattering is relevant for measuring the orientational dynamics of the protein molecules.
This result clearly shows that photoselectively aligned molecules give rise to anisotropic scattering patterns and the time evolution of these patterns can be used to monitor the orientational dynamics of excited molecules. Since TRXL using third-generation synchrotrons has a time resolution of only 100 ps, the anisotropy effect on the X-ray scattering pattern has not been seriously considered so far because rotational diffusion of small molecules commonly occurs on a much shorter time scale than 100 ps. However, when using femtosecond X-ray pulses generated from X-ray free electron laser (XFEL), it will be needed to appropriately analyze anisotropic scattering patterns. Recently, using the same approach, femtosecond orientational dynamics of a metal complex were investigated by performing the TRXL experiment at XFEL.224
4. Comparison of TRXL and TRXAS
It is worthwhile to compare the two representative time-resolved X-ray methods, TRXL and TRXAS techniques. These two different but closely related techniques have been successful in investigating structural dynamics in the solution phase by making use of the direct relationship of X-ray scattering and absorption with global and local molecular structures, respectively. These techniques are common in that they use a pump laser pulse to initiate the chemical reaction and the induced changes are subsequently probed by a time-delayed X-ray pulse. However, they are based on different types of phenomena (scattering and absorption) and have their own limitations and advantages for probing structural dynamics of solvated molecules.Since X-rays are scattered off all atom–atom pairs and chemical species within the sample, direct information on the global molecular structure can be retrieved from the measured X-ray scattering patterns. Therefore, TRXL is well suited for probing global structural dynamics of reacting species present in the solution. However, due to relatively weak contribution of solutes to the TRXL signal from the solution sample, it is crucial to perform accurate model simulations such as a global-fitting procedure to distinguish structural changes of solutes from other solvent-related contributions. Also, the application of TRXL is generally limited to only molecules containing heavy atoms with high scattering power.
On the other hand, TRXAS can provide more selective information than TRXL due to element-specific nature of the energy levels of core electronic states. Also, TRXAS simultaneously probes the electronic and local geometric structure of solvated chromophores in solution. However, since TRXAS probes only the local structure around specific atoms absorbing X-rays, it is not sensitive to the overall structure of solute molecules and solvation shells that are present at a long distance from the absorbing atoms. Therefore, TRXAS is particularly suited for probing ultrafast structural dynamics at short distances from the absorbing atoms. From recent studies, it has been demonstrated that TRXAS can uniquely monitor the dynamics associated with d-orbitals of the transition metal complex, for example, changes of valence bond occupancy and charge transfer. Since these orbitals are mainly responsible for the chemistry of this class of molecules, TRXAS measurements can give insight into chemical bonding in the transition-metal coordination compounds. In a broader sense, the prevalence of transition metals in biological molecules underscores the importance of TRXAS for understanding the evolution of the electronic structure associated with chemical reaction dynamics of such systems. For example, a recent TRXAS measurement on ligand dissociation of MbCO225,226 demonstrates the potential of TRXAS for investigating biological samples.
Considering the pros and cons of TRXL and TRXAS, an approach of combining the two complementary techniques may be the most effective way of elucidating the dynamics and mechanism of chemical reactions accurately. A recent study on the photolysis of iodoform (CHI3) is a good example demonstrating the power of such a combined approach.227 From two previous studies on the photolysis of CHI3 using TRXL98 or transient absorption,228 there has been a strong debate about the identity of the major reaction intermediate species among three candidate intermediates: CHI2 radical (TRXL) vs. CHI2–I isomer and CH2+ ion (transient absorption). To resolve this conflict, we used both TRXL and TRXAS to measure the reaction dynamics of iodoform photolysis on a sub-nanosecond time scale.
Fig. 11(A) shows the difference X-ray scattering curves measured at the time delays of −20 ps and 100 ps using the time-slicing scheme. The time-slicing scheme allows us to achieve the effective time resolution of ∼20 ps by deconvolution of the instrument response function from the scattering curves measured at time delays earlier than the X-ray pulse width (∼100 ps).132,133 The two scattering curves are identical to each other within the noise level, suggesting that the major intermediate species is generated within the time resolution of our measurement. The temporal change of the peak intensity at q = 2.205 Å−1 shown in the inset of Fig. 11(A) gives a clear view of the reaction kinetics and its rise is well described by a single error function with a width of 80 ps full-width-at-half-maximum (FWHM), which corresponds to the instrument response function (IRF) of the TRXL setup. This result indicates that no further chemical reaction occurs on the sub-nanosecond time scale and is in contrast to the result from a previous transient absorption study, which identified the dissociation of the CHI2–I isomer with an ∼700 ps time constant. To identify the major intermediate formed within the time resolution, we analyzed the difference scattering curve at −20 ps as shown in Fig. 11(B). We note that the TRXL measurement should be equally sensitive to the structure of the CHI2 radical and the CHI2–I isomer because X-rays scatter from every atom in the molecules. Judging by the χ2 value, the CHI2 + I channel (χ2 = 1.6) fits the experimental curve much better than the CHI2–I isomer channel (χ2 = 4.9) or the CHI2+ + I− channel (χ2 = 4.7). These results from the TRXL measurement shows that the CHI2 radical, rather than the isomer or the CHI2+ ion, is the major intermediate of the iodoform photolysis with the estimated yield of ∼8%.
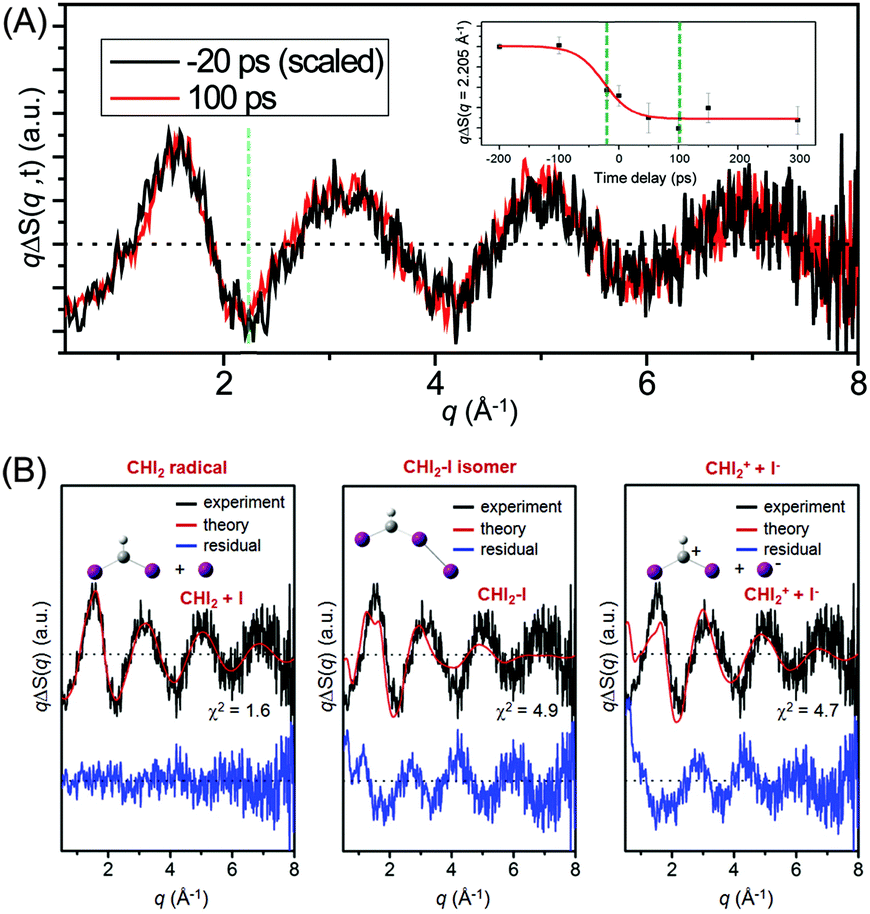 | ||
| Fig. 11 (A) Time-resolved X-ray solution scattering curves of CHI3 photolysis at the time delays of −20 ps (black) and 100 ps (red). (inset) Temporal change of the intensity at a negative peak position (q = 2.205 Å−1, marked with a green dashed line in the main figure) displayed from −200 ps to 300 ps. (B) Theoretical difference scattering curves at −20 ps time delay for three candidate reaction pathways, (i) dissociation into the CHI2 radical and I, (ii) CHI2–I isomer formation and (iii) dissociation into CHI2+ and I− ions. Experimental (black) and theoretical (red) curves are compared at the top and the residual (blue) obtained by subtracting the two curves is displayed at the bottom. | ||
Fig. 12(A) shows the difference XANES spectra measured around the iodine L1 edge at the time delays of 100 ps and 300 ps. The feature peaked at 5.1857 keV is located at a lower energy than the main edge and thus corresponds to the 2s → 5p transition. The peak positions, amplitudes and shapes of the two transient spectra are almost identical to each other within the noise level, indicating that the major intermediate species are generated within the first 100 ps and remain constant thereafter. We examined the kinetics on the sub-nanosecond time scale by performing a time scan from −1 to 2 ns at the peak position (5.1857 keV) of the difference XANES spectrum. As shown in the inset of Fig. 12(A), the rise of the time trace is governed by a single error function with a width of 93 ps FWHM, which corresponds to the IRF of our measurement, while no other kinetic component was identified. The same dynamics were obtained from the difference XANES spectra measured at the L3 edge of the iodine atom. To examine the identity of the major intermediate, as shown in Fig. 12(B), we performed a fitting analysis of the difference XANES spectrum at the L1 edge and 100 ps time delay using the static spectra of iodine-containing analogue species (I2, CHI3, I atom) measured from previous TRXAS studies as a basis set. To generate theoretical difference XANES spectra from these static spectra, we considered (1) the depletion of the –C–I group and the formation of the free I species for the CHI2 + I channel and (2) the depletion of the –C–I group and the formation of the –I–I group for the CHI2–I isomer channel. Although neither of the fits shown in Fig. 12(B) are perfect, probably due to the limitation of the analysis using the analogue species, we clearly see that the CHI2 + I channel fits the experimental curve much better than the CHI2–I isomer channel. These results from the TRXAS measurement confirm that the CHI2 radical is the major intermediate of the iodoform photolysis in agreement with the TRXL results. Considering that the X-ray-based methods are equally sensitive to both the CHI2 radical and CHI2–I isomer species, the missing signature of the CH2–I isomer in the TRXL and TRXAS measurements suggest that the yield of the CH2–I isomer, which was detected by optical spectroscopy, might be too low to be detected by TRXL or TRXAS.
