
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegioselective modification of unprotected glycosides
Manuel
Jäger
and
Adriaan J.
Minnaard
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 7, 9747 AG Groningen, The Netherlands. E-mail: a.j.minnaard@rug.nl
First published on 9th November 2015
Abstract
Selective modification of unprotected carbohydrates is difficult due to the similar reactivity of the hydroxyl groups. In carbohydrate synthesis, therefore, even straightforward transformations often require multiple synthetic steps. The development of selective methods for carbohydrate modification is consequently highly desired. This review describes the methods for the regio- and chemoselective carbohydrate modification, with a focus on novel approaches that mainly apply transition metal catalysis and organocatalysis, and discusses the challenges and opportunities in this field.
 Manuel Jäger | Manuel grew up in Metelen (Germany) and studied Chemistry at the Saxion Hogeschool in Enschede. He received his bachelor’s degree with Prof. von Kiedrowski at the University of Bochum, where he worked on the synthesis and the oligomerisation of a 5′-aminodidesoxynucleotide-3′-phosphate. In 2007 he started to work as a research chemist at Syncom B.V., Groningen. Manuel joined the Minnaard group in 2010 as a PhD-student, where he studied catalytic selective oxidation of carbohydrates. In 2015 he joined the Hirsch group as a postdoctoral researcher and is currently working on the design and synthesis of inhibitors for glucansucrases. |
 Adriaan J. Minnaard | Adriaan J. Minnaard completed his PhD in 1997 in the group of Prof. De Groot at the Agricultural University Wageningen, The Netherlands. After two years as a scientist at DSM in Geleen, he became an assistant professor at the University of Groningen, The Netherlands. He has been there ever since, with a break in the group of Prof. Waldmann at the Max Planck Institute in Dortmund. He is a full professor and head of the department of Chemical Biology. |
Introduction
Carbohydrate chemistry is, because of its importance for biology, a vivid field of research. The chemical synthesis of carbohydrates is, next to glycosidic bond formation, dominated by hydroxyl group protection/deprotection strategies. These strategies are essential for the synthesis of oligosaccharides and carbohydrate-conjugates and as part of this steering the regio- and stereochemical outcome in glycosylation.1 Although often masterpieces of control on chemical reactivity, carbohydrate synthesis frequently takes a considerable number of steps, also for the straightforward manipulation of commercially available monosaccharides. The requirement for protection hampers in addition the modification of naturally occurring oligosaccharides and conjugation reactions, for example to proteins, in an aqueous environment. In order to trim these long synthetic routes, and to allow the application of available (oligo)saccharides, strategies for the selective modification of (partly) unprotected carbohydrates have been developed over recent years. This review gives an overview of these methods and highlights the trends and challenges. The selective protection and non-protective modifications, in particular oxidation, of carbohydrate substrates will be discussed and the main focus lies on, but is not limited to, the protection, modification and oxidation of secondary hydroxyl groups, since their regioselective conversion is particularly challenging. Two reviews on the modification of unprotected carbohydrates have appeared, one from Taylor and Lee2 focusing on catalyst controlled reactions and a short review from Saloranta et al.3 dealing with the potential of unprotected carbohydrates as starting materials in synthesis.Although glycosylation of unprotected carbohydrates, either chemical or enzymatic, is an important topic in carbohydrate chemistry, it is outside the scope of this review. A review4 and recent examples can be found elsewhere.5–13 Furthermore, the modification of aminoglycoside antibiotics is not covered as reviews14–16 including examples of impressive selectivities17,18 can be found elsewhere.
Selective protection of glycosides
Here, the concepts used in selective protection, e.g. the inherent reactivity difference of the hydroxyl groups, and the application of metal-catalysis, organocatalysis and enzyme-mediated selective protection of carbohydrates and glycosides are discussed.Inherent reactivity differences in glycoside hydroxyl groups
Primary hydroxyl groups are sterically less hindered compared to their secondary and tertiary counterparts and this can be used to selectively protect the primary hydroxyl group in glycosides. Bulky protecting groups such as tert-butyldimethylsilyl, tert-butyldiphenylsilyl,19 triphenylmethyl (trityl)20 and pivaloyl21 react with good to high selectivity with primary hydroxyl groups in the presence of secondary ones.Steric hindrance can also be utilized to some extent to discriminate between secondary hydroxyl groups. Jiang et al. have described a selective double acylation of several glycosides using pivaloyl chloride.22 The isolated yields were generally high and selective protection of the primary hydroxyl group in combination with esterification at either the 2- or 3-position of several glycosides was reported (Scheme 1). The observed higher reactivity of equatorial hydroxyl groups adjacent to axial hydroxyl or alkoxy groups was rationalized with the argument that the bulky pivaloyl chloride approaches the molecule for steric reasons rather sideways than from the bottom or top. Equatorial hydroxyl groups adjacent to axial substituents would therefore be more accessible than equatorial ones flanked by other equatorial hydroxyl groups. Interestingly, when no axial hydroxyl group was present, as in 8, selective protection of C6 and C3 was observed to give 9.
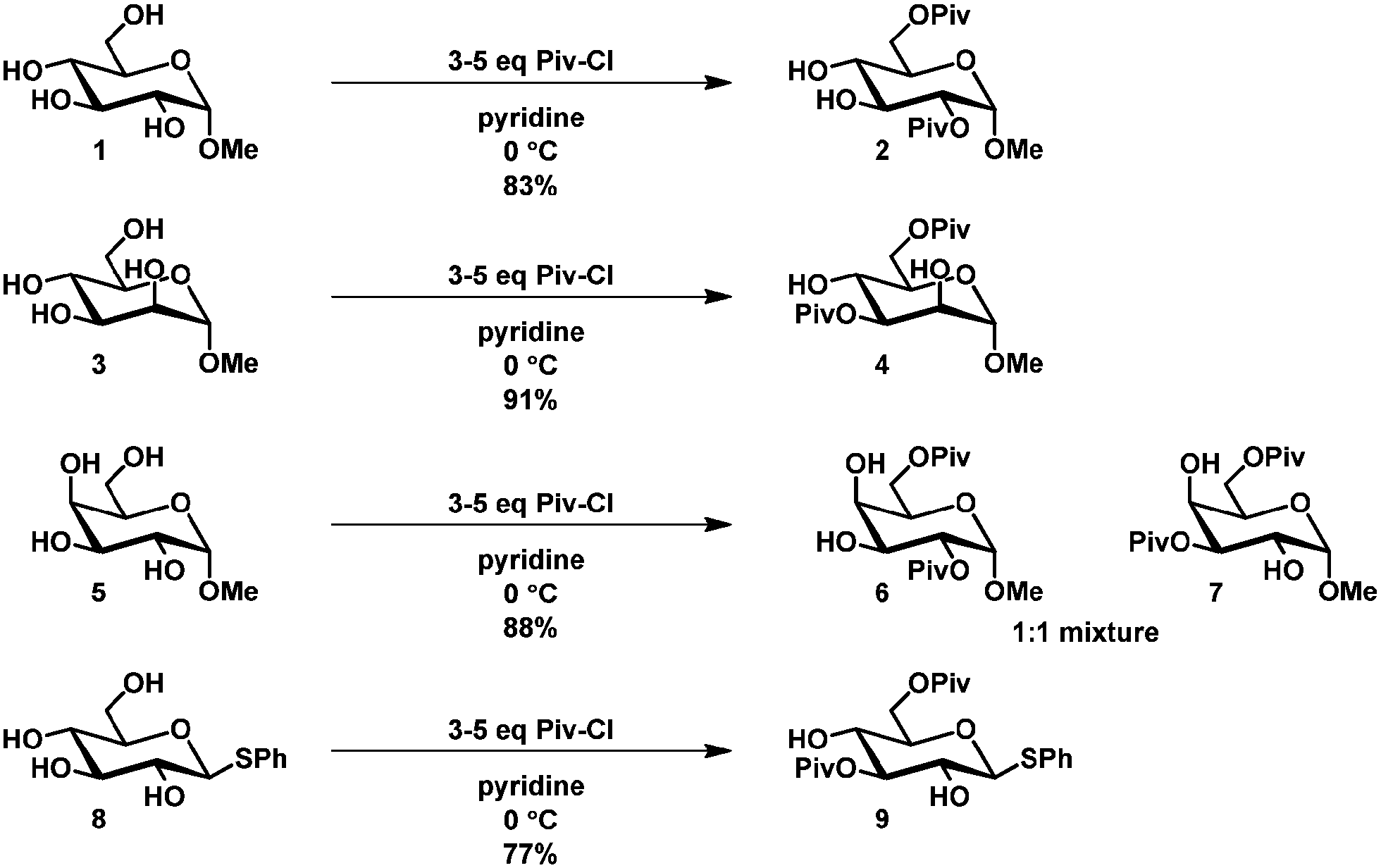 | ||
| Scheme 1 Selective double protection of glucopyranosides. | ||
Hydrogen bond networks as a rationale for selective acylation have been proposed by Kurahashi et al.23 Treatment of octyl α- and β-glucopyranosides with a sub-stoichiometric amount of acetic anhydride and catalytic DMAP in chloroform gave the 3-, 4- and 6-acetyl protected glucosides 11–14 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1 ratio (Scheme 2). It was rationalized that the higher reactivity of the C3- and C4-hydroxyl group is due to the ability to spread the developing positive charge over a larger hydrogen bond network (15) in contrast to the C2-hydroxyl group.
:2:1 ratio (Scheme 2). It was rationalized that the higher reactivity of the C3- and C4-hydroxyl group is due to the ability to spread the developing positive charge over a larger hydrogen bond network (15) in contrast to the C2-hydroxyl group.
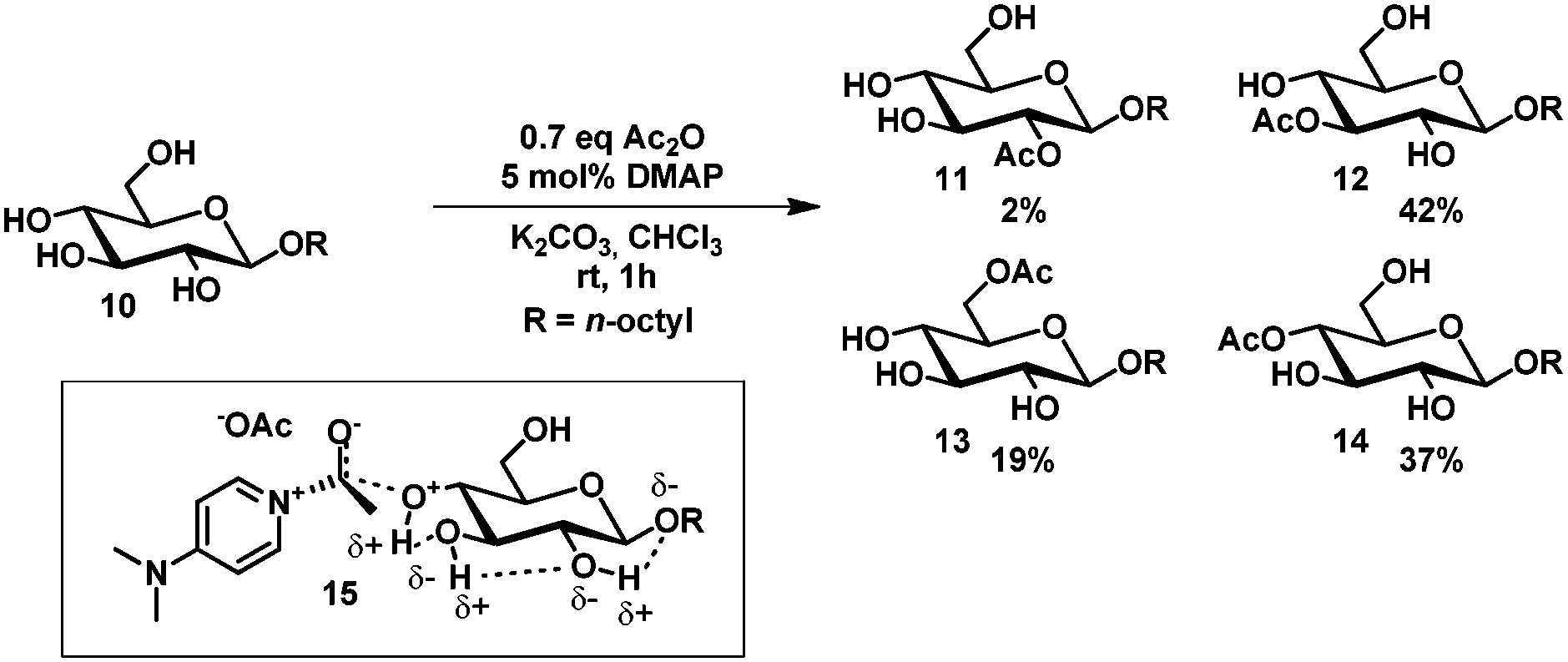 | ||
| Scheme 2 The effect of intramolecular hydrogen bonds on the acetylation of glucosides. | ||
Hydrogen bond networks play probably only a significant role in nonpolar solvents (and solvents that are no hydrogen bond acceptors or donors); Moitessier et al. showed a decrease in selectivity by switching from dichloromethane to tetrahydrofuran.24 In solvents typically used in carbohydrate synthesis, like DMSO, DMF, MeOH and water, intramolecular hydrogen bonds are probably of minor importance due to hydrogen bonding to the solvent.
