
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanoelectrodes reveal the electrochemistry of single nickelhydroxide nanoparticles†
Jan
Clausmeyer
*,
Justus
Masa
,
Edgar
Ventosa
,
Dennis
Öhl
and
Wolfgang
Schuhmann
*
Analytical Chemistry – Center for Electrochemical Sciences (CES), Ruhr-Universität Bochum, Universitätsstrasse 150, D-44780, Bochum, Germany. E-mail: jan.clausmeyer@rub.de; wolfgang.schuhmann@rub.de
First published on 22nd December 2015
Abstract
Individual Ni(OH)2 nanoparticles deposited on carbon nanoelectrodes are investigated in non-ensemble measurements with respect to their energy storage properties and electrocatalysis for the oxygen evolution reaction (OER). Charging by oxidation of Ni(OH)2 is limited by the diffusion of protons into the particle bulk and the OER activity is independent of the particle size.
Electrochemical techniques have empowered us to analyse single nanoparticles without averaging over large statistical ensembles. Stochastic collisions of individual particles with microelectrodes as demonstrated by Bard's1 and Compton's2 groups have provided information concerning the particle size3 as well as their physical4 and electrocatalytic properties.5 However, the particle collision method is an intrinsically transient method concerning the number of analysed particles at a given time, i.e. single particles cannot be studied over a long time in steady-state measurements. Alternatively, scanning electrochemical microscopy (SECM) or scanning electrochemical droplets allow for the analysis of the particle size and detection of the products of catalytic conversion.6,7 The deposition of electroactive materials on nanoelectrodes has allowed the study of the activity of individual nanoparticles towards reactions that are relevant for electrochemical energy conversion such as the hydrogen evolution reaction (HER)8 or the oxygen reduction reaction (ORR).9,10 Here, we evaluate the oxygen evolution reaction (OER) as well as charge storage by proton insertion at single Ni(OH)2 particle modified electrodes. In the quest to develop competitive technologies for the storage of electrical energy, Ni(OH)2 has proved to be both a suitable catalyst for electrochemical water oxidation11 as well as a material in aqueous batteries12 and supercapacitors.13 In particular, nanostructured materials show a superior performance compared to their bulk counterparts.14–17
However, while understanding the nature of the enhanced electrocatalytic activity of nanoparticles, it is difficult to distinguish between the specific activity and geometric effects such as increased surface area or increased mass transport rates. Moreover, there is an ongoing debate concerning the mechanism of energy storage in nanomaterials for batteries and supercapacitors.13,18 As the dimensions of nanostructured materials decrease, electrochemical processes proximal to the surface and thus capacitive and pseudo-capacitive contributions become more important due to the high surface area.
We use nanoelectrodes fabricated using a facile and effective method19,20 and decorated with a single nanoparticle to demonstrate a range of dynamic experiments to assess size, composition and catalytic activity of nanoparticles at high mass transport rates. For the OER, we deduce novel but counter-intuitive observations about the relationship between size and activity of catalytic particles. Likewise unforeseen, we find that the charge storage in single medium-scale Ni(OH)2 nanoparticles is limited by diffusion and thus does not show pseudo-capacitive behaviour.
Carbon nanoelectrodes are fabricated by the pyrolytic decomposition of butane gas inside pulled quartz glass nanopipettes. This time-effective (ca. 1 min per electrode) method produces disk-shaped carbon electrodes with dimensions of a few nanometres surrounded by a thin glass sheath19 (see the ESI† for the fabrication and characterization of nanoelectrodes). Then, Ni(OH)2 is cathodically deposited on the carbon nanoelectrodes from NiCl2 solutions, yielding single Ni(OH)2 nanoparticles attached to the nanoelectrodes. Due to the small dimensions of the carbon nanoelectrodes, which are smaller or of a comparable size to the deposited particles, the Ni(OH)2 nanoparticles exhibit an approximately spherical geometry as confirmed by SEM imaging (Fig. 1). The radii of the deposited particles range from 20 nm to 500 nm as determined electrochemically and confirmed by SEM (see ESI†).
 | ||
| Fig. 1 Nanoparticle-modified electrodes allow electrochemical characterization of individual Ni(OH)2 nanoparticles. SEM images show two individual nanoparticles deposited on carbon nanoelectrodes and a magnified image of the smaller particle. | ||
The Ni(OH)2/NiOOH redox couple has been widely used in highly dispersed materials for energy storage in aqueous batteries and pseudo-capacitors. The general paradigm is that the surface or near-surface reactions, i.e. pseudo-capacitive contributions to the charge storage, become more important with decreasing material dimensions. In contrast to previous studies,13,15 by investigating the oxidation of single Ni(OH)2 particles we observe that the process is entirely limited by diffusion.