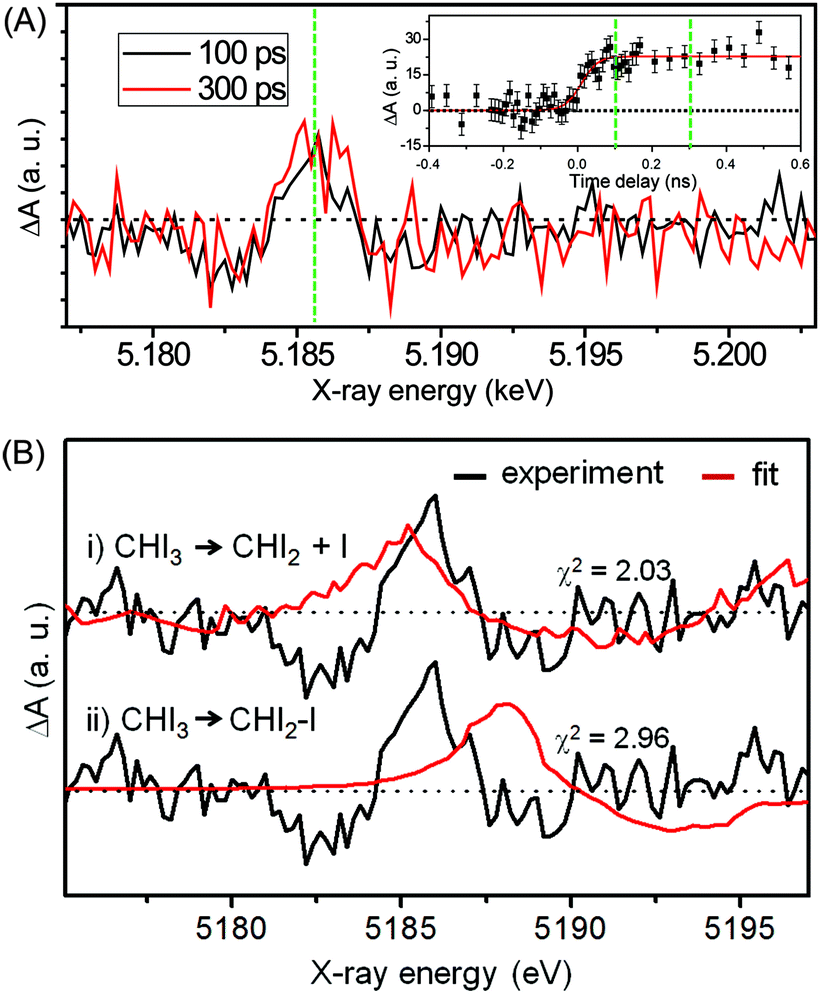 | ||
| Fig. 12 (A) Time-resolved X-ray absorption spectra of the iodoform photolysis measured at the L1 edge of the iodine atom and at the time delays of 100 ps (black) and 300 ps (red). (inset) Temporal change of the difference absorption intensity at a peak position of 5.1857 keV (green dashed line) in the time range from −0.4 to 0.6 ns. (B) The fitting analysis of the L1-edge difference XANES spectrum measured at 100 ps for two candidate reaction pathways, (i) CHI2 radical + I and (ii) CHI2–I isomer formation. Experimental (black) and theoretical (red) difference XANES spectra are compared with each other. | ||
5. Future prospects of TRXL and TRXAS
In this article, we reviewed the principles and experimental details of TRXL and TRXAS along with recent studies of photoinduced chemical and biological reactions in the liquid phase using these techniques. As described above, TRXL and TRXAS are powerful tools for monitoring the changes in geometrical and electronic structures involved in photoinduced processes in the solution phase. Importantly, these time-resolved techniques based on X-rays can provide direct insight into atomic-scale structural changes following photoexcitation. Due to its sensitivity to the global molecular structure, TRXL addresses detailed structural changes of both solute and solvent molecules over a broad range of length scales and reaction kinetics related with hydrodynamics of solvents. However, we note that TRXL exhibits lower spatial resolution than time-resolved X-ray diffraction applied to solid crystalline samples (that is, time-resolved X-ray crystallography) because random orientation of sample molecules in the solution phase wipes out some of the structural information contained in the scattering pattern. In contrast, TRXAS can selectively probe the changes in local geometry due to the element-specific nature of core-level transitions. Simultaneously, TRXAS can provide the changes in the electronic structure of atomic elements, as demonstrated by the Fe L2,3-edge measurements of spin crossover dynamics. Ultimately, the approach of applying both TRXL and TRXAS to the same system will give more information on the reaction dynamics as was witnessed in recent studies: photolysis of CHI3227 and photoinduced electron transfer in a bimetallic RuCo complex.141 This combined approach can be even extended to structural transitions of biological macromolecules. For example, local structural changes of photoactive proteins such as Mb and Hb can be revealed by TRXAS,225,226 whereas overall secondary and tertiary structural changes can be elucidated by TRXL as demonstrated by recent studies.101,127,134Recently, X-ray science has made a major breakthrough with the advent of XFEL and energy-recovery linac (ERL) that deliver femtosecond X-ray pulses with increased photon flux and spatial coherence. As a result, TRXL and TRXAS are now being used to probe ultrafast dynamics on femtosecond time scales,139–142,171–174 catching up with their optical spectroscopy counterparts. On the other hand, these femtosecond X-ray sources pose experimental and theoretical challenges in the measurement and analysis of the data acquired by the two techniques. Once these challenges are overcome, TRXL and TRXAS will become more common structural techniques for investigating ultrafast dynamics in a broad range of molecular systems.
Acknowledgements
We greatly appreciate our co-workers listed in many references of this article. We acknowledge other research groups who have made significant contributions to the advance of TRXL and TRXAS as well as other related X-ray techniques. This work was supported by IBS-R004-G2. This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF-2014R1A1A1002511, 2013R1A1A2009575, 2014R1A4A1001690, and 2011-0031558). This research has been supported by the TJ Park Science Fellowship of POSCO TJ Park Foundation.References
- G. R. Fleming, Annu. Rev. Phys. Chem., 1986, 37, 81–104 CrossRef CAS.
- N. F. Scherer, R. J. Carlson, A. Matro, M. Du, A. J. Ruggiero, V. Romero-Rochin, J. A. Cina, G. R. Fleming and S. A. Rice, J. Chem. Phys., 1991, 95, 1487–1511 CrossRef CAS.
- C. E. Crespo-Hernandez, B. Cohen and B. Kohler, Nature, 2005, 436, 1141–1144 CrossRef CAS PubMed.