More selective acetylations of glycosides in water have been described by Lu et al.25 The primary hydroxyl group in a range of unprotected glycosides, triols and diols could be acetylated selectively using acetyl imidazole, with tetramethyl ammonium hydroxide as the catalyst, and isolated in moderate to good yields. Recently, Ren et al. described a method for the selective acetylation of glycosides using acetate catalysis.26 A range of glycosides could be acetylated at either C6 in combination with C3 or at C3 in the case of C6 protected glycosides and isolated in good yields. In order to understand the origin of the selectivity, the authors conducted intensive theoretical studies and propose that a bifurcated hydrogen bond together with the geometry in the transition state are responsible for the selectivity. In other words, stereo electronic effects and the inherent structure of the acetate/diol complex determine the selectivity (Scheme 3). In general, care should be taken in the analysis of the formed products as intramolecular acyl-shifts can be fast in particular with acetyl groups. In this case the authors could rule out acyl migration since it would lead to a different product.
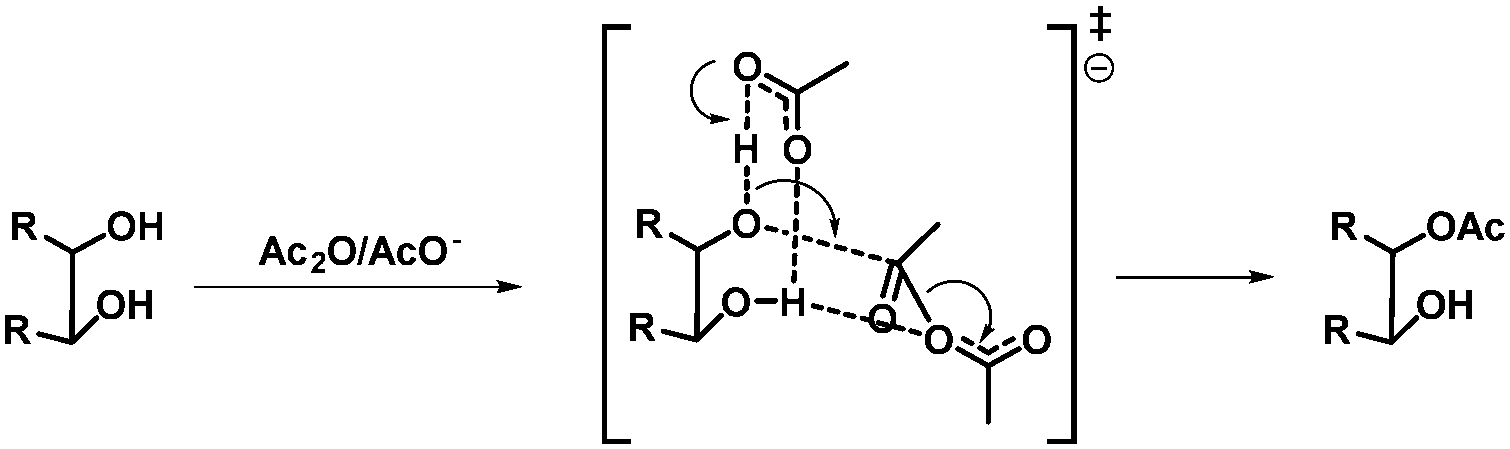 | ||
| Scheme 3 Proposed transition state, representing the bifurcated H-bond, for the acetate catalysed acetylation of diols. | ||
Tin mediated protection of glycosides
Regioselective protection mediated by organotin compounds like tributyltin oxide and dibutyltin oxide is well-known.27,28 Treatment of glycosides with e.g. dibutyltin oxide gives stannylene acetal 17 and subsequent treatment with an electrophile gives the modified glycoside 18 (Scheme 4).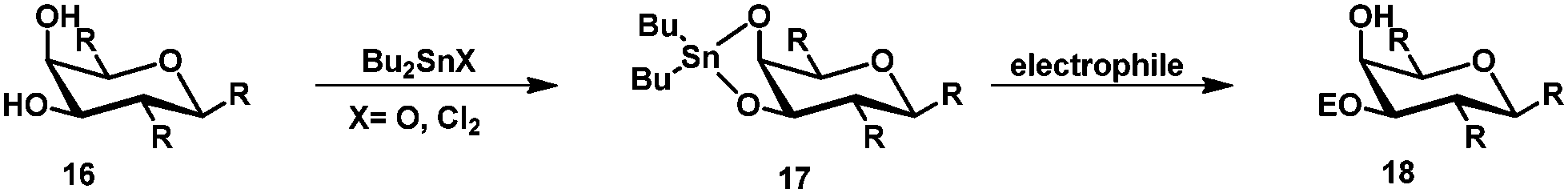 | ||
| Scheme 4 General tin mediated protection of diols. | ||
Using this strategy, regioselective acylation,29,30 alkylation,31,32 silylation,33 sulfonylation30,34,35 and glycosylation36,37 is possible.
In general, protection via dibutyltin acetals gives (1) for cis-diols a selective protection of the equatorial hydroxyl group29,38,39 (2) for trans-diols, with adjacent axial or equatorial substituents, a less pronounced selectivity,40,41 (3) for trans-diols, with one axial and one equatorial adjacent substituent, selective protection of the equatorial hydroxyl group next to the axial substituent42,43 and (4) a hydroxyl group next to a deoxy center is preferably protected (Table 1).44
| cis | trans | trans | cis/transd | |
|---|---|---|---|---|
| a cis-Diol. b trans-Diol adjacent to both equatorial and axial substituents. c trans-Diol adjacent to one axial and one equatorial substituent. d cis-or trans-diol adjacent to a deoxy-center, eq.: equatorial, ax.: axial, P: protecting group. | ||||
| Diol |
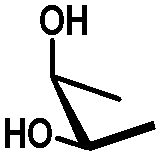
|
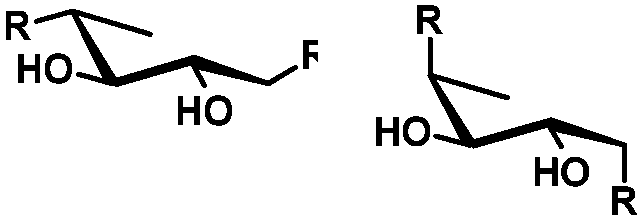
|
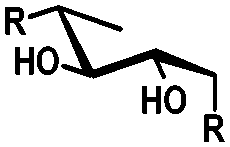
|
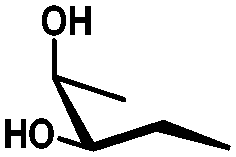
|
| Selectivity |
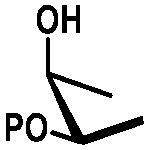
|
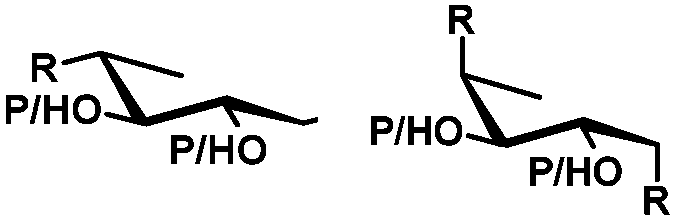
|
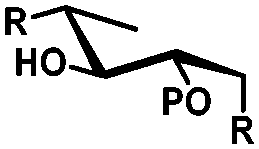
|
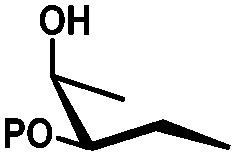
|
| eq. | Less selective | Next to adjacent ax. | Next to deoxy | |
Recent studies by the group of Ramström45 suggest that the selectivities in the organotin-mediated protection of carbohydrates are not related (as assumed before) to complex stannylene structures such as dimers and oligomers.27 Ramström and coworkers studied the steric and stereo-electronic effects controlling the geometry of the five-membered ring intermediates (Scheme 5) in acyl migration, neighboring group participation, and orthoester transesterification reactions in pyranose rings. It turned out that these effects result in the same product, showing the same selectivity (e.g. acyl migration towards the axial hydroxyl group). Furthermore, the selectivity appeared to be contrary to the above mentioned selectivity in tin-mediated protection. As example, acyl migration as shown in Scheme 5via the intermediate five-membered ring leads to the axial acylated glycoside, which is contrary to the equatorial acetylated glucoside obtained from the tin-mediated protection. The bond in the intermediate five-membered ring, which is broken in the tin-mediated protection and acyl migration is the same, is the equatorial bond. This leads to the experimentally observed opposite selectivities and indicates that the bond breakage is dependent on the stereo-electronic effects of the carbohydrate.45
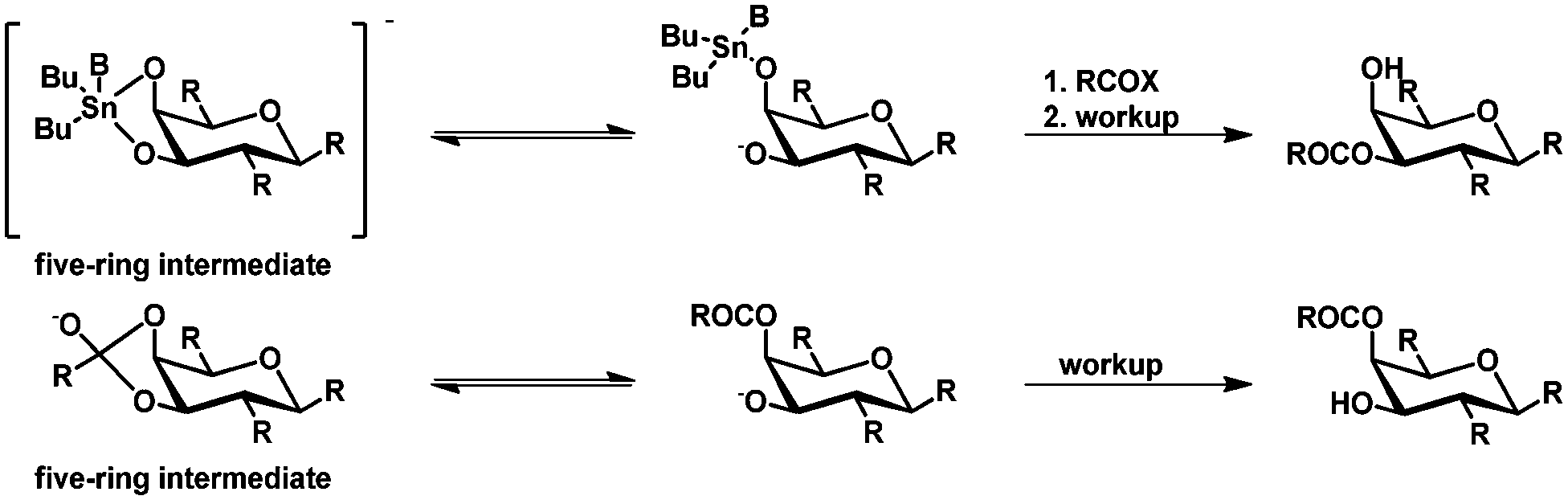 | ||
| Scheme 5 Comparison of tin-mediated selective protection and acyl migration. | ||
It is worthwhile mentioning that the general trend observed for the selective protection mediated by organotin compounds is in accordance with the idea that the electrophile approaches the molecule from the side rather than from the top or bottom. This indicates a dependency on sterics rather than on electronics for the protection,22 next to the formation of the stannylene acetals of triols or polyols which is probably dependent on the stereo-electronic effects in the carbohydrate.