Voltammetric analysis of the Ni(OH)2 → NiOOH + H+ + e− transition reveals a linear dependence of the corresponding anodic peak currents with respect to the scan rate (Fig. 2). The trend line of the log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ipavs. logv plot shows a slope close to the theoretical value of 0.5 expected for a diffusion-controlled process, as opposed to a value of 1 for surface-confined species (i.e. a pseudo-capacitive process). It is very important to note that due to the small dimensions of the single particle electrode, the diffusion of the involved species in the electrolyte occurs extremely fast. Hence, it is the diffusion of protons inside the bulk of the Ni(OH)2 nanoparticle that limits the overall oxidation reaction. Due to the analogy to a macroscopic electrode with diffusion-limitation in the electrolyte, the Randles–Sevcik equation can be used to describe the voltammetric peak currents at the nanoelectrode with diffusion-limitation in the particle bulk.12
ipavs. logv plot shows a slope close to the theoretical value of 0.5 expected for a diffusion-controlled process, as opposed to a value of 1 for surface-confined species (i.e. a pseudo-capacitive process). It is very important to note that due to the small dimensions of the single particle electrode, the diffusion of the involved species in the electrolyte occurs extremely fast. Hence, it is the diffusion of protons inside the bulk of the Ni(OH)2 nanoparticle that limits the overall oxidation reaction. Due to the analogy to a macroscopic electrode with diffusion-limitation in the electrolyte, the Randles–Sevcik equation can be used to describe the voltammetric peak currents at the nanoelectrode with diffusion-limitation in the particle bulk.12
 | ||
| Fig. 2 The oxidation of Ni(OH)2 nanoparticles is limited by proton diffusion in the particle bulk. Cyclic voltammograms at a single Ni(OH)2 nanoparticle (r = 75 nm) electrode at varying scan rates from 0.01 to 0.5 V s−1 in 0.1 M KOH (a) and analysis of anodic peak currents as a function of the scan rate (b). | ||
Proton diffusion coefficients for single Ni(OH)2 nanoparticles calculated from the Randles–Sevcik equation using the electrochemically estimated particle surface area and assuming stoichiometric proton release (resulting in a bulk proton concentration of 44.7 M in Ni(OH)2) range around 2 × 10−11 cm2 s−1 for particles smaller than r = 75 nm. Slower apparent diffusion is observed for larger particles. This might be due to the contribution of poor electronic conductivity of Ni(OH)2, which becomes more important for long electron-transfer paths inside larger particles. The values for the proton diffusion coefficient in Ni(OH)2 observed by other authors range from 10−11 to 10−7,12,21 which might be due to the difficulty in investigating thick porous film electrodes.
Upon sweeping the potential anodically to investigate the OER, single Ni(OH)2 particle electrodes exhibit defined redox signals for the oxidation of Ni(OH)2 to NiOOH, which are well separated from the onset of the anodic current for the OER (Fig. 3a). The voltammetric peak for the oxidation of Ni(OH)2 (inset) is suitable for the precise electrochemical determination of the catalyst amount.3,16 From the amount of deposited Ni(OH)2 the particle size is calculated, assuming a spherical geometry (see ESI†). To assess the catalyst activity, the turnover frequencies (TOF) for the OER at high overpotentials are analysed as a function of particle size (Fig. 3b). The TOF indicates the rate of evolved oxygen molecules per catalyst active centre (see the ESI†). Generally, single nanoparticle electrodes can be driven to high turnover frequencies at high potentials of 1.88 V vs. RHE (η = 0.65). Since no binders or conductive additives as typically used to formulate porous electrodes are necessary, the degradation of those components at high anodic potentials is excluded. Also, in contrast to thick porous films and powder electrodes, the particles are directly contacted by the nanoelectrodes to reduce the influence of electric resistance in the film. For single particles a relatively high variability of the TOF is observed, which may reflect information concerning the heterogeneity of individual particles that is normally lost when studying large ensembles of particles (Fig. 3b).