- D. Polli, P. Altoe, O. Weingart, K. M. Spillane, C. Manzoni, D. Brida, G. Tomasello, G. Orlandi, P. Kukura, R. A. Mathies, M. Garavelli and G. Cerullo, Nature, 2010, 467, 440–443 CrossRef CAS PubMed.
- J. Herbst, K. Heyne and R. Diller, Science, 2002, 297, 822–825 CrossRef CAS PubMed.
- E. T. J. Nibbering, H. Fidder and E. Pines, Annu. Rev. Phys. Chem., 2005, 56, 337–367 CrossRef CAS PubMed.
- W. J. Schreier, T. E. Schrader, F. O. Koller, P. Gilch, C. E. Crespo-Hernández, V. N. Swaminathan, T. Carell, W. Zinth and B. Kohler, Science, 2007, 315, 625–629 CrossRef CAS PubMed.
- J. M. Friedman, D. L. Rousseau and M. R. Ondrias, Annu. Rev. Phys. Chem., 1982, 33, 471–491 CrossRef CAS.
- H. Hamaguchi and T. L. Gustafson, Annu. Rev. Phys. Chem., 1994, 45, 593–622 CrossRef CAS.
- P. Kukura, D. W. McCamant, S. Yoon, D. B. Wandschneider and R. A. Mathies, Science, 2005, 310, 1006–1009 CrossRef CAS PubMed.
- P. Kukura, D. W. McCamant and R. A. Mathies, Annu. Rev. Phys. Chem., 2007, 58, 461–488 CrossRef CAS PubMed.
- D. M. Jonas, Annu. Rev. Phys. Chem., 2003, 54, 425–463 CrossRef CAS PubMed.
- T. Brixner, T. Mančal, I. V. Stiopkin and G. R. Fleming, J. Chem. Phys., 2004, 121, 4221–4236 CrossRef CAS PubMed.
- M. Cho, Chem. Rev., 2008, 108, 1331–1418 CrossRef CAS PubMed.
- B. A. West and A. M. Moran, J. Phys. Chem. Lett., 2012, 3, 2575–2581 CrossRef CAS PubMed.
- C. Consani, G. Auböck, F. van Mourik and M. Chergui, Science, 2013, 339, 1586–1589 CrossRef CAS PubMed.
- M. C. Asplund, M. T. Zanni and R. M. Hochstrasser, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8219–8224 CrossRef CAS PubMed.
- M. L. Cowan, B. D. Bruner, N. Huse, J. R. Dwyer, B. Chugh, E. T. J. Nibbering, T. Elsaesser and R. J. D. Miller, Nature, 2005, 434, 199–202 CrossRef CAS PubMed.
- P. Hamm, J. Helbing and J. Bredenbeck, Annu. Rev. Phys. Chem., 2008, 59, 291–317 CrossRef CAS PubMed.
- M. D. Fayer, Annu. Rev. Phys. Chem., 2009, 60, 21–38 CrossRef CAS PubMed.
- A. H. Zewail, J. Phys. Chem. A, 2000, 104, 5660–5694 CrossRef CAS.
- A. A. Ischenko, V. V. Golubkov, V. P. Spiridonov, A. V. Zgurskii, A. S. Akhmanov, M. G. Vabischevich and V. N. Bagratashvili, Appl. Phys. B: Photophys. Laser Chem., 1983, 32, 161–163 CrossRef.
- J. C. Williamson and A. H. Zewail, J. Phys. Chem., 1994, 98, 2766–2781 CrossRef CAS.
- J. C. Williamson, J. M. Cao, H. Ihee, H. Frey and A. H. Zewail, Nature, 1997, 386, 159–162 CrossRef CAS.
- A. H. Zewail, Angew. Chem., Int. Ed., 2001, 40, 4371–4375 CrossRef CAS.
- R. C. Dudek and P. M. Weber, J. Phys. Chem. A, 2001, 105, 4167–4171 CrossRef CAS.
- H. Ihee, J. Cao and A. H. Zewail, Angew. Chem., Int. Ed., 2001, 40, 1532–1536 CrossRef CAS.
- H. Ihee, V. A. Lobastov, U. M. Gomez, B. M. Goodson, R. Srinivasan, C. Y. Ruan and A. H. Zewail, Science, 2001, 291, 458–462 CrossRef CAS PubMed.
- C. Y. Ruan, V. A. Lobastov, R. Srinivasan, B. M. Goodson, H. Ihee and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7117–7122 CrossRef CAS PubMed.
- H. Ihee, J. S. Feenstra, J. M. Cao and A. H. Zewail, Chem. Phys. Lett., 2002, 353, 325–334 CrossRef CAS.
- B. J. Siwick, J. R. Dwyer, R. E. Jordan and R. J. D. Miller, Science, 2003, 302, 1382–1385 CrossRef CAS PubMed.
- R. Srinivasan, V. A. Lobastov, C. Y. Ruan and A. H. Zewail, Helv. Chim. Acta, 2003, 86, 1763–1838 CrossRef CAS.
- V. A. Lobastov, R. Srinivasan and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7069–7073 CrossRef CAS PubMed.
- R. Srinivasan, J. S. Feenstra, S. T. Park, S. Xu and A. H. Zewail, Science, 2005, 307, 558–563 CrossRef CAS PubMed.
- S. T. Park, J. S. Feenstra and A. H. Zewail, J. Chem. Phys., 2006, 124, 174707 CrossRef PubMed.
- A. H. Zewail, Annu. Rev. Phys. Chem., 2006, 57, 65–103 CrossRef CAS PubMed.
- V. A. Lobastov, J. Weissenrieder, J. Tang and A. H. Zewail, Nano Lett., 2007, 7, 2552–2558 CrossRef CAS PubMed.
- D. Shorokhov and A. H. Zewail, Phys. Chem. Chem. Phys., 2008, 10, 2879–2893 RSC.
- M. Chergui and A. H. Zewail, ChemPhysChem, 2009, 10, 28–43 CrossRef CAS PubMed.
- R. Ernstorfer, M. Harb, C. T. Hebeisen, G. Sciaini, T. Dartigalongue and R. J. D. Miller, Science, 2009, 323, 1033–1037 CrossRef CAS PubMed.
- G. Sciaini, M. Harb, S. G. Kruglik, T. Payer, C. T. Hebeisen, F.-J. M. z. Heringdorf, M. Yamaguchi, M. H.-v. Hoegen, R. Ernstorfer and R. J. D. Miller, Nature, 2009, 458, 56–59 CrossRef CAS PubMed.
- M. Eichberger, H. Schafer, M. Krumova, M. Beyer, J. Demsar, H. Berger, G. Moriena, G. Sciaini and R. J. D. Miller, Nature, 2010, 468, 799–802 CrossRef CAS PubMed.
- M. Gao, C. Lu, H. Jean-Ruel, L. C. Liu, A. Marx, K. Onda, S.-y. Koshihara, Y. Nakano, X. Shao, T. Hiramatsu, G. Saito, H. Yamochi, R. R. Cooney, G. Moriena, G. Sciaini and R. J. D. Miller, Nature, 2013, 496, 343–346 CrossRef CAS PubMed.
- V. R. Morrison, R. P. Chatelain, K. L. Tiwari, A. Hendaoui, A. Bruhács, M. Chaker and B. J. Siwick, Science, 2014, 346, 445–448 CrossRef CAS PubMed.
- M. W. van Mourik, W. J. Engelen, E. J. D. Vredenbregt and O. J. Luiten, Struct. Dyn., 2014, 1, 034302 CrossRef.
- A. Feist, K. E. Echternkamp, J. Schauss, S. V. Yalunin, S. Schafer and C. Ropers, Nature, 2015, 521, 200–203 CrossRef CAS PubMed.
- C. Gerbig, A. Senftleben, S. Morgenstern, C. Sarpe and T. Baumert, New J. Phys., 2015, 17, 043050 CrossRef.
- L. Waldecker, T. A. Miller, M. Rude, R. Bertoni, J. Osmond, V. Pruneri, R. E. Simpson, R. Ernstorfer and S. Wall, Nat. Mater., 2015, 14, 991–995 CrossRef CAS PubMed.
- V. Srajer, T. Y. Teng, T. Ursby, C. Pradervand, Z. Ren, S. Adachi, W. Schildkamp, D. Bourgeois, M. Wulff and K. Moffat, Science, 1996, 274, 1726–1729 CrossRef CAS PubMed.
- C. Rischel, A. Rousse, I. Uschmann, P. A. Albouy, J. P. Geindre, P. Audebert, J. C. Gauthier, E. Forster, J. L. Martin and A. Antonetti, Nature, 1997, 390, 490–492 CrossRef CAS.