Whereas the classical selective protection uses stoichiometric amounts of the organotin reagent to generate the stannylene acetal, recently several catalytic systems for the selective sulfonylation34,35 and benzylation38 have been described. The same selectivity as in the stoichiometric methods was observed, but the amount of toxic organotin reagents was reduced to 2–10 mol%.34,35,38
Metal- and metalloid-mediated protection of glycosides other than tin
Other systems for the regioselective protection of diols have been reported based on organoboron,46–48 organosilicon,49,50 copper(II),51–55 silver(I),56 nickel(II)57 and molybdenum(V) and (II) (Table 2).58,59 These systems show often a similar selectivity as observed in tin mediated protections. This could indicate that the selectivity also depends on stereo-electronic effects in the carbohydrate45 or as mentioned before solely depends on steric interactions between the carbohydrate and electrophile.22|
|
||||
|---|---|---|---|---|
| Method | Electrophile | Yield | Ref. | |
| A | (1) 10 mol% Bu2SnO, toluene, 100 °C, 1 h (2) K2CO3, TBAB, MeCN: DMF 10:1, 80 °C 3 h, P = TBDMS |
BnBr | 90 | 38 |
| B | 5–10 mol% 19, iPr2NEt, MeCN, 20–40 °C, P = TBS | TsCl | Ts: 99% | Ts47 |
| BnBr | Bn: 74% | Bn48 | ||
| C | 10 mol% 20, iPr2NEt, MeCN, −5 °C, 16 h, P = TBDMS | BzCl | 80% | 53 |
| D | 2 mol% MoO2(acac)2, sym-collidine, dioxane, r.t., 6 h, P = Tr | BzCl | 87% | 59 |
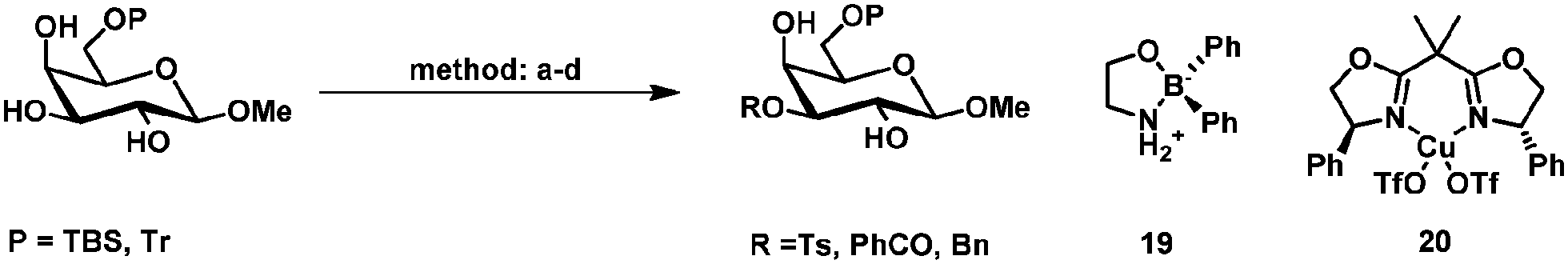
Method c (Table 2) developed by Dong and coworkers showed apart from the usual selectivity for a range of C6-TBS-protected methyl- and thioglycopyranosides, also a ligand-controlled protection of glycosides. The selectivity could be inverted by using tetramethylethylenediamine (TMEDA) instead of Ph-box ligand 20 (Scheme 6).53 The authors reasoned that the complex with the amine-based ligand TMEDA prefers the 1,2-dioxy motif and the complex with the imine-ligand (20) the 3,4-diol motif, probably based on the change in electron density of the complex (Scheme 6).
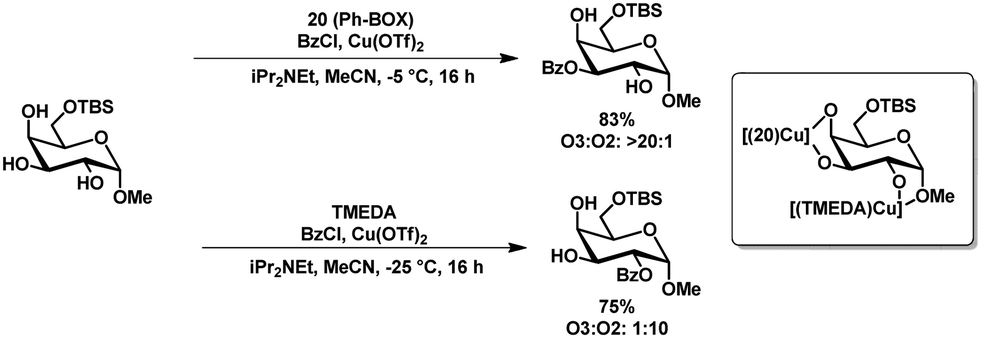 | ||
| Scheme 6 Ligand controlled inversion of selectivity in the regioselective benzoylation of methyl glucopyranoside. | ||
Biomimetic and stereoselective catalysis in glycoside protection
Recently, new biomimetic approaches for selective protection of carbohydrates have been developed. Miller et al. developed a “kinase-mimic” for the asymmetric phosphorylation of myo-inositol (which is a meso compound).60,61 Catalyst 21, a small peptide containing a histidine residue, was found through exhaustive screening of an elaborate peptide library. By varying the catalyst, either position 1 or 3 of myo-inositol was phosphorylated in excellent enantiomeric excess and good isolated yields (Scheme 7).60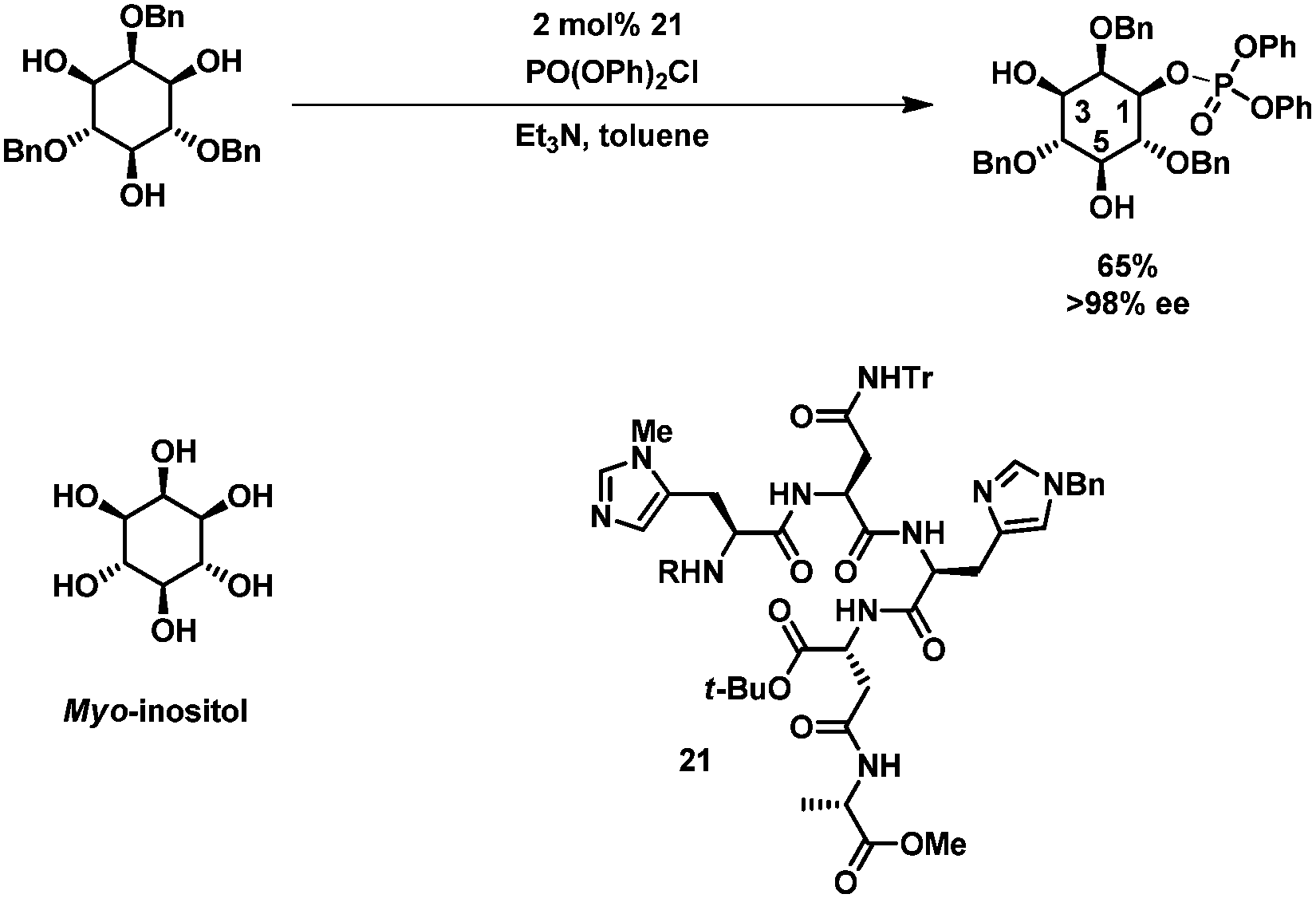 | ||
| Scheme 7 Peptide-catalyzed phosphorylation of myo-inositol. | ||
The concept was further expanded to accomplish the selective acylation of carbohydrate derivatives. For the acylation of 22 one catalyst (from a library of 186) was found which allowed the selective acylation at C3 (97:3 in C3 versus C4) and for 23 the best catalyst gave a mixture of roughly 6:1:1:2 with acylation on C4 as the major product (Scheme 8).62
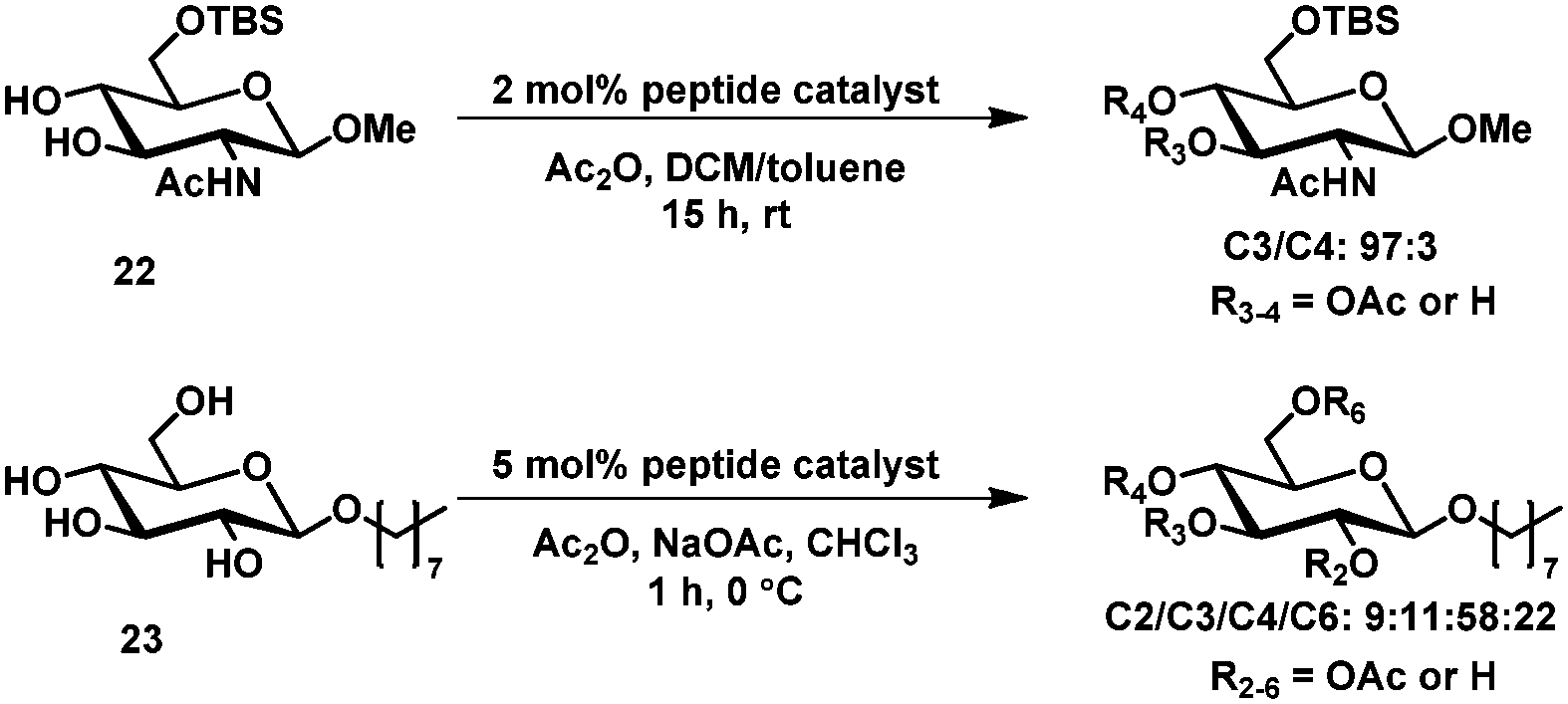 | ||
| Scheme 8 Peptide catalyzed acylation of glucopyranosyl derivatives. | ||
In future the substrate scope should be extended to identify the success rate of this approach.