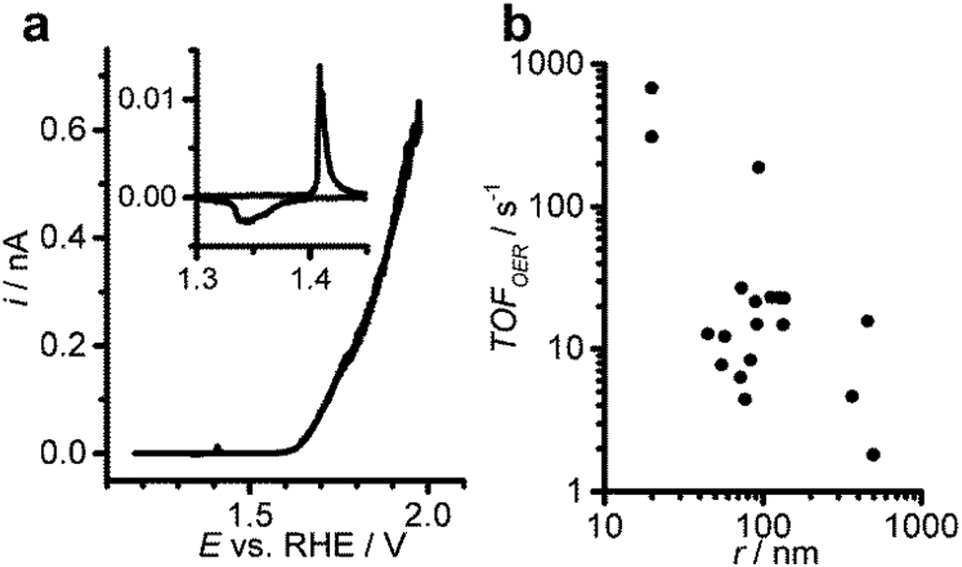 | ||
| Fig. 3 The OER is performed at single Ni(OH)2 nanoparticles to investigate the relationship between activity and particle size. Cyclic voltammogram (10 mV s−1) at a single particle electrode (r = 83 nm) in 0.1 M KOH (a) and turnover frequencies for the OER at 1.88 V vs. RHE for differently sized particles (b). | ||
Nevertheless, the analysis of the TOF with respect to the particle size shows a general decrease of the TOF with increasing particle radius. Since the TOF is considered to be an intrinsic material property, this observation may be used to support the claim that smaller Ni(OH)2 particles are intrinsically more active with respect to the OER than larger particles. However, we attribute this finding to geometric effects, namely a higher surface-to-bulk ratio of the smaller particles that allows more precise quantification of the active centres. Additionally, in the high overpotential region the poor electronic conductivity of the material may hamper the reaction rate at larger particles which have a longer electron path. We can exclude the increased specific activity of small particles by comparing the electrode kinetics of small and large particles. Tafel analysis in the low overpotential region of the OER at single Ni(OH)2 particles reveals that the electrode kinetics are largely invariant with the particle size (Fig. 4). The Tafel plots for small and large particles all exhibit similarly low slopes in the low overpotential region between 1.57 and 1.66 vs. RHE (η = 0.34 and η = 0.43), followed by larger values at higher potentials (Fig. 4a). Comparing the values for the Tafel slope over the whole population of individually analysed particles (Fig. 4b) yields a slope of 0.05 ± 0.01 V dec−1 as expected for highly active OER catalysts.14,16,17 This observation contradicts the assumption that small particles are intrinsically more active than large particles. The apparent increase in activity as concluded from the turnover frequencies is rather a result of geometric effects. For small particles the number of truly active sites can be estimated with higher precision because of the increased fraction of active centres located in direct contact between the electrode and the electrolyte solution. These findings highlight that the TOF is generally underestimated due to the difficulties in determining the number of active sites, especially for large particles or thick porous catalyst films on macroscopic electrodes. Also, in thick films or powder electrodes, electronic conductivity may have a detrimental influence on the reaction rate, which might be already observed for larger nanoparticles.