- U. K. Genick, G. E. O. Borgstahl, K. Ng, Z. Ren, C. Pradervand, P. M. Burke, V. Šrajer, T.-Y. Teng, W. Schildkamp, D. E. McRee, K. Moffat and E. D. Getzoff, Science, 1997, 275, 1471–1475 CrossRef CAS PubMed.
- U. K. Genick, S. M. Soltis, P. Kuhn, I. L. Canestrelli and E. D. Getzoff, Nature, 1998, 392, 206–209 CrossRef CAS PubMed.
- A. Ostermann, R. Waschipky, F. G. Parak and G. U. Nienhaus, Nature, 2000, 404, 205–208 CrossRef CAS PubMed.
- K. Moffat, Chem. Rev., 2001, 101, 1569–1581 CrossRef CAS PubMed.
- S. Techert, F. Schotte and M. Wulff, Phys. Rev. Lett., 2001, 86, 2030–2033 CrossRef CAS PubMed.
- E. Collet, M. H. Lemee-Cailleau, M. Buron-Le Cointe, H. Cailleau, M. Wulff, T. Luty, S. Y. Koshihara, M. Meyer, L. Toupet, P. Rabiller and S. Techert, Science, 2003, 300, 612–615 CrossRef CAS PubMed.
- P. Coppens, Chem. Commun., 2003, 1317–1320 RSC.
- F. Schotte, M. Lim, T. A. Jackson, A. V. Smirnov, J. Soman, J. S. Olson, G. N. Phillips, Jr., M. Wulff and P. A. Anfinrud, Science, 2003, 300, 1944–1947 CrossRef CAS PubMed.
- P. Coppens, I. I. Vorontsov, T. Graber, A. Y. Kovalevsky, Y. S. Chen, G. Wu, M. Gembicky and I. V. Novozhilova, J. Am. Chem. Soc., 2004, 126, 5980–5981 CrossRef CAS PubMed.
- M. Schmidt, R. Pahl, V. Srajer, S. Anderson, Z. Ren, H. Ihee, S. Rajagopal and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4799–4804 CrossRef CAS PubMed.
- F. Schotte, P. A. Anfinrud, G. Hummer and M. Wulff, Biophys. J., 2004, 86, 525a Search PubMed.
- M. Schmidt, K. Nienhaus, R. Pahl, A. Krasselt, S. Anderson, F. Parak, G. U. Nienhaus and V. Srajer, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11704–11709 CrossRef CAS PubMed.
- H. Ihee, S. Rajagopal, V. Srajer, R. Pahl, S. Anderson, M. Schmidt, F. Schotte, P. A. Anfinrud, M. Wulff and K. Moffat, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 7145–7150 CrossRef CAS PubMed.
- M. Bargheer, N. Zhavoronkov, M. Woerner and T. Elsaesser, ChemPhysChem, 2006, 7, 783–792 CrossRef CAS PubMed.
- E. Collet, M. B. L. Cointe, M. Lorenc and H. Cailleau, Z. Kristallogr., 2008, 223, 272–282 CrossRef CAS.
- E. Collet, M. L. Boillot, J. Hebert, N. Moisan, M. Servol, M. Lorenc, L. Toupet, M. Buron-Le Cointe, A. Tissot and J. Sainton, Acta Crystallogr., Sect. B: Struct. Sci., 2009, 65, 474–480 CAS.
- I. I. Vorontsov, T. Graber, A. Y. Kovalevsky, I. V. Novozhilova, M. Gembicky, Y.-S. Chen and P. Coppens, J. Am. Chem. Soc., 2009, 131, 6566–6573 CrossRef CAS PubMed.
- S.-L. Zheng, Y. Wang, Z. Yu, Q. Lin and P. Coppens, J. Am. Chem. Soc., 2009, 131, 18036–18037 CrossRef CAS PubMed.
- A. B. Wöhri, G. Katona, L. C. Johansson, E. Fritz, E. Malmerberg, M. Andersson, J. Vincent, M. Eklund, M. Cammarata, M. Wulff, J. Davidsson, G. Groenhof and R. Neutze, Science, 2010, 328, 630–633 CrossRef PubMed.
- H. Cailleau, M. Lorenc, L. Guerin, M. Servol, E. Collet and M. Buron-Le Cointe, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 189–197 CrossRef CAS PubMed.
- J. B. Benedict, A. Makal, J. D. Sokolow, E. Trzop, S. Scheins, R. Henning, T. Graber and P. Coppens, Chem. Commun., 2011, 47, 1704–1706 RSC.
- P. Coppens, J. Phys. Chem. Lett., 2011, 2, 616–621 CrossRef CAS.
- X. Yang, Z. Ren, J. Kuk and K. Moffat, Nature, 2011, 479, 428–432 CrossRef CAS PubMed.
- E. Collet, M. Lorenc, M. Cammarata, L. Guérin, M. Servol, A. Tissot, M.-L. Boillot, H. Cailleau and M. Buron-LeCointe, Chem. – Eur. J., 2012, 18, 2051–2055 CrossRef CAS PubMed.
- E. Collet, N. Moisan, C. Balde, R. Bertoni, E. Trzop, C. Laulhe, M. Lorenc, M. Servol, H. Cailleau, A. Tissot, M.-L. Boillot, T. Graber, R. Henning, P. Coppens and M. B.-L. Cointe, Phys. Chem. Chem. Phys., 2012, 14, 6192–6199 RSC.
- A. Makal, J. Benedict, E. Trzop, J. Sokolow, B. Fournier, Y. Chen, J. A. Kalinowski, T. Graber, R. Henning and P. Coppens, J. Phys. Chem. A, 2012, 116, 3359–3365 CrossRef CAS PubMed.
- Y. O. Jung, J. H. Lee, J. Kim, M. Schmidt, K. Moffat, V. Šrajer and H. Ihee, Nat. Chem., 2013, 5, 212–220 CrossRef CAS PubMed.
- K. N. Jarzembska, R. Kamiński, B. Fournier, E. Trzop, J. D. Sokolow, R. Henning, Y. Chen and P. Coppens, Inorg. Chem., 2014, 53, 10594–10601 CrossRef CAS PubMed.
- R. Neutze, R. Wouts, S. Techert, J. Davidsson, M. Kocsis, A. Kirrander, F. Schotte and M. Wulff, Phys. Rev. Lett., 2001, 87, 195508 CrossRef CAS PubMed.
- S. Bratos, F. Mirloup, R. Vuilleumier and M. Wulff, J. Chem. Phys., 2002, 116, 10615–10625 CrossRef CAS.
- S. Bratos, F. Mirloup, R. Vuilleumier, M. Wulff and A. Plech, Chem. Phys., 2004, 304, 245–251 CrossRef CAS.
- A. Plech, M. Wulff, S. Bratos, F. Mirloup, R. Vuilleumier, F. Schotte and P. A. Anfinrud, Phys. Rev. Lett., 2004, 92, 125505 CrossRef PubMed.
- A. Plech, V. Kotaidis, S. Gresillon, C. Dahmen and G. von Plessen, Phys. Rev. B: Condens. Matter Mater. Phys., 2004, 70, 195423 CrossRef.
- H. Ihee, M. Lorenc, T. K. Kim, Q. Y. Kong, M. Cammarata, J. H. Lee, S. Bratos and M. Wulff, Science, 2005, 309, 1223–1227 CrossRef CAS PubMed.
- J. Davidsson, J. Poulsen, M. Cammarata, P. Georgiou, R. Wouts, G. Katona, F. Jacobson, A. Plech, M. Wulff, G. Nyman and R. Neutze, Phys. Rev. Lett., 2005, 94, 245503 CrossRef.
- A. Plech, V. Kotaidis, M. Lorenc and M. Wulff, Chem. Phys. Lett., 2005, 401, 565–569 CrossRef CAS.
- M. Cammarata, M. Lorenc, T. K. Kim, J. H. Lee, Q. Y. Kong, E. Pontecorvo, M. Lo Russo, G. Schiro, A. Cupane, M. Wulff and H. Ihee, J. Chem. Phys., 2006, 124, 124504 CrossRef CAS PubMed.
- P. Georgiou, J. Vincent, M. Andersson, A. B. Wohri, P. Gourdon, J. Poulsen, J. Davidsson and R. Neutze, J. Chem. Phys., 2006, 124, 234507 CrossRef PubMed.
- T. K. Kim, M. Lorenc, J. H. Lee, M. Russo, J. Kim, M. Cammarata, Q. Y. Kong, S. Noel, A. Plech, M. Wulff and H. Ihee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 9410–9415 CrossRef CAS PubMed.