Another approach in the selective protection of carbohydrates by mimicking the active site of an enzyme has been reported by Sun et al.63 Here, selective silylation, acylation and sulfonylation of a range of C6 protected carbohydrate derivatives could be achieved by using catalyst 26 or 27. Furthermore, catalyst-controlled protection of either the C2 or C3 position of C6-TBS protected methyl α-D-mannopyranoside was demonstrated (24/25, Scheme 9). Excellent isolated yields and selectivities were achieved.
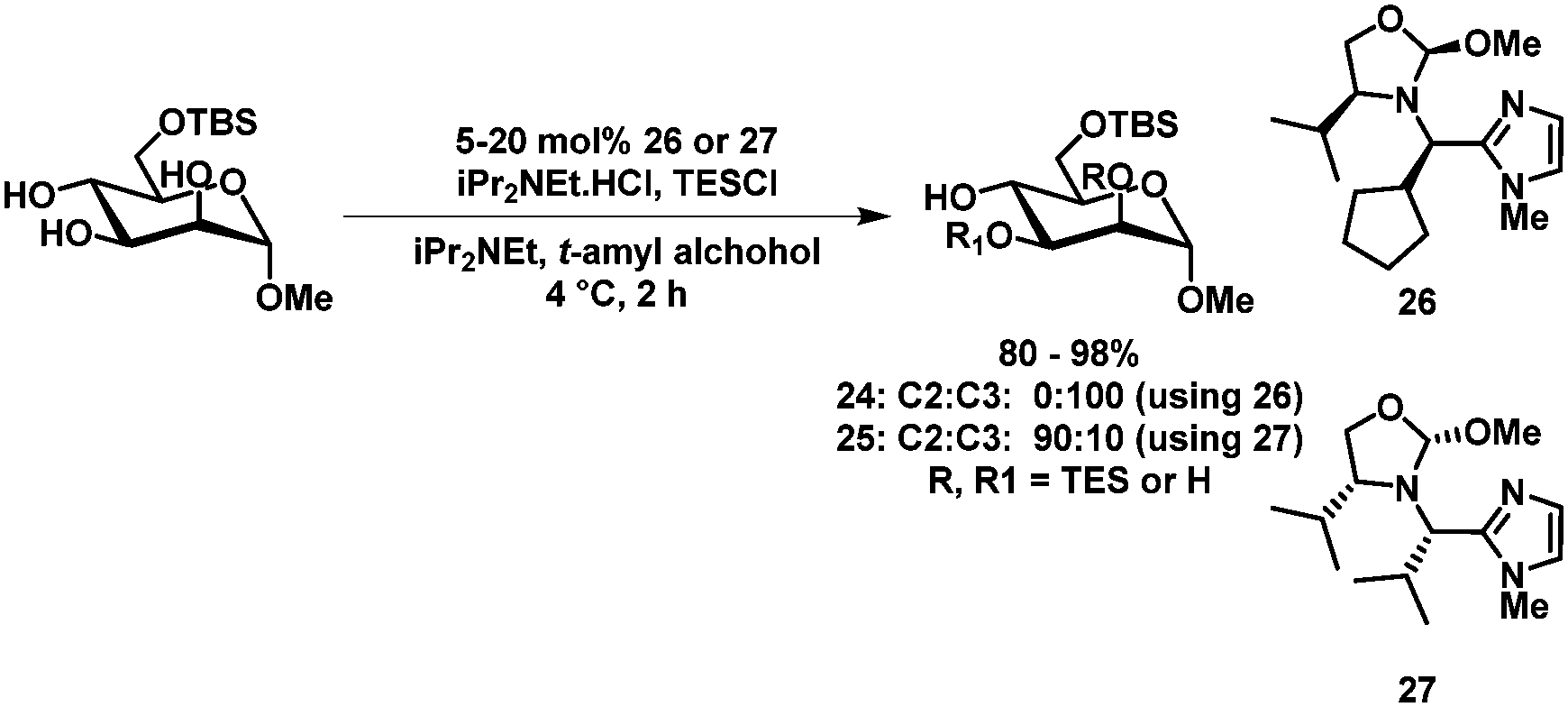 | ||
| Scheme 9 Selective silylation as described by Tan et al.63 | ||
Organocatalytic regioselective acylation at C4 of octyl-α glucopyranoside has been achieved by Kawabata et al.64,65 with excellent selectivity and excellent isolated yield. High selectivities could be obtained only with chloroform as solvent, which requires the presence of an octyl or related substituent on the carbohydrate to ensure solubility (Scheme 10).
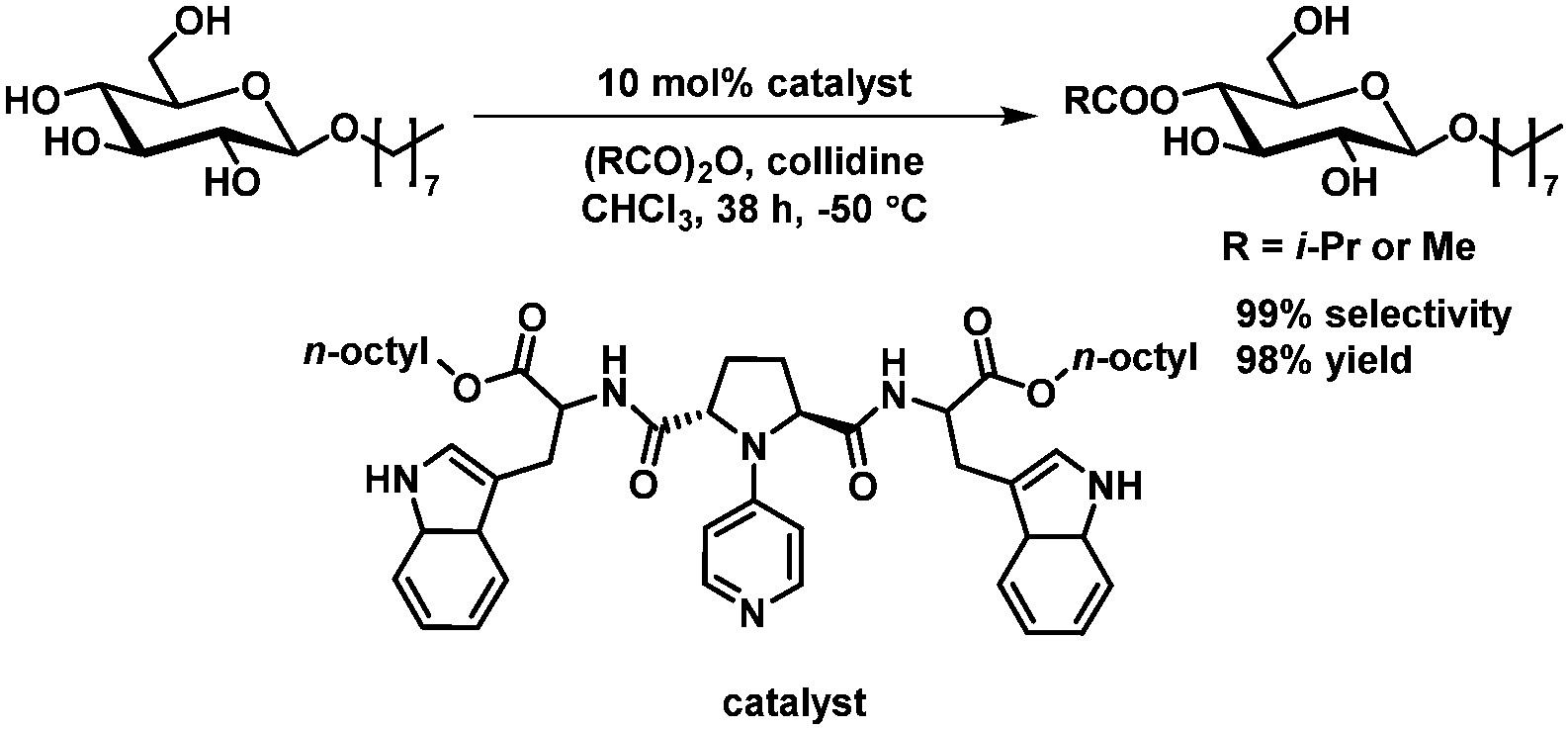 | ||
| Scheme 10 Organocatalytic acetylation of C4 as described by Kawabata et al.64,65 | ||
Enzyme-catalyzed protection of carbohydrates
Also enzyme-catalyzed methods for selective protection of carbohydrates are well-described and reviewed in the literature,66–68 some examples will be discussed to illustrate trends and challenges.Enzymatic acylation of carbohydrates is usually carried out by irreversible transesterification in organic solvents (pyridine, THF, and dioxane) using enol esters,69 trihaloethyl esters70 and oxime esters.71 Almost exclusive protection of the primary alcohol is obtained using lipases such as porcine pancreas lipase,70Candida antartica lipase71 or Candia cylindracea lipase69 for glycosides and reducing carbohydrates. Whereas for primary alcohol protected reducing carbohydrates the selectivity depends on the enzyme (reaction takes place at C2 or C3),72 in the case of primary alcohol protected glycosides the general trend is that α-glycosides are protected at C2 and β-glycosides at C3.66 Di- and oligosaccharides can in some cases be selectively acetylated, here discrimination between several primary hydroxyl groups is possible also. Klibanov and coworkers described the protection of a range of di- and oligosaccharides using subtilisin (a non-specific protease) in DMF.73
The acylation of glucose, maltose, cellobiose and maltotriose is selective for the terminal C6OH, a similar preference is observed using chemical acylation.74 The acylation of sucrose, however, is selective at the C1′ hydroxyl group (Fig. 1), contrary to what has been observed using chemical acylation.75
 | ||
| Fig. 1 Regioselective acylation catalyzed by subtilisin as described by Klibanov and coworkers.73 The site of acylation is indicated by an arrow. | ||
Selective non-protective modification of carbohydrates
The selective modification of single hydroxyl groups of unprotected carbohydrates is particularly difficult. Due to the inherent difference in steric hindrance, most studies concentrate on the modification of the primary hydroxyl group. Introduction of halogens is one of the most popular modifications of unprotected (or partially protected) carbohydrates. Here typical (but often modified) Appel and Mitsunobu conditions show good selectivities.Halogen introduction