 | ||
| Fig. 4 Electron transfer kinetics for the OER at single Ni(OH)2 nanoparticles show no activity dependence on particle size. Tafel plots for two selected particles with radii of 20 nm (solid line) and 365 nm (dotted line) (a) and analysis of Tafel slopes between 1.57 and 1.66 vs. RHE as a function of particle size (b). | ||
In conclusion, studying the electrochemistry of single nanoparticles deposited on carbon nanoelectrodes has several advantages: (1) extremely fast mass transport rates are obtained which by far exceed those accessible in rotating ring–disk electrode experiments.9 (2) The particles are directly contacted with low resistance and in addition the degradation of non-active components while applying high potentials necessary for the OER can be excluded. (3) The characterization of individual particles provides precise knowledge of the particle size and thus allows for deducing size–activity relations. Using these advantages, we find that the oxidation of Ni(OH)2, a typical reaction representative of charge storage in aqueous batteries and supercapacitors, is entirely limited by proton diffusion inside the particle bulk even for relatively small particles. The capacitive behaviour due to charging of the electrochemical double layer or pseudo-capacitive contributions due to faradaic reactions at or near the surface are insignificant.
For the electrocatalytic evolution of oxygen we investigate the size-dependent catalytic turnover rate of individual Ni(OH)2 particles. In contrast to the notion that a small particle size results in high catalytic activity, we observe constant reaction kinetics for particles with radii between 20 nm and 500 nm. The analysis of single active nanoparticles using nanoelectrodes can be used to develop a powerful tool to elucidate more size-specific phenomena in catalysis at the nanoscale and thus will help in understanding size–structure–activity relationships.
The contribution of Sandra Schmidt for assistance with SEM/EDX measurements is acknowledged. This work was financially supported by the DFG (Cluster of Excellence RESOLV; EXC 1069) and by the BMBF (NeMeZu; FKZ 03SF0497B).
Notes and references
- X. Xiao and A. J. Bard, J. Am. Chem. Soc., 2007, 129, 9610 CrossRef CAS PubMed; S. J. Kwon, F.-R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 2010, 132, 13165 CrossRef PubMed.
- W. Cheng and R. G. Compton, Trends Anal. Chem., 2014, 58, 79 CrossRef CAS.
- Y.-G. Zhou, N. V. Rees and R. G. Compton, Angew. Chem., Int. Ed., 2011, 50, 4219 CrossRef CAS PubMed.
- K. Tschulik and R. G. Compton, Phys. Chem. Chem. Phys., 2014, 16, 13909 RSC.
- H. Zhou, J. H. Park, F.-R. F. Fan and A. J. Bard, J. Am. Chem. Soc., 2012, 134, 13212 CrossRef CAS PubMed; P. Sun, F. Li, C. Yang, T. Sun, I. Kady, B. Hunt and J. Zhuang, J. Phys. Chem. C, 2013, 117, 6120 Search PubMed.
- M. A. O'Connell and A. J. Wain, Anal. Chem., 2014, 86, 12100 CrossRef PubMed.
- Y.-R. Kim, S. C. S. Lai, K. McKelvey, G. Zhang, D. Perry, T. S. Miller and P. R. Unwin, J. Phys. Chem. C, 2015, 119, 17389 Search PubMed; S. E. F. Kleijn, S. C. S. Lai, T. S. Miller, A. I. Yanson, M. T. M. Koper and P. R. Unwin, J. Am. Chem. Soc., 2012, 134, 18558 CrossRef CAS PubMed.
- X. Shan, I. Díez-Pérez, L. Wang, P. Wiktor, Y. Gu, L. Zhang, W. Wang, J. Lu, S. Wang, Q. Gong, J. Li and N. Tao, Nat. Nanotechnol., 2012, 7, 668 CrossRef CAS PubMed; Q. Chen, L. Luo, H. Faraji, S. W. Feldberg and H. S. White, J. Phys. Chem. Lett., 2014, 5, 3539 CrossRef PubMed; Y. Yu, Y. Gao, K. Hu, P.-Y. Blanchard, J.-M. Noël, T. Nareshkumar, K. L. Phani, G. Friedman, Y. Gogotsi and M. V. Mirkin, ChemElectroChem, 2015, 2, 58 CrossRef.