- V. Kotaidis, C. Dahmen, G. von Plessen, F. Springer and A. Plech, J. Chem. Phys., 2006, 124, 184702 CrossRef CAS PubMed.
- A. Plech, V. Kotaidis, M. Lorenc and J. Boneberg, Nat. Phys., 2006, 2, 44–47 CrossRef CAS.
- M. Wulff, S. Bratos, A. Plech, R. Vuilleumier, F. Mirloup, M. Lorenc, Q. Kong and H. Ihee, J. Chem. Phys., 2006, 124, 034501 CrossRef CAS PubMed.
- Q. Y. Kong, M. Wulff, J. H. Lee, S. Bratos and H. Ihee, J. Am. Chem. Soc., 2007, 129, 13584–13591 CrossRef CAS PubMed.
- A. Plech, R. Cerna, V. Kotaidis, F. Hudert, A. Bartels and T. Dekorsy, Nano Lett., 2007, 7, 1026–1031 CrossRef CAS PubMed.
- A. Plech, V. Kotaidis, K. Istomin and M. Wulff, J. Synchrotron Radiat., 2007, 14, 288–294 CrossRef CAS PubMed.
- M. Cammarata, M. Levantino, F. Schotte, P. A. Anfinrud, F. Ewald, J. Choi, A. Cupane, M. Wulff and H. Ihee, Nat. Methods, 2008, 5, 881–886 CrossRef CAS PubMed.
- Q. Y. Kong, J. H. Lee, A. Plech, M. Wulff, H. Ihee and M. H. J. Koch, Angew. Chem., Int. Ed., 2008, 47, 5550–5553 CrossRef CAS PubMed.
- J. H. Lee, J. Kim, M. Cammarata, Q. Kong, K. H. Kim, J. Choi, T. K. Kim, M. Wulff and H. Ihee, Angew. Chem., Int. Ed., 2008, 47, 1047–1050 CrossRef CAS PubMed.
- J. H. Lee, T. K. Kim, J. Kim, Q. Kong, M. Cammarata, M. Lorenc, M. Wulff and H. Ihee, J. Am. Chem. Soc., 2008, 130, 5834–5835 CrossRef CAS PubMed.
- A. Plech, V. Kotaidis, A. Siems and M. Sztucki, Phys. Chem. Chem. Phys., 2008, 10, 3888–3894 RSC.
- S. Ahn, K. H. Kim, Y. Kim, J. Kim and H. Ihee, J. Phys. Chem. B, 2009, 113, 13131–13133 CrossRef CAS PubMed.
- M. Andersson, E. Malmerberg, S. Westenhoff, G. Katona, M. Cammarata, A. B. Wohri, L. C. Johansson, F. Ewald, M. Eklund, M. Wulff, J. Davidsson and R. Neutze, Structure, 2009, 17, 1265–1275 CrossRef CAS PubMed.
- M. Christensen, K. Haldrup, K. Bechgaard, R. Feidenhans'l, Q. Y. Kong, M. Cammarata, M. Lo Russo, M. Wulff, N. Harrit and M. M. Nielsen, J. Am. Chem. Soc., 2009, 131, 502–508 CrossRef CAS PubMed.
- K. Haldrup, M. Christensen, M. Cammarata, Q. Kong, M. Wulff, S. O. Mariager, K. Bechgaard, R. Feidenhans'l, N. Harrit and M. M. Nielsen, Angew. Chem., Int. Ed., 2009, 48, 4180–4184 CrossRef CAS PubMed.
- H. Ihee, Acc. Chem. Res., 2009, 42, 356–366 CrossRef CAS PubMed.
- T. K. Kim, J. H. Lee, M. Wulff, Q. Y. Kong and H. Ihee, ChemPhysChem, 2009, 10, 1958–1980 CrossRef CAS PubMed.
- J. Vincent, M. Andersson, M. Eklund, A. B. Wöhri, M. Odelius, E. Malmerberg, Q. Kong, M. Wulff, R. Neutze and J. Davidsson, J. Chem. Phys., 2009, 130, 154502 CrossRef PubMed.
- H. S. Cho, N. Dashdorj, F. Schotte, T. Graber, R. Henning and P. Anfinrud, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7281–7286 CrossRef CAS PubMed.
- M. Christensen, K. Haldrup, K. S. Kjaer, M. Cammarata, M. Wulff, K. Bechgaard, H. Weihe, N. H. Harrit and M. M. Nielsen, Phys. Chem. Chem. Phys., 2010, 12, 6921–6923 RSC.
- H. Ihee, M. Wulff, J. Kim and S. Adachi, Int. Rev. Phys. Chem., 2010, 29, 453–520 CrossRef CAS.
- S. Jun, J. H. Lee, J. Kim, J. Kim, K. H. Kim, Q. Y. Kong, T. K. Kim, M. Lo Russo, M. Wulff and H. Ihee, Phys. Chem. Chem. Phys., 2010, 12, 11536–11547 RSC.
- J. Kim, K. H. Kim, J. H. Lee and H. Ihee, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 270–280 CrossRef CAS PubMed.
- Q. Kong, J. H. Lee, K. H. Kim, J. Kim, M. Wulff, H. Ihee and M. H. J. Koch, J. Am. Chem. Soc., 2010, 132, 2600–2607 CrossRef CAS PubMed.
- Q. Y. Kong, J. H. Lee, M. Lo Russo, T. K. Kim, M. Lorenc, M. Cammarata, S. Bratos, T. Buslaps, V. Honkimaki, H. Ihee and M. Wulff, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 252–260 CrossRef CAS PubMed.
- J. H. Lee and H. Ihee, Struct. Chem., 2010, 21, 37–42 CrossRef CAS.
- L. Salassa, E. Borfecchia, T. Ruiu, C. Garino, D. Gianolio, R. Gobetto, P. J. Sadler, M. Cammarata, M. Wulff and C. Lamberti, Inorg. Chem., 2010, 49, 11240–11248 CrossRef CAS PubMed.
- S. Westenhoff, E. Malmerberg, D. Arnlund, L. Johansson, E. Nazarenko, M. Cammarata, J. Davidsson, V. Chaptal, J. Abramson, G. Katona, A. Menzel and R. Neutze, Nat. Methods, 2010, 7, 775–776 CrossRef CAS PubMed.
- S. Westenhoff, E. Nazarenko, E. Malmerberg, J. Davidsson, G. Katona and R. Neutze, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 207–219 CrossRef CAS PubMed.
- K. Haldrup, T. Harlang, M. Christensen, A. Dohn, T. B. van Driel, K. S. Kjaer, N. Harrit, J. Vibenholt, L. Guerin, M. Wulff and M. M. Nielsen, Inorg. Chem., 2011, 50, 9329–9336 CrossRef CAS PubMed.
- S. Ibrahimkutty, J. Kim, M. Cammarata, F. Ewald, J. Choi, H. Ihee and A. Plech, ACS Nano, 2011, 5, 3788–3794 CrossRef CAS PubMed.
- K. H. Kim, K. Y. Oang, J. Kim, J. H. Lee, Y. Kim and H. Ihee, Chem. Commun., 2011, 47, 289–291 RSC.
- J. Kim, K. H. Kim, J. G. Kim, T. W. Kim, Y. Kim and H. Ihee, J. Phys. Chem. Lett., 2011, 2, 350–356 CrossRef CAS PubMed.
- E. Malmerberg, Z. Omran, J. S. Hub, X. Li, G. Katona, S. Westenhoff, L. C. Johansson, M. Andersson, M. Cammarata, M. Wulff, D. van der Spoel, J. Davidsson, A. Specht and R. Neutze, Biophys. J., 2011, 101, 1345–1353 CrossRef CAS PubMed.
- K. Haldrup, G. Vanko, W. Gawelda, A. Galler, G. Doumy, A. M. March, E. P. Kanter, A. Bordage, A. Dohn, T. B. van Driel, K. S. Kjaer, H. T. Lemke, S. E. Canton, J. Uhlig, V. Sundstrom, L. Young, S. H. Southworth, M. M. Nielsen and C. Bressler, J. Phys. Chem. A, 2012, 116, 9878–9887 CrossRef CAS PubMed.
- S. Ibrahimkutty, P. Wagener, A. Menzel, A. Plech and S. Barcikowski, Appl. Phys. Lett., 2012, 101, 103104 CrossRef.