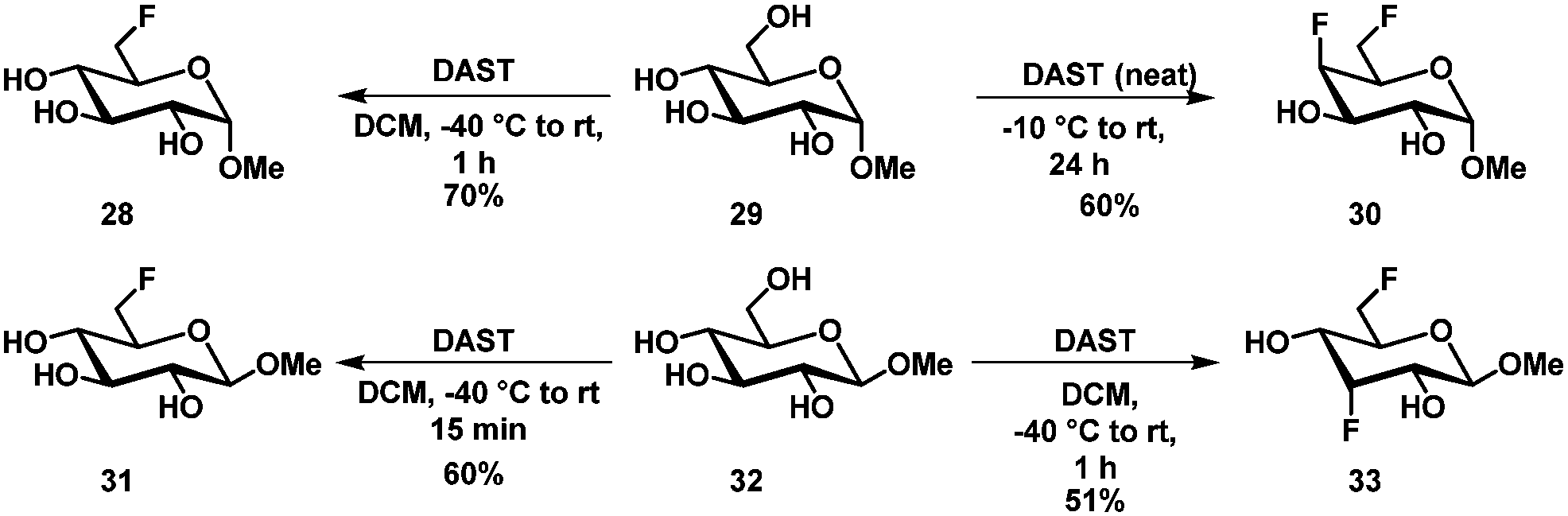 | ||
| Scheme 11 Selective fluorination of methyl glucopyranoside. | ||
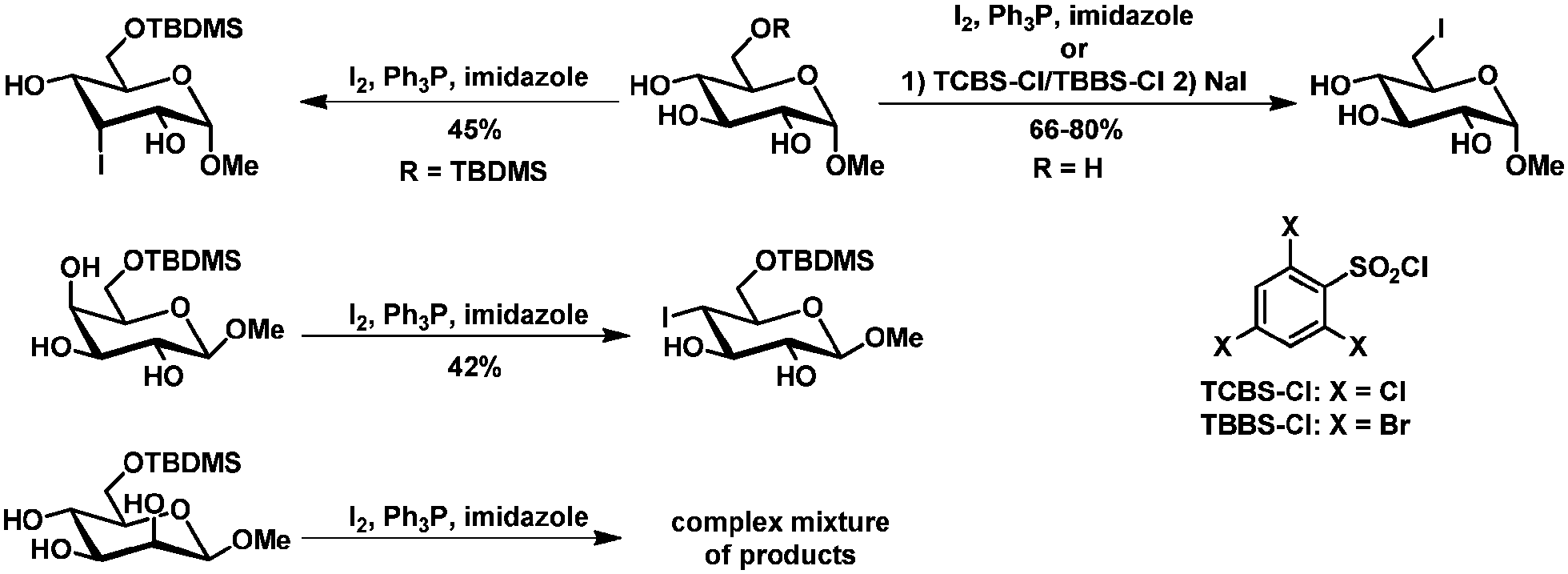 | ||
| Scheme 12 Regioselective iodination of primary and secondary methyl glucosides. | ||
Regioselective iodination of secondary hydroxyl groups has been described for 1,2-isopropylidene protected fructopyranoside and psicopyranose and for C6 protected methyl-galacto- and -glucopyranoside.80 The yields however were moderated at most and the corresponding mannopyranoside showed no selectivity (Scheme 12).
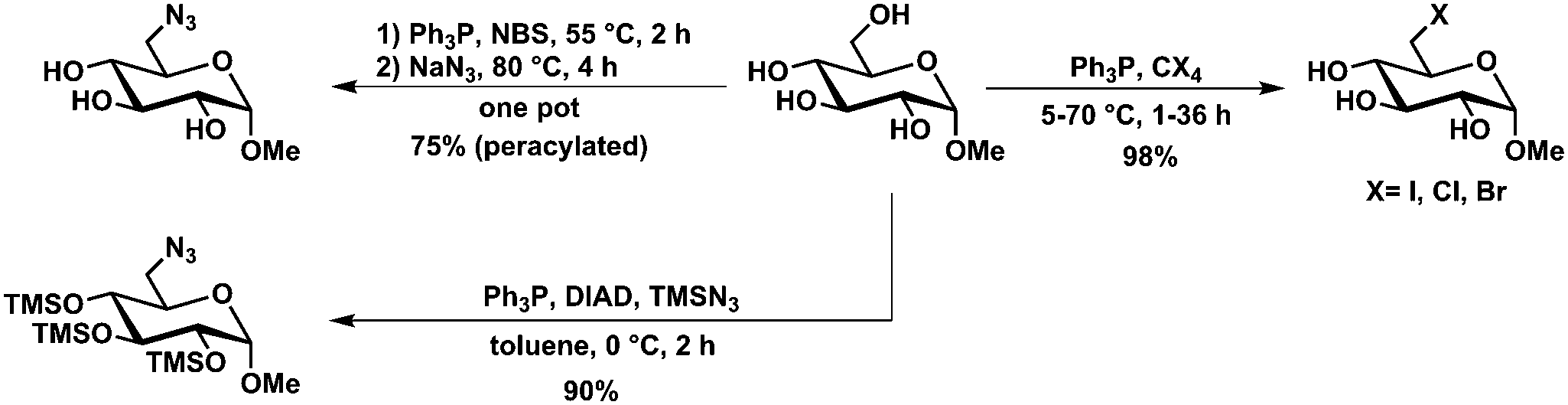 | ||
| Scheme 13 Selective Appel- and Mitsunobu-type halogen and azido introduction. | ||
Azide introduction
Further development of the method described by Hanessian et al. for the selective bromination of glycosides84 allowed the selective in situ transformation into the C6-azide using N-bromosuccinimide, triphenylphosphine and sodium azide. Extension of this method towards fully unprotected carbohydrates (reducing sugars) was done by Gouin et al. Here, glycosyl azides are obtained also in combination with substitution on C6, however the isolated yields are rather poor (13–49%).85 Next to typical Appel reaction-type modifications of glycosides and other carbohydrates, Mitsunobu conditions also allowed azidotrimethylsilylation of glycosides with azide introduction selectively on C6 and TMS-protection of the remaining secondary hydroxyl groups.86Deoxygenation and epimerization of glycosides
A selective thiofunctionalization and subsequent desulfurization towards C6 deoxyglycosides has been published by Thiem et al.87,88 Typical Mitsunobu conditions using diethylazodicarboxylate (DEAD), triphenylphosphine and 2-mercaptobenzothiazole as the nucleophile afforded thioether 35 selectively, which allowed further desulfurization to the C6 deoxy glycoside (Scheme 14).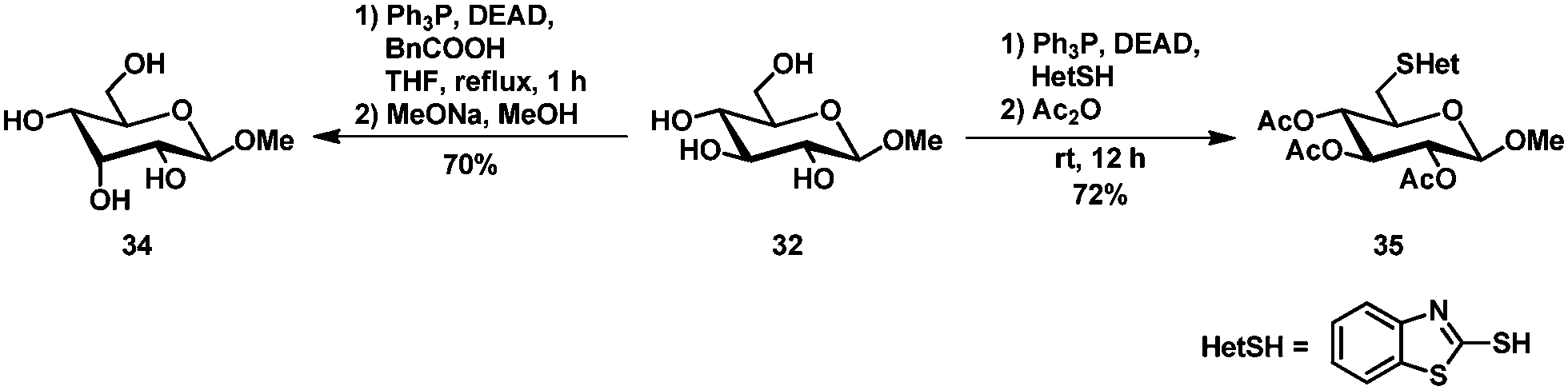 | ||
| Scheme 14 Epimerisation and thiofunctionalization by Mitsunobu reaction. | ||
Epimerisation could be achieved by the Mitsunobu reaction using benzoic acid as the nucleophile and subsequent saponification. Selective epimerization at C3 was observed to allose derivative 34 (the parallel Mitsunobu reaction at C6 is inconsequential). The substrate scope is limited to methyl β-glucopyranoside, as the α-anomer gave a mixture of products89 and the epimerisation of methyl β-xylopyranoside was incomplete and the product could only be isolated in 20% yield.90
Selective oxidation of carbohydrates
The selective oxidation of carbohydrates is a valuable modification since it is a direct approach and allows in principle a range of further modifications without the need for protection of the remaining hydroxyl groups. Subsequent reactions of the carbonyl group, such as reductive amination, epimerization and deoxygenation, lead to carbohydrate derivatives without a protection/deprotection protocol. Several chemical and enzymatic methods are discussed here.Chemical oxidation of glycosides