- S. Chen and A. Kucernak, J. Phys. Chem. B, 2004, 108, 3262 CrossRef CAS.
- Y. Li, J. T. Cox and B. Zhang, J. Am. Chem. Soc., 2010, 132, 3047 CrossRef CAS PubMed; Y. Li, Q. Wu, S. Jiao, C. Xu and L. Wang, Anal. Chem., 2013, 85, 4135 CrossRef PubMed.
- R. D. L. Smith, M. S. Prévot, R. D. Fagan, Z. Zhang, P. A. Sedach, M. K. J. Siu, S. Trudel and C. P. Berlinguette, Science, 2013, 340, 60 CrossRef CAS PubMed; C. C. L. McCrory, S. Jung, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2013, 135, 16977 CrossRef PubMed.
- M. A. Kiani, M. F. Mousavi and S. Ghasemi, J. Power Sources, 2010, 195, 5794 CrossRef CAS.
- T. Brousse, D. Belanger and J. W. Long, J. Electrochem. Soc., 2015, 162, A5185–A5189 CrossRef CAS.
- M. Gao, W. Sheng, Z. Zhuang, Q. Fang, S. Gu, J. Jiang and Y. Yan, J. Am. Chem. Soc., 2014, 136, 7077 CrossRef CAS PubMed.
- H. B. Li, M. H. Yu, F. X. Wang, P. Liu, Y. Liang, J. Xiao, C. X. Wang, Y. X. Tong and G. W. Yang, Nat. Commun., 2013, 4, 1894 CrossRef CAS PubMed.
- D. Cibrev, M. Jankulovska, T. Lana-Villarreal and R. Gómez, Int. J. Hydrogen Energy, 2013, 38, 2746 CrossRef CAS.
- L. Trotochaud, J. K. Ranney, K. N. Williams and S. W. Boettcher, J. Am. Chem. Soc., 2012, 134, 17253 CrossRef CAS PubMed.
- H.-S. Kim, T. Itoh, M. Nishizawa, M. Mohamedi, M. Umeda and I. Uchida, Int. J. Hydrogen Energy, 2002, 27, 295 CrossRef.
- P. Actis, S. Tokar, J. Clausmeyer, B. Babakinejad, S. Mikhaleva, R. Cornut, Y. Takahashi, A. López Córdoba, P. Novak, A. I. Shevchuck, J. A. Dougan, S. G. Kazarian, P. V. Gorelkin, A. S. Erofeev, I. V. Yaminsky, P. R. Unwin, W. Schuhmann, D. Klenerman, D. A. Rusakov, E. V. Sviderskaya and Y. E. Korchev, ACS Nano, 2014, 8, 875 CrossRef CAS PubMed.
- J. Clausmeyer, P. Actis, A. López Córdoba, Y. Korchev and W. Schuhmann, Electrochem. Commun., 2014, 40, 28 CrossRef CAS; Y. Takahashi, A. I. Shevchuk, P. Novak, Y. Zhang, N. Ebejer, J. V. Macpherson, P. R. Unwin, A. J. Pollard, D. Roy, C. A. Clifford, H. Shiku, T. Matsue, D. Klenerman and Y. E. Korchev, Angew. Chem., Int. Ed., 2011, 50, 9638 CrossRef PubMed.
- J. W. Weidner and P. Timmermann, J. Electrochem. Soc., 1994, 141, 346 CrossRef CAS; K. Watanabe, T. Kikuoka and N. Kumagai, J. Appl. Electrochem., 1995, 25, 219 CrossRef; G. Briggs and P. R. Snodin, Electrochim. Acta, 1982, 27, 565 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, nanoelectrode characterization, electrochemical estimation of particle sizes, additional SEM images, EDX analysis, and OER polarization curves. See DOI: 10.1039/c5cc08796a |
| This journal is © The Royal Society of Chemistry 2016 |