- T. W. Kim, J. H. Lee, J. Choi, K. H. Kim, L. J. van Wilderen, L. Guerin, Y. Kim, Y. O. Jung, C. Yang, J. Kim, M. Wulff, J. J. van Thor and H. Ihee, J. Am. Chem. Soc., 2012, 134, 3145–3153 CrossRef CAS PubMed.
- K. H. Kim, S. Muniyappan, K. Y. Oang, J. G. Kim, S. Nozawa, T. Sato, S.-y. Koshihara, R. Henning, I. Kosheleva, H. Ki, Y. Kim, T. W. Kim, J. Kim, S.-i. Adachi and H. Ihee, J. Am. Chem. Soc., 2012, 134, 7001–7008 CrossRef CAS PubMed.
- J. Kim, J. H. Lee, J. Kim, S. Jun, K. H. Kim, T. W. Kim, M. Wulff and H. Ihee, J. Phys. Chem. A, 2012, 116, 2713–2722 CrossRef CAS PubMed.
- A. Spilotros, M. Levantino, G. Schiro, M. Cammarata, M. Wulff and A. Cupane, Soft Matter, 2012, 8, 6434–6437 RSC.
- K. H. Kim, J. H. Lee, J. Kim, S. Nozawa, T. Sato, A. Tomita, K. Ichiyanagi, H. Ki, J. Kim, S.-i. Adachi and H. Ihee, Phys. Rev. Lett., 2013, 110, 165505 CrossRef PubMed.
- K. H. Kim, H. Ki, K. Y. Oang, S. Nozawa, T. Sato, J. Kim, T. K. Kim, J. Kim, S.-i. Adachi and H. Ihee, ChemPhysChem, 2013, 14, 3687–3697 CrossRef CAS PubMed.
- J. H. Lee, M. Wulff, S. Bratos, J. Petersen, L. Guerin, J.-C. Leicknam, M. Cammarata, Q. Kong, J. Kim, K. B. Møller and H. Ihee, J. Am. Chem. Soc., 2013, 135, 3255–3261 CrossRef CAS PubMed.
- K. H. Kim, J. Kim, J. H. Lee and H. Ihee, Struct. Dyn., 2014, 1, 011301 CrossRef.
- K. Y. Oang, J. G. Kim, C. Yang, T. W. Kim, Y. Kim, K. H. Kim, J. Kim and H. Ihee, J. Phys. Chem. Lett., 2014, 5, 804–808 CrossRef CAS PubMed.
- K. Y. Oang, K. H. Kim, J. Jo, Y. Kim, J. G. Kim, T. W. Kim, S. Jun, J. Kim and H. Ihee, Chem. Phys., 2014, 442, 137–142 CrossRef CAS PubMed.
- K. H. Kim, H. Ki, J. H. Lee, S. Park, Q. Kong, J. Kim, J. Kim, M. Wulff and H. Ihee, Phys. Chem. Chem. Phys., 2015, 17, 8633–8637 RSC.
- J. G. Kim, T. W. Kim, J. Kim and H. Ihee, Acc. Chem. Res., 2015, 48, 2200–2208 CrossRef CAS PubMed.
- E. Malmerberg, P. H. M. Bovee-Geurts, G. Katona, X. Deupi, D. Arnlund, C. Wickstrand, L. C. Johansson, S. Westenhoff, E. Nazarenko, G. F. X. Schertler, A. Menzel, W. J. de Grip and R. Neutze, Sci. Signaling, 2015, 8, ra26 CrossRef PubMed.
- D. Arnlund, L. C. Johansson, C. Wickstrand, A. Barty, G. J. Williams, E. Malmerberg, J. Davidsson, D. Milathianaki, D. P. DePonte, R. L. Shoeman, D. J. Wang, D. James, G. Katona, S. Westenhoff, T. A. White, A. Aquila, S. Bari, P. Berntsen, M. Bogan, T. B. van Driel, R. B. Doak, K. S. Kjaer, M. Frank, R. Fromme, I. Grotjohann, R. Henning, M. S. Hunter, R. A. Kirian, I. Kosheleva, C. Kupitz, M. N. Liang, A. V. Martin, M. M. Nielsen, M. Messerschmidt, M. M. Seibert, J. Sjohamn, F. Stellato, U. Weierstall, N. A. Zatsepin, J. C. H. Spence, P. Fromme, I. Schlichting, S. Boutet, G. Groenhof, H. N. Chapman and R. Neutze, Nat. Methods, 2014, 11, 923–926 CrossRef CAS PubMed.
- K. H. Kim, J. G. Kim, S. Nozawa, T. Sato, K. Y. Oang, T. W. Kim, H. Ki, J. Jo, S. Park, C. Song, K. Ogawa, T. Togashi, K. Tono, M. Yabashi, T. Ishikawa, J. Kim, R. Ryoo, H. Ihee and S. Adachi, Nature, 2015, 518, 385–389 CrossRef CAS PubMed.
- S. E. Canton, K. S. Kjær, G. Vankó, T. B. van Driel, S.-i. Adachi, A. Bordage, C. Bressler, P. Chabera, M. Christensen, A. O. Dohn, A. Galler, W. Gawelda, D. Gosztola, K. Haldrup, T. Harlang, Y. Liu, K. B. Møller, Z. Németh, S. Nozawa, M. Pápai, T. Sato, T. Sato, K. Suarez-Alcantara, T. Togashi, K. Tono, J. Uhlig, D. A. Vithanage, K. Wärnmark, M. Yabashi, J. Zhang, V. Sundström and M. M. Nielsen, Nat. Commun., 2015, 6, 6359 CrossRef CAS PubMed.
- M. Levantino, G. Schirò, H. T. Lemke, G. Cottone, J. M. Glownia, D. Zhu, M. Chollet, H. Ihee, A. Cupane and M. Cammarata, Nat. Commun., 2015, 6, 6772 CrossRef CAS PubMed.
- J. Tenboer, S. Basu, N. Zatsepin, K. Pande, D. Milathianaki, M. Frank, M. Hunter, S. Boutet, G. J. Williams, J. E. Koglin, D. Oberthuer, M. Heymann, C. Kupitz, C. Conrad, J. Coe, S. Roy-Chowdhury, U. Weierstall, D. James, D. J. Wang, T. Grant, A. Barty, O. Yefanov, J. Scales, C. Gati, C. Seuring, V. Srajer, R. Henning, P. Schwander, R. Fromme, A. Ourmazd, K. Moffat, J. J. Van Thor, J. C. H. Spence, P. Fromme, H. N. Chapman and M. Schmidt, Science, 2014, 346, 1242–1246 CrossRef CAS PubMed.
- L. X. Chen, W. J. H. Jager, G. Jennings, D. J. Gosztola, A. Munkholm and J. P. Hessler, Science, 2001, 292, 262–264 CrossRef CAS PubMed.
- L. X. Chen, G. B. Shaw, I. Novozhilova, T. Liu, G. Jennings, K. Attenkofer, G. J. Meyer and P. Coppens, J. Am. Chem. Soc., 2003, 125, 7022–7034 CrossRef CAS PubMed.
- L. X. Chen, Faraday Discuss., 2003, 122, 315–329 RSC.
- M. Saes, C. Bressler, R. Abela, D. Grolimund, S. L. Johnson, P. A. Heimann and M. Chergui, Phys. Rev. Lett., 2003, 90, 047403 CrossRef PubMed.
- C. Bressler and M. Chergui, Chem. Rev., 2004, 104, 1781–1812 CrossRef CAS PubMed.
- L. X. Chen, Angew. Chem., Int. Ed., 2004, 43, 2886–2905 CrossRef CAS PubMed.
- P. Wernet, D. Nordlund, U. Bergmann, M. Cavalleri, M. Odelius, H. Ogasawara, L. Å. Näslund, T. K. Hirsch, L. Ojamäe, P. Glatzel, L. G. M. Pettersson and A. Nilsson, Science, 2004, 304, 995–999 CrossRef CAS PubMed.
- L. X. Chen, Annu. Rev. Phys. Chem., 2005, 56, 221–254 CrossRef CAS PubMed.
- M. Benfatto, S. Della Longa, K. Hatada, K. Hayakawa, W. Gawelda, C. Bressler and M. Chergui, J. Phys. Chem. B, 2006, 110, 14035–14039 CrossRef CAS PubMed.
- W. Gawelda, M. Johnson, F. M. F. de Groot, R. Abela, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2006, 128, 5001–5009 CrossRef CAS PubMed.
- M. Khalil, M. A. Marcus, A. L. Smeigh, J. K. McCusker, H. H. W. Chong and R. W. Schoenlein, J. Phys. Chem. A, 2006, 110, 38–44 CrossRef CAS PubMed.