The selective chemical oxidation of the primary hydroxy group in pyranosides to the corresponding acid,91,92 aldehyde93 or internal acetal94 using the 2,2,6,6-tetramethyl-1-piperidinyloxy radical (TEMPO) has been well described. The selectivity is solely based on steric hindrance.The group of Grützmayer reported a rhodium catalyzed oxidation of primary hydroxyl groups in the presence of secondary hydroxyl groups. In that way also methyl β-D-glucopyranoside could be oxidized selectively to the glucoronic acid (Scheme 15).95
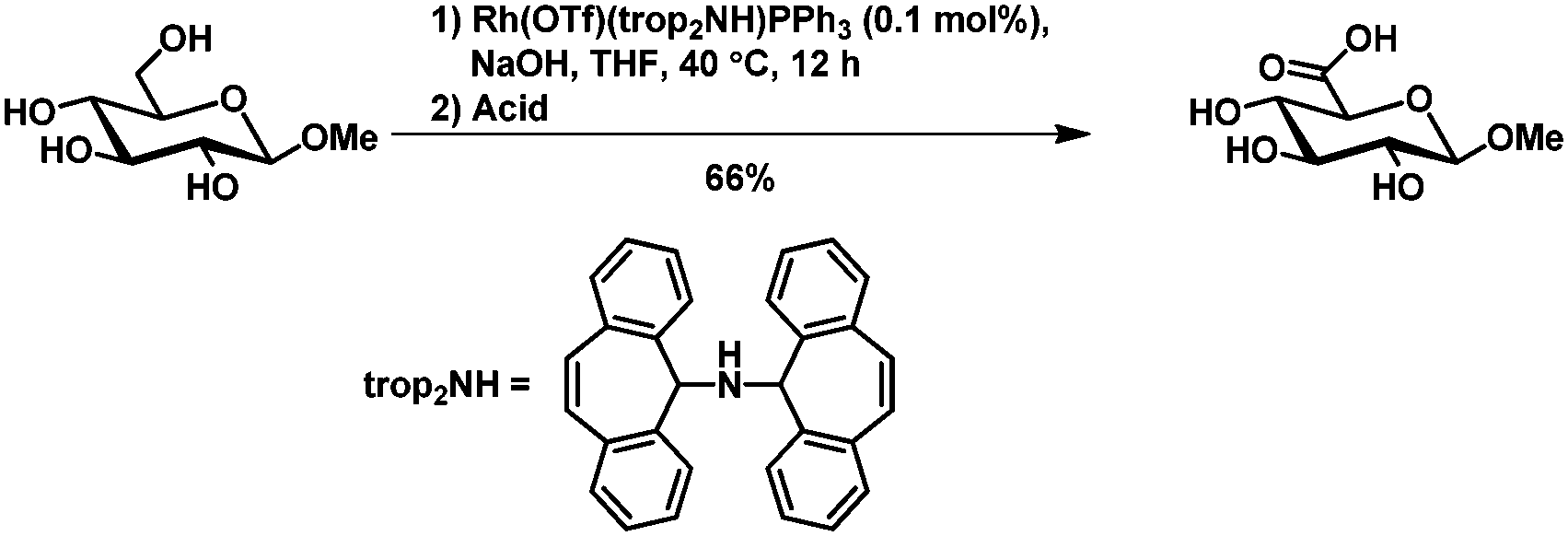 | ||
| Scheme 15 Rhodium catalyzed oxidation of methyl β-D-glucopyranoside. | ||
In contrast, the selective oxidation of the secondary hydroxy groups is extremely difficult and barely known.2 Tsuda et al. have described the selective oxidation of several methyl glycosides with stoichiometric bistributyltin oxide and bromine.96,97 Although good yields were obtained and the regioselectivity could be steered by choice of the substrate, the use of stoichiometric amounts of organotin reagents limits the use of this approach. Recently a catalytic version of this reaction was published by Muramatsu using 2 mol% of dioctyltin dichloride and trimethylphenylammonium tribromide as the oxidant.98 The selectivity is the same as described for the stoichiometric oxidation and isolated yields are high for galacto- but only mediocre for gluco-, manno- and rhamno-derivatives. Remarkably, the selectivity for the tin mediated oxidation appears to be in most cases reversed compared to the previously described tin mediated selective protection. This could be explained by the theory mentioned earlier, where the electrophile or the oxidant enters rather from the side than from the top or bottom of the substrate (Scheme 16).22
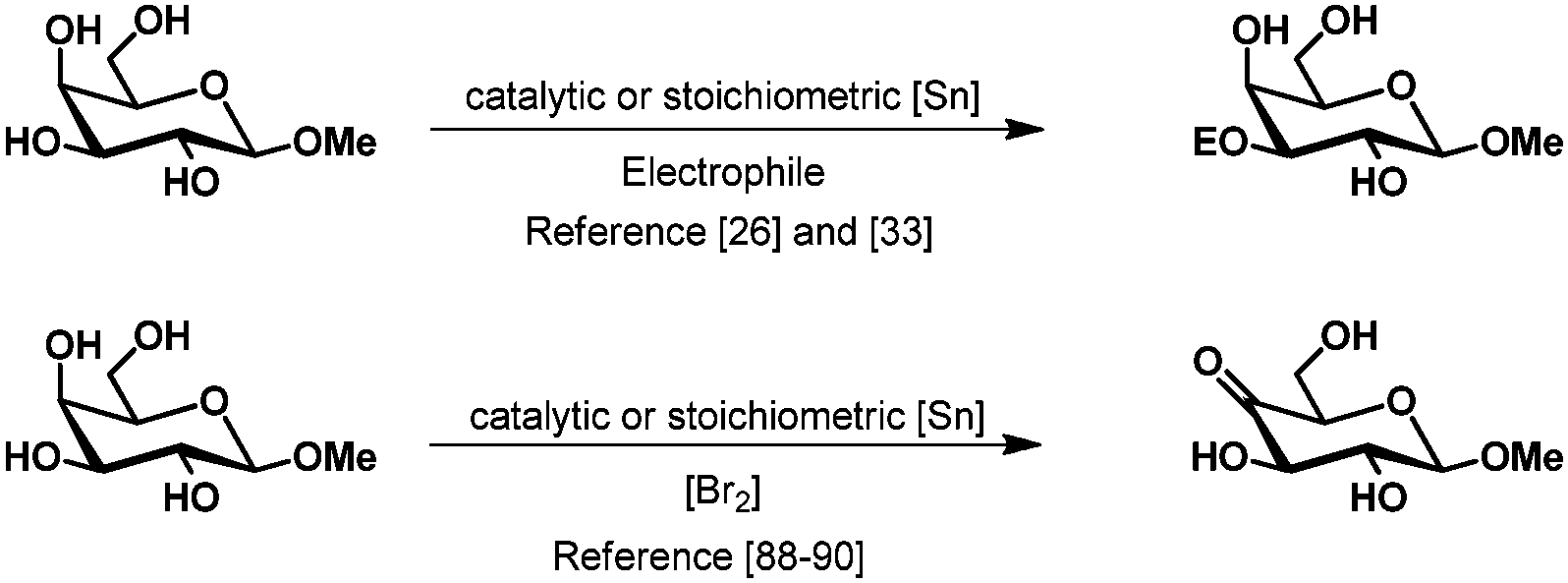 | ||
| Scheme 16 Reversed selectivity in the protection and oxidation of methyl β-D-galactopyranoside using tin acetals. | ||
Recently our lab has developed a method for the palladium catalyzed oxidation of glucosides.99 A range of glucosides including two disaccharides could be oxidized selectively in good to excellent isolated yields, with oxidation at C3 as the sole product (Scheme 17).
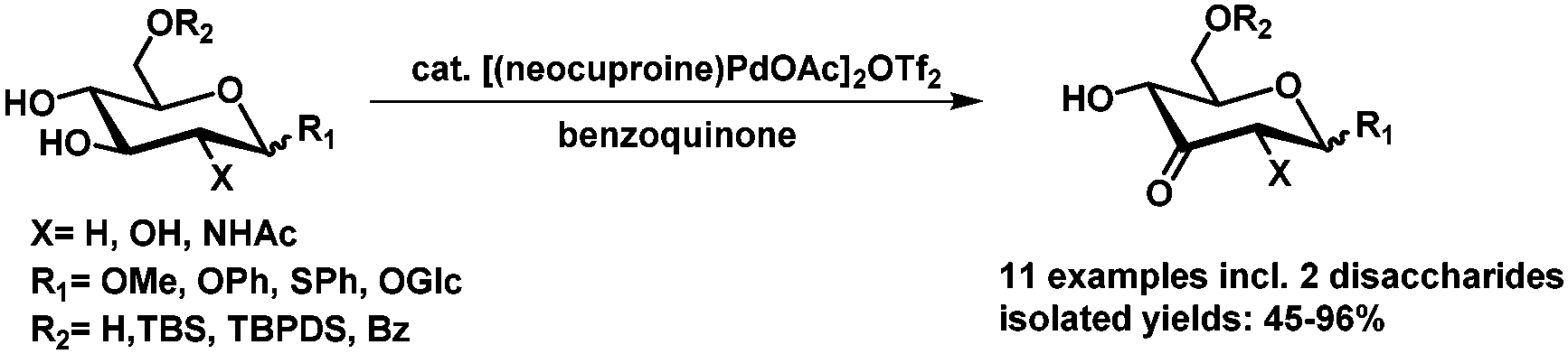 | ||
| Scheme 17 Palladium catalyzed oxidation of glucosides.99 | ||
Enzymatic oxidation of carbohydrates
Enzymatic oxidation of primary hydroxyl groups of carbohydrates has been described using uridine 5′-diphosphoglucose dehydrogenase100 and galactose oxidase.101 The oxidation of uridine 5′-diphosphoglucose by uridine 5′-diphosphoglucose dehydrogenase has been described by Whitesides et al. Unfortunately, the substrate scope has not been investigated and the rather specific substrate limits the general use of this approach. The oxidation by galactose oxidase as described by Kieboom et al. allowed selective oxidation to the C6-aldehyde. The scope of the reaction, however is limited to galactose derivatives (including the disaccharides lactose and melibiose).101The enzymatic oxidation of secondary alcohols of several carbohydrates, including glycosides, has been described by Köpper and co-workers.102 By using pyranose oxidase, selective oxidation at C2 and C3 was achieved, depending on the substrate. For reducing carbohydrates, the yields were generally high, but for glycosides low yields were observed. The activity of this enzyme is rather low (Scheme 18). This and the substrates being restricted to the β-anomer have prohibited the application of this method.
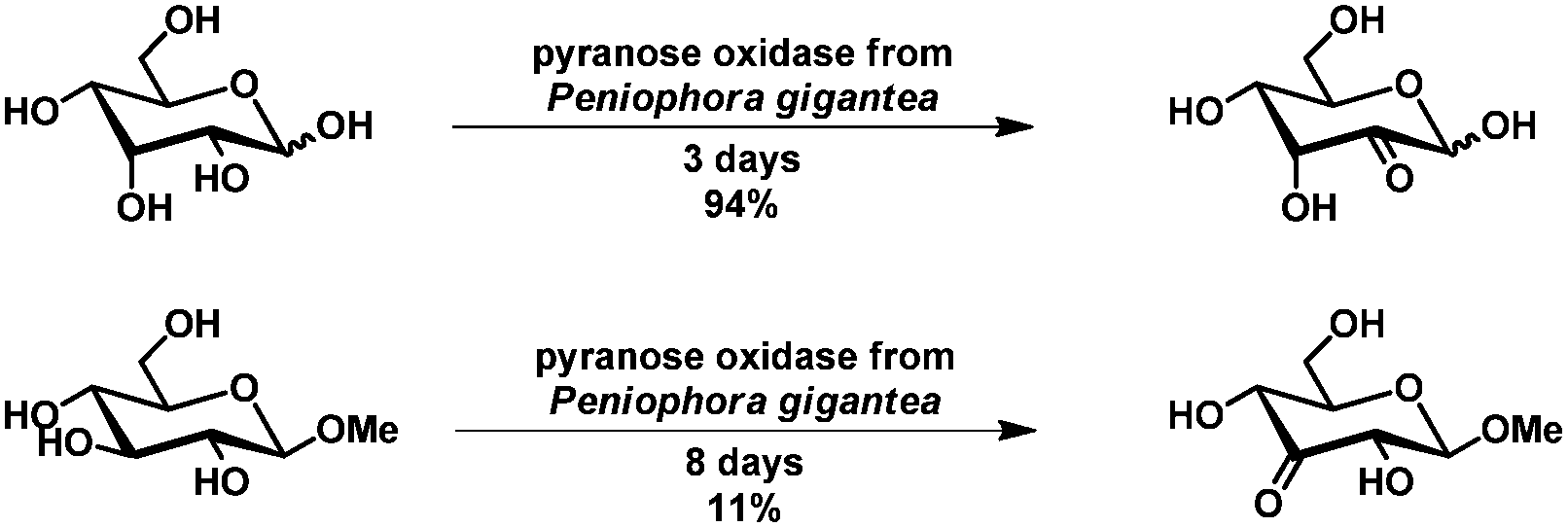 | ||
| Scheme 18 Oxidation of allose and methyl-β-D-glucopyranoside by pyranose oxidase. | ||
Another enzymatic approach has been described in the group of Haltrich, in which a fungal pyranose dehydrogenase was able to oxidize a series of carbohydrates at C1, C2, C3, C1,3′, or C2,3′.103,104 Yields of isolated products were not reported, however.
The first selective enzymatic oxidation of disaccharides was achieved by De Ley and coworkers in 1960.105 Among others, sucrose could be oxidized selectively at C3 by fermentation with Agrobacterium tumefaciens. The group of Buchholz elaborated on this and optimized this fermentation process to yield 3-ketosucrose 36 in 70% on the kilogram scale.106 Furthermore, a series of modifications of 3-ketosucrose including the conversion into allosucrose 37,107 allose,107 3-amino-3-deoxy-α-D-allopyranosyl β-D-fructofuranoside (38),108,109 corresponding polymer building blocks (39),109 surfactants (39),109 sucrose–amino acid conjugates (39),108 alkylated sucrose via Grignard-reaction (40)109 and the cyanohydrin 41110 could be obtained. The derivatives were synthesized usually without the use of protecting groups in 2–4 steps and could be isolated in good yields. This cluster of reports forms a rare example of the power of a selective oxidation method for carbohydrates (Scheme 19).
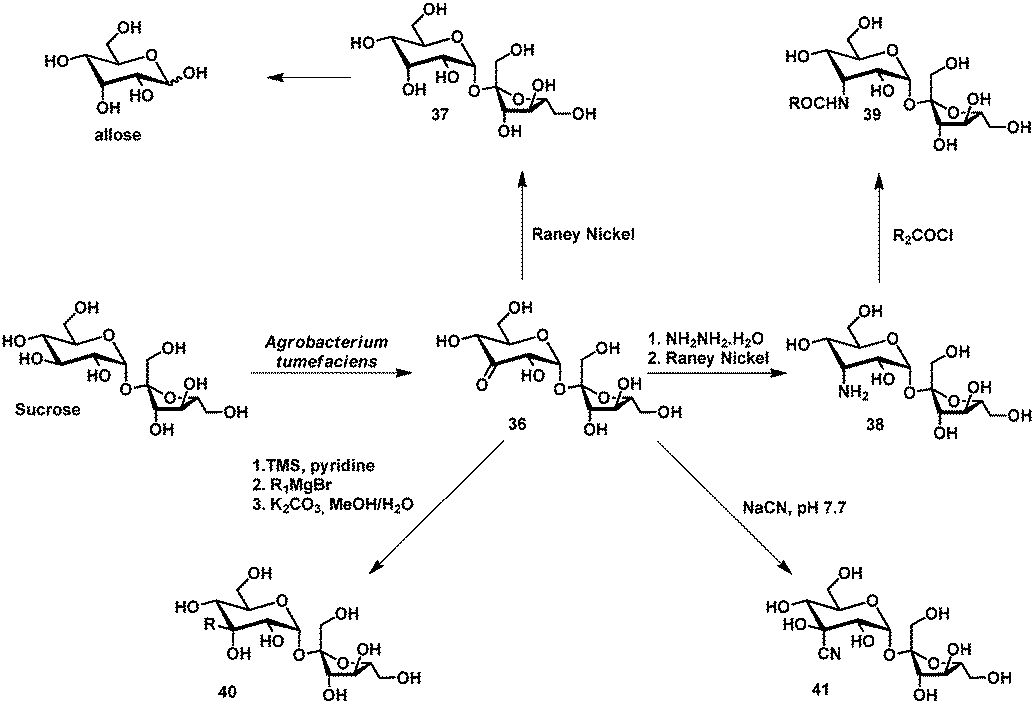 | ||
| Scheme 19 Derivatization of 3-ketosucrose. | ||
The substrate scope comprises of disaccharides sucrose, isomaltulose,111 leucrose,112 maltose and lactose.113
Conclusions
Considerable steps have been made in recent years regarding the selective modification of carbohydrates. However, although different methods and catalysts are used, the results, and in particular the selectivities, are mostly identical. So, the modification of the C6 position is, due to the smaller steric hindrance, a rather straightforward task, but the modification of C4 is hardly observed. This makes some derivatives readily accessible by a variety of methods whereas others only by long and time consuming synthetic routes.Part of the challenge faced is that apparently the regioselective functionalization of various secondary hydroxyl groups depends on stereo-electronic effects of the carbohydrate. From a synthetic point of view a rather unfortunate situation since it seems to hamper the possibility for catalyst control. The Holy Grail in carbohydrate functionalization; a general strategy for the discrimination of any specific hydroxyl group in a carbohydrate is therefore still a huge challenge and it has to been shown whether it is possible at all.
Acknowledgements
This research was performed within the framework of the Catch-Bio Program. The authors gratefully acknowledge support from the SmartMix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science.Notes and references
- J. Guo and X.-S. Ye, Molecules, 2010, 15, 7235–7265 CrossRef CAS PubMed.
- D. Lee and M. Taylor, Synthesis, 2012, 3421–3431 CAS.
- T. Saloranta and R. Leino, Synlett, 2015, 26, 421–425 CrossRef CAS.
- S. Hanessian and B. Lou, Chem. Rev., 2000, 100, 4443–4464 CrossRef CAS PubMed.
- L. J. G. Edgar, S. Dasgupta and M. Nitz, Org. Lett., 2012, 14, 4226–4229 CrossRef CAS PubMed.
- A. V Gudmundsdottir and M. Nitz, Org. Lett., 2008, 10, 3461–3463 CrossRef PubMed.
- T. Tanaka, W. C. Huang, M. Noguchi, A. Kobayashi and S. Shoda, Tetrahedron Lett., 2009, 50, 2154–2157 CrossRef CAS.
- P. Rawat, M. Kumar, N. Rahuja, D. S. Lal Srivastava, A. K. Srivastava and R. Maurya, Bioorg. Med. Chem. Lett., 2011, 21, 228–233 CrossRef CAS PubMed.
- Y. Ichikawa, S. Kusaba, T. Minami, Y. Tomita, K. Nakano and H. Kotsuki, Synlett, 2011, 1462–1466 CrossRef CAS.
- E. Brachet, J.-D. Brion, M. Alami and S. Messaoudi, Chem. – Eur. J., 2013, 19, 15276–15280 CrossRef CAS PubMed.
- T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Commun., 2009, 3378–3379 RSC.
- N. Yoshida, M. Noguchi, T. Tanaka, T. Matsumoto, N. Aida, M. Ishihara, A. Kobayashi and S. Shoda, Chem. – Asian J., 2011, 6, 1876–1885 CrossRef CAS PubMed.
- D. Lim, M. A. Brimble, R. Kowalczyk, A. J. A. Watson and A. J. Fairbanks, Angew. Chem., Int. Ed., 2014, 53, 11907–11911 CrossRef CAS PubMed.
- J. L. Houghton, K. D. Green, W. Chen and S. Garneau-Tsodikova, ChemBioChem, 2010, 11, 880–902 CrossRef CAS PubMed.
- D. P. Arya, Aminoglycoside antibiotics: from chemical biology to drug discovery, WILEY-VCH Verlag, New Jersey, 2007 Search PubMed.
- J. Zhou, G. Wang, L.-H. Zhang and X.-S. Ye, Med. Res. Rev., 2007, 27, 279–316 CrossRef CAS PubMed.
- A. A. Bastian, A. Marcozzi and A. Herrmann, Nat. Chem., 2012, 4, 789–793 CrossRef CAS PubMed.
- A. A. Bastian, E. M. Warszawik, P. Panduru, C. Arenz and A. Herrmann, Chem. – Eur. J., 2013, 19, 9151–9154 CrossRef CAS PubMed.
- I. Fokt, S. Skora, C. Conrad, T. Madden, M. Emmett and W. Priebe, Carbohydr. Res., 2013, 368, 111–119 CrossRef CAS PubMed.
- L. Zou, R. B. Zheng and T. L. Lowary, Beilstein J. Org. Chem., 2012, 8, 1219–1226 CrossRef CAS PubMed.
- P. J. Card and G. S. Reddy, J. Org. Chem., 1983, 48, 4734–4743 CrossRef CAS.
- L. Jiang and T.-H. Chan, J. Org. Chem., 1998, 63, 6035–6038 CrossRef CAS PubMed.
- T. Kurahashi, T. Mizutani and J. Yoshida, J. Chem. Soc., Perkin Trans. 1, 1999, 465–474 RSC.
- N. Moitessier, P. Englebienne and Y. Chapleur, Tetrahedron, 2005, 61, 6839–6853 CrossRef CAS.
- Y. Lu, P. Wei, Y. Pei, H. Xu, X. Xin and Z. Pei, Green Chem., 2014, 16, 4510–4514 RSC.