- L. X. Chen, X. Y. Zhang, E. C. Wasinger, K. Attenkofer, G. Jennings, A. Z. Muresan and J. S. Lindsey, J. Am. Chem. Soc., 2007, 129, 9616–9618 CrossRef CAS PubMed.
- W. Gawelda, V.-T. Pham, M. Benfatto, Y. Zaushitsyn, M. Kaiser, D. Grolimund, S. L. Johnson, R. Abela, A. Hauser, C. Bressler and M. Chergui, Phys. Rev. Lett., 2007, 98, 057401 CrossRef PubMed.
- V.-T. Pham, W. Gawelda, Y. Zaushitsyn, M. Kaiser, D. Grolimund, S. L. Johnson, R. Abela, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 1530–1531 CrossRef CAS PubMed.
- C. Bressler, R. Abela and M. Chergui, Z. Kristallogr., 2008, 223, 307–321 CrossRef CAS.
- C. Bressler and M. Chergui, Actual. Chim., 2008, 59–61 CAS.
- C. Bressler, C. Milne, V. T. Pham, A. ElNahhas, R. M. van der Veen, W. Gawelda, S. Johnson, P. Beaud, D. Grolimund, M. Kaiser, C. N. Borca, G. Ingold, R. Abela and M. Chergui, Science, 2009, 323, 489–492 CrossRef CAS PubMed.
- S. Della-Longa, L. X. Chen, P. Frank, K. Hayakawa, K. Hatada and M. Benfatto, Inorg. Chem., 2009, 48, 3934–3942 CrossRef CAS PubMed.
- N. Huse, M. Khalil, T. K. Kim, A. L. Smeigh, L. Jamula, J. K. McCusker and R. W. Schoenlein, J. Phys.: Conf. Ser., 2009, 148, 012043 CrossRef.
- N. Huse, H. D. Wen, D. Nordlund, E. Szilagyi, D. Daranciang, T. A. Miller, A. Nilsson, R. W. Schoenlein and A. M. Lindenberg, Phys. Chem. Chem. Phys., 2009, 11, 3951–3957 RSC.
- R. M. van der Veen, C. J. Milne, A. El Nahhas, F. A. Lima, V. T. Pham, J. Best, J. A. Weinstein, C. N. Borca, R. Abela, C. Bressler and M. Chergui, Angew. Chem., Int. Ed., 2009, 48, 2711–2714 CrossRef CAS PubMed.
- C. Bressler and M. Chergui, Annu. Rev. Phys. Chem., 2010, 61, 263–282 CrossRef CAS PubMed.
- L. X. Chen, X. Y. Zhang, E. C. Wasinger, J. V. Lockard, A. B. Stickrath, M. W. Mara, K. Attenkofer, G. Jennings, G. Smolentsev and A. Soldatov, Chem. Sci., 2010, 1, 642–650 RSC.
- L. X. Chen, X. Y. Zhang, J. V. Lockard, A. B. Stickrath, K. Attenkofer, G. Jennings and D. J. Liu, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 240–251 CrossRef CAS PubMed.
- M. Chergui, Acta Crystallogr., Sect. A: Found. Crystallogr., 2010, 66, 229–239 CrossRef CAS PubMed.
- N. Huse, T. K. Kim, L. Jamula, J. K. McCusker, F. M. F. de Groot and R. W. Schoenlein, J. Am. Chem. Soc., 2010, 132, 6809–6816 CrossRef CAS PubMed.
- N. Huse, H. Cho, K. Hong, L. Jamula, F. M. F. de Groot, T. K. Kim, J. K. McCusker and R. W. Schoenlein, J. Phys. Chem. Lett., 2011, 2, 880–884 CrossRef CAS PubMed.
- H. T. Lemke, C. Bressler, L. X. Chen, D. M. Fritz, K. J. Gaffney, A. Galler, W. Gawelda, K. Haldrup, R. W. Hartsock, H. Ihee, J. Kim, K. H. Kim, J. H. Lee, M. M. Nielsen, A. B. Stickrath, W. Zhang, D. Zhu and M. Cammarata, J. Phys. Chem. A, 2013, 117, 735–740 CrossRef CAS PubMed.
- W. Zhang, R. Alonso-Mori, U. Bergmann, C. Bressler, M. Chollet, A. Galler, W. Gawelda, R. G. Hadt, R. W. Hartsock, T. Kroll, K. S. Kjaer, K. Kubicek, H. T. Lemke, H. W. Liang, D. A. Meyer, M. M. Nielsen, C. Purser, J. S. Robinson, E. I. Solomon, Z. Sun, D. Sokaras, T. B. van Driel, G. Vanko, T.-C. Weng, D. Zhu and K. J. Gaffney, Nature, 2014, 509, 345–348 CrossRef CAS PubMed.
- M. Cammarata, R. Bertoni, M. Lorenc, H. Cailleau, S. Di Matteo, C. Mauriac, S. F. Matar, H. Lemke, M. Chollet, S. Ravy, C. Laulhé, J.-F. Létard and E. Collet, Phys. Rev. Lett., 2014, 113, 227402 CrossRef PubMed.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grubel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. F. de Groot, K. J. Gaffney, S. Techert, M. Odelius and A. Fohlisch, Nature, 2015, 520, 78–81 CrossRef CAS PubMed.
- C. Reich, C. M. Laperle, X. Li, B. Ahr, F. Benesch and C. G. Rose-Petruck, Opt. Lett., 2007, 32, 427–429 CrossRef CAS PubMed.
- L. Miaja-Avila, G. C. Neil, J. Uhlig, C. L. Cromer, M. L. Dowell, R. Jimenez, A. S. Hoover, K. L. Silverman and J. N. Ullom, Struct. Dyn., 2015, 2, 024301 CrossRef.
- T. Elsaesser and M. Woerner, J. Chem. Phys., 2014, 140, 020901 CrossRef CAS PubMed.
- A. Oguz Er, J. Chen and P. M. Rentzepis, J. Appl. Phys., 2012, 112, 031101 CrossRef.
- J. Chen, H. Zhang, I. V. Tomov, X. Ding and P. M. Rentzepis, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15235–15240 CrossRef CAS PubMed.
- F. Ráksi, K. R. Wilson, Z. Jiang, A. Ikhlef, C. Y. Côté and J.-C. Kieffer, J. Chem. Phys., 1996, 104, 6066–6069 CrossRef.
- J. Vura-Weis, C.-M. Jiang, C. Liu, H. Gao, J. M. Lucas, F. M. F. de Groot, P. Yang, A. P. Alivisatos and S. R. Leone, J. Phys. Chem. Lett., 2013, 4, 3667–3671 CrossRef CAS.
- J. Chen and P. M. Rentzepis, J. Phys. Chem. Lett., 2014, 5, 225–232 CrossRef CAS PubMed.
- F. de Groot, Chem. Rev., 2001, 101, 1779–1808 CrossRef CAS PubMed.
- J. J. Rehr and R. C. Albers, Rev. Mod. Phys., 2000, 72, 621–654 CrossRef CAS.
- M. Rini, R. Tobey, N. Dean, J. Itatani, Y. Tomioka, Y. Tokura, R. W. Schoenlein and A. Cavalleri, Nature, 2007, 449, 72–74 CrossRef CAS PubMed.
- M. Rini, Z. Hao, R. W. Schoenlein, C. Giannetti, F. Parmigiani, S. Fourmaux, J. C. Kieffer, A. Fujimori, M. Onoda, S. Wall and A. Cavalleri, Appl. Phys. Lett., 2008, 92, 181904 CrossRef.
- M. Rini, Y. Zhu, S. Wall, R. I. Tobey, H. Ehrke, T. Garl, J. W. Freeland, Y. Tomioka, Y. Tokura, A. Cavalleri and R. W. Schoenlein, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 155113 CrossRef.
- D. Kraemer, M. L. Cowan, A. Paarmann, N. Huse, E. T. J. Nibbering, T. Elsaesser and R. J. D. Miller, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 437–442 CrossRef CAS PubMed.
- M. Nagasaka, T. Hatsui, T. Horigome, Y. Hamamura and N. Kosugi, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 130–134 CrossRef CAS.
- C. Mueller, M. Harb, J. R. Dwyer and R. J. D. Miller, J. Phys. Chem. Lett., 2013, 4, 2339–2347 CrossRef CAS.
- J. E. Katz, X. Zhang, K. Attenkofer, K. W. Chapman, C. Frandsen, P. Zarzycki, K. M. Rosso, R. W. Falcone, G. A. Waychunas and B. Gilbert, Science, 2012, 337, 1200–1203 CrossRef CAS PubMed.