- Y. Zhou, M. Rahm, B. Wu, X. Zhang, B. Ren and H. Dong, J. Org. Chem., 2013, 78, 11618–11622 CrossRef CAS PubMed.
- S. David and S. Hanessian, Tetrahedron, 1985, 41, 643–663 CrossRef CAS.
- T. B. Grindley, Advances in Carbohydrate Chemistry and Biochemistry, Elsevier, 1998, vol. 53 Search PubMed.
- F. Peri, L. Cipolla and F. Nicotra, Tetrahedron Lett., 2000, 41, 8587–8590 CrossRef CAS.
- K. Takeo and K. Shibata, Carbohydr. Res., 1984, 133, 147–151 CrossRef CAS.
- V. K. Srivastava and C. Schuerch, Tetrahedron Lett., 1979, 20, 3269–3272 CrossRef.
- L. Ballell, J. A. F. Joosten, F. A. el Maate, R. M. J. Liskamp and R. J. Pieters, Tetrahedron Lett., 2004, 45, 6685–6687 CrossRef CAS.
- A. Glen, D. A. Leigh, R. P. Martin, J. P. Smart and A. M. Truscello, Carbohydr. Res., 1993, 248, 365–369 CrossRef CAS PubMed.
- M. J. Martinelli, R. Vaidyanathan, J. M. Pawlak, N. K. Nayyar, U. P. Dhokte, C. W. Doecke, L. M. H. Zollars, E. D. Moher, V. Van Khau and B. Košmrlj, J. Am. Chem. Soc., 2002, 124, 3578–3585 CrossRef CAS PubMed.
- M. J. Martinelli, R. Vaidyanathan and V. Van Khau, Tetrahedron Lett., 2000, 41, 3773–3776 CrossRef CAS.
- G. Hodosi and P. Kováč, J. Am. Chem. Soc., 1997, 119, 2335–2336 CrossRef CAS.
- G. Hodosi and P. Kováč, Carbohydr. Res., 1997, 303, 239–243 CrossRef CAS.
- H. Xu, Y. Lu, Y. Zhou, B. Ren, Y. Pei, H. Dong and Z. Pei, Adv. Synth. Catal., 2014, 356, 1735–1740 CrossRef CAS.
- F.-Y. Dupradeau, J. Prandi and J.-M. Beau, Tetrahedron, 1995, 51, 3205–3220 CrossRef CAS.
- F. Dasgupta and P. J. Garegg, Synthesis, 1994, 1121–1123 CrossRef CAS.
- H. Qin and T. B. Grindley, J. Carbohydr. Chem., 1996, 15, 95–108 CrossRef CAS.
- D. D. Manning, X. Hu, P. Beck and L. L. Kiessling, J. Am. Chem. Soc., 1997, 119, 3161–3162 CrossRef CAS.
- S. David and A. Malleron, Carbohydr. Res., 2000, 329, 215–218 CrossRef CAS PubMed.
- R. S. Coleman and J. R. Fraser, J. Org. Chem., 1993, 58, 385–392 CrossRef CAS.
- H. Dong, Y. Zhou, X. Pan, F. Cui, W. Liu, J. Liu and O. Ramström, J. Org. Chem., 2012, 77, 1457–1467 CrossRef CAS PubMed.
- D. Lee and M. S. Taylor, J. Am. Chem. Soc., 2011, 133, 3724–3727 CrossRef CAS PubMed.
- D. Lee, C. L. Williamson, L. Chan and M. S. Taylor, J. Am. Chem. Soc., 2012, 134, 8260–8267 CrossRef CAS PubMed.
- L. Chan and M. S. Taylor, Org. Lett., 2011, 13, 3090–3093 CrossRef CAS PubMed.
- Y. Zhou, O. Ramström and H. Dong, Chem. Commun., 2012, 48, 5370–5372 RSC.
- C.-C. Wang, J.-C. Lee, S.-Y. Luo, S. S. Kulkarni, Y.-W. Huang, C.-C. Lee, K.-L. Chang and S.-C. Hung, Nature, 2007, 446, 896–899 CrossRef CAS PubMed.
- H. M. Osborn, V. A. Brome, L. M. Harwood and W. G. Suthers, Carbohydr. Res., 2001, 332, 157–166 CrossRef CAS PubMed.
- C. L. Allen and S. J. Miller, Org. Lett., 2013, 15, 6178–6181 CrossRef CAS PubMed.
- I.-H. Chen, K. G. M. Kou, D. N. Le, C. M. Rathbun and V. M. Dong, Chem. – Eur. J., 2014, 20, 5013–5018 CrossRef CAS PubMed.
- E. V Evtushenko, Carbohydr. Res., 2012, 359, 111–119 CrossRef PubMed.
- E. V. Evtushenko, J. Carbohydr. Chem., 2015, 34, 41–54 CrossRef CAS.
- S. Malik, V. A. Dixit, P. V Bharatam and K. P. R. Kartha, Carbohydr. Res., 2010, 345, 559–564 CrossRef CAS PubMed.
- U. B. Gangadharmath and A. V Demchenko, Synlett, 2004, 2191–2193 CAS.
- E. V. Evtushenko, Synth. Commun., 2006, 36, 1593–1599 CrossRef CAS.
- E. V. Evtushenko, J. Carbohydr. Chem., 2010, 29, 369–378 CrossRef CAS.
- B. R. Sculimbrene, A. J. Morgan and S. J. Miller, J. Am. Chem. Soc., 2002, 124, 11653–11656 CrossRef CAS PubMed.
- B. R. Sculimbrene and S. J. Miller, J. Am. Chem. Soc., 2001, 123, 10125–10126 CrossRef CAS PubMed.
- K. S. Griswold and S. J. Miller, Tetrahedron, 2003, 59, 8869–8875 CrossRef CAS.
- X. Sun, H. Lee, S. Lee and K. L. Tan, Nat. Chem., 2013, 5, 790–795 CrossRef CAS PubMed.
- T. Kawabata, W. Muramatsu, T. Nishio, T. Shibata and H. Schedel, J. Am. Chem. Soc., 2007, 129, 12890–12895 CrossRef CAS PubMed.
- H. Takeuchi, K. Mishiro, Y. Ueda, Y. Fujimori, T. Furuta and T. Kawabata, Angew. Chem., 2015, 127, 6275–6278 CrossRef.
- D. Kadereit and H. Waldmann, Chem. Rev., 2001, 101, 3367–3396 CrossRef CAS PubMed.
- B. La Ferla, Monatsh. Chem., 2002, 133, 351–368 CrossRef CAS.
- H. G. Park, J. H. Do and H. N. Chang, Biotechnol. Bioprocess Eng., 2003, 8, 1–8 CrossRef CAS.
- E. W. Holla, Angew. Chem., Int. Ed. Engl., 1989, 28, 220–221 CrossRef.
- W. J. Hennen, H. M. Sweers, Y. F. Wang and C. H. Wong, J. Org. Chem., 1988, 53, 4939–4945 CrossRef CAS.
- R. Pulido and V. Gotor, Carbohydr. Res., 1994, 252, 55–68 CAS.
- M. Therisod and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 3977–3981 CrossRef CAS.
- S. Riva, J. Chopineau, A. P. G. Kieboom and A. M. Klibanov, J. Am. Chem. Soc., 1988, 110, 584–589 CrossRef CAS.
- R. Khan, The Chemistry of Maltose, Elsevier, 1981, vol. 39 Search PubMed.
- R. Khan, Pure Appl. Chem., 1984, 56, 833–844 CrossRef CAS.
- C. W. Somawardhana and E. G. Brunngraber, Carbohydr. Res., 1983, 121, 51–60 CrossRef CAS PubMed.
- P. J. Card, J. Org. Chem., 1983, 48, 393–395 CrossRef CAS.
- P. J. Garegg, Pure Appl. Chem., 1984, 56, 845–858 CrossRef CAS.
- P. R. Skaanderup, C. S. Poulsen, L. Hyldtoft, M. R. Jørgensen and R. Madsen, Synthesis, 2002, 1721–1727 CAS.
- A.-C. Simao, S. Silva, A. P. Rauter, P. Rollin and A. Tatibouët, Eur. J. Org. Chem., 2011, 2286–2292 CrossRef CAS.
- W. Xue and L. Zhang, Chin. J. Chem., 2012, 30, 1429–1432 CrossRef CAS.
- A. Kashem, M. Anisuzzaman and R. L. Whistler, Carbohydr. Res., 1978, 61, 511–518 CrossRef.
- B. Classon, P. J. Garegg and B. Samuelsson, Can. J. Chem., 1981, 59, 339–343 CrossRef CAS.
- S. Hanessian, M. M. Ponpipom and P. Lavallee, Carbohydr. Res., 1972, 24, 45–56 CrossRef CAS.
- S. G. Gouin and J. Kovensky, Tetrahedron Lett., 2007, 48, 2875–2879 CrossRef CAS.
- M. R. L, S. K. Yousuf, D. Mukherjee and S. C. Taneja, Org. Biomol. Chem., 2012, 10, 9090–9098 Search PubMed.
- I. Dancy, L. Laupichler, P. Rollin and J. Thiem, Liebigs Ann. Chem., 1993, 1993, 343–350 CrossRef.
- I. Dancy, L. Laupichler, P. Rollin and J. Thiem, Synlett, 1992, 283–284 CrossRef CAS.
- M. Kim, B. Grzeszczyk and A. Zamojski, Carbohydr. Res., 1999, 320, 244–249 CrossRef CAS.
- K. Weinges, S. Haremsa and W. Maurer, Carbohydr. Res., 1987, 164, 453–458 CrossRef CAS.
- N. J. Davis and S. L. Flitsch, Tetrahedron Lett., 1993, 34, 1181–1184 CrossRef CAS.
- A. De Nooy, A. Besemer and H. Van Bekkum, Tetrahedron, 1995, 51, 8023–8032 CrossRef CAS.
- T. Breton, G. Bashiardes, J.-M. Léger and K. B. Kokoh, Eur. J. Org. Chem., 2007, 1567–1570 CrossRef CAS.
- A. Gassama and N. Hoffmann, Adv. Synth. Catal., 2008, 350, 35–39 CrossRef CAS.
- M. Trincado, K. Kühlein and H. Grützmacher, Chem. – Eur. J., 2011, 17, 11905–11913 CrossRef CAS PubMed.
- Y. Tsuda, M. Hanajima, N. Matsuhira, Y. Okuno and K. Kanemitsu, Chem. Pharm. Bull., 1989, 37, 2344–2350 CrossRef CAS.
- H.-M. Lui, Y. Sato and Y. Tsuda, Chem. Pharm. Bull., 1996, 41, 491–501 Search PubMed.
- W. Muramatsu, Org. Lett., 2014, 16, 4846–4849 CrossRef CAS PubMed.
- M. Jäger, M. Hartmann, J. G. de Vries and A. J. Minnaard, Angew. Chem., Int. Ed., 2013, 52, 7809–7812 CrossRef PubMed.
- E. J. Toone, E. S. Simon and G. M. Whitesides, J. Org. Chem., 1991, 56, 5603–5606 CrossRef CAS.
- A. Siebum, A. van Wijk, R. Schoevaart and T. Kieboom, J. Mol. Catal. B: Enzym., 2006, 41, 141–145 CrossRef CAS.
- S. Freimund, A. Huwig, F. Giffhorn and S. Köpper, Chem. – Eur. J., 1998, 4, 2442–2455 CrossRef CAS.
- J. Volc, P. Sedmera, P. Halada, G. Daniel, V. Přikrylová and D. Haltrich, J. Mol. Catal. B: Enzym., 2002, 17, 91–100 CrossRef CAS.
- P. Sedmera, P. Halada, E. Kubátová, D. Haltrich, V. Přikrylová and J. Volc, J. Mol. Catal. B: Enzym., 2006, 41, 