- S. E. Canton, X. Zhang, L. M. Lawson Daku, A. L. Smeigh, J. Zhang, Y. Liu, C.-J. Wallentin, K. Attenkofer, G. Jennings, C. A. Kurtz, D. Gosztola, K. Wärnmark, A. Hauser and V. Sundström, J. Phys. Chem. C, 2014, 118, 4536–4545 CAS.
- C. J. Milne, T. J. Penfold and M. Chergui, Coord. Chem. Rev., 2014, 277–278, 44–68 CrossRef CAS.
- G. Smolentsev, A. A. Guda, M. Janousch, C. Frieh, G. Jud, F. Zamponi, M. Chavarot-Kerlidou, V. Artero, J. A. van Bokhoven and M. Nachtegaal, Faraday Discuss., 2014, 171, 259–273 RSC.
- S. Yao, K. Mudiyanselage, W. Xu, A. C. Johnston-Peck, J. C. Hanson, T. Wu, D. Stacchiola, J. A. Rodriguez, H. Zhao, K. A. Beyer, K. W. Chapman, P. J. Chupas, A. Martínez-Arias, R. Si, T. B. Bolin, W. Liu and S. D. Senanayake, ACS Catal., 2014, 4, 1650–1661 CrossRef CAS.
- M. W. Mara, K. A. Fransted and L. X. Chen, Coord. Chem. Rev., 2015, 282–283, 2–18 CrossRef CAS.
- H. Cho, M. L. Strader, K. Hong, L. Jamula, E. M. Gullikson, T. K. Kim, F. M. F. de Groot, J. K. McCusker, R. W. Schoenlein and N. Huse, Faraday Discuss., 2012, 157, 463–474 RSC.
- W. Gawelda, A. Cannizzo, V.-T. Pham, F. van Mourik, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2007, 129, 8199–8206 CrossRef CAS PubMed.
- J. E. Monat and J. K. McCusker, J. Am. Chem. Soc., 2000, 122, 4092–4097 CrossRef CAS.
- M. M. N. Wolf, R. Groß, C. Schumann, J. A. Wolny, V. Schunemann, A. Dossing, H. Paulsen, J. J. McGarvey and R. Diller, Phys. Chem. Chem. Phys., 2008, 10, 4264–4273 RSC.
- A. L. Smeigh, M. Creelman, R. A. Mathies and J. K. McCusker, J. Am. Chem. Soc., 2008, 130, 14105–14107 CrossRef CAS PubMed.
- C. Consani, M. Prémont-Schwarz, A. ElNahhas, C. Bressler, F. vanMourik, A. Cannizzo and M. Chergui, Angew. Chem., Int. Ed., 2009, 48, 7184–7187 CrossRef CAS PubMed.
- J. J. Rehr, J. J. Kas, F. D. Vila, M. P. Prange and K. Jorissen, Phys. Chem. Chem. Phys., 2010, 12, 5503–5513 RSC.
- S. Nozawa, T. Sato, M. Chollet, K. Ichiyanagi, A. Tomita, H. Fujii, S.-i. Adachi and S.-y. Koshihara, J. Am. Chem. Soc., 2010, 132, 61–63 CrossRef CAS PubMed.
- M. H. Rittmann-Frank, C. J. Milne, J. Rittmann, M. Reinhard, T. J. Penfold and M. Chergui, Angew. Chem., 2014, 126, 5968–5972 CrossRef.
- G. Capano, T. J. Penfold, N. A. Besley, C. J. Milne, M. Reinhard, H. Rittmann-Frank, P. Glatzel, R. Abela, U. Rothlisberger, M. Chergui and I. Tavernelli, Chem. Phys. Lett., 2013, 580, 179–184 CrossRef CAS.
- F. M. F. de Groot and A. Kotani, Core Level Spectroscopy of Solids; Advances in Condensed Matter Sciences, Taylor & Francis, New York, 2008 Search PubMed.
- R. M. van der Veen, J. J. Kas, C. J. Milne, V. T. Pham, A. El Nahhas, F. A. Lima, D. A. Vithanage, J. J. Rehr, R. Abela and M. Chergui, Phys. Chem. Chem. Phys., 2010, 12, 5551–5561 RSC.
- B. E. Van Kuiken, N. Huse, H. Cho, M. L. Strader, M. S. Lynch, R. W. Schoenlein and M. Khalil, J. Phys. Chem. Lett., 2012, 3, 1695–1700 CrossRef CAS PubMed.
- P. Wernet, G. Gavrila, K. Godehusen, C. Weniger, E. T. J. Nibbering, T. Elsaesser and W. Eberhardt, Appl. Phys. A: Mater. Sci. Process., 2008, 92, 511–516 CrossRef CAS.
- H. Wen, N. Huse, R. W. Schoenlein and A. M. Lindenberg, J. Chem. Phys., 2009, 131, 234505 CrossRef PubMed.
- M. Wulff, F. Schotte, G. Naylor, D. Bourgeois, K. Moffat and G. Mourou, Nucl. Instrum. Methods Phys. Res., Sect. A, 1997, 398, 69–84 CrossRef CAS.
- H. Frauenfelder, S. G. Sligar and P. G. Wolynes, Science, 1991, 254, 1598–1603 CAS.
- T. S. Li, M. L. Quillin, G. N. Phillips and J. S. Olson, Biochemistry, 1994, 33, 1433–1446 CrossRef CAS PubMed.
- M. H. Lim, T. A. Jackson and P. A. Anfinrud, J. Chem. Phys., 1995, 102, 4355–4366 CrossRef CAS.
- K. Nienhaus, J. S. Olson, S. Franzen and G. U. Nienhaus, J. Am. Chem. Soc., 2005, 127, 40–41 CrossRef CAS PubMed.
- J. B. Johnson, D. C. Lamb, H. Frauenfelder, J. D. Muller, B. McMahon, G. U. Nienhaus and R. D. Young, Biophys. J., 1996, 71, 1563–1573 CrossRef CAS PubMed.
- H. Ishikawa, K. Kwak, J. K. Chung, S. Kim and M. D. Fayer, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8619–8624 CrossRef CAS PubMed.
- J. S. Baskin and A. H. Zewail, ChemPhysChem, 2005, 6, 2261–2276 CrossRef CAS PubMed.
- J. S. Baskin and A. H. Zewail, ChemPhysChem, 2006, 7, 1562–1574 CrossRef CAS PubMed.
- P. Reckenthaeler, M. Centurion, W. Fuß, S. A. Trushin, F. Krausz and E. E. Fill, Phys. Rev. Lett., 2009, 102, 213001 CrossRef PubMed.
- D. J. Wilbur, R. S. Norton, A. O. Clouse, R. Addleman and A. Allerhand, J. Am. Chem. Soc., 1976, 98, 8250–8254 CrossRef CAS PubMed.
- D. Wang, U. Kreutzer, Y. Chung and T. Jue, Biophys. J., 1997, 73, 2764–2770 CrossRef CAS PubMed.
- J. G. Kim, K. H. Kim, K. Y. Oang, T. W. Kim, H. Ki, J. Jo, J. Kim, T. Sato, S. Nozawa, S.-i. Adachi and H. Ihee, J. Phys. B: At., Mol. Opt. Phys., 2015, 48, 244005 CrossRef.
- M. Chergui, C. J. Milne, R. M. Van der Veen, V. T. Pham, F. A. Lima, H. Rittmann-Frank, M. Reinhard, F. van Mourik, S. Karlsson and T. J. Penfold, Chimia, 2011, 65, 303–307 CrossRef.
- F. A. Lima, C. J. Milne, D. C. V. Amarasinghe, M. H. Rittmann-Frank, R. M. van der Veen, M. Reinhard, V. T. Pham, S. Karlsson, S. L. Johnson, D. Grolimund, C. Borca, T. Huthwelker, M. Janousch, F. van Mourik, R. Abela and M. Chergui, Rev. Sci. Instrum., 2011, 82, 063111 CrossRef PubMed.
- K. H. Kim, J. Kim, K. Y. Oang, J. H. Lee, D. Grolimund, C. J. Milne, T. J. Penfold, S. L. Johnson, A. Galler, T. W. Kim, J. G. Kim, D. Suh, J. Moon, J. Kim, K. Hong, L. Guerin, T. K. Kim, M. Wulff, C. Bressler and H. Ihee, Phys. Chem. Chem. Phys., 2015, 17, 23298–23302 RSC.
- P. Z. El-Khoury, W. M. Kwok, X. Guan, C. Ma, D. L. Phillips and A. N. Tarnovsky, ChemPhysChem, 2009, 10, 1895–1900 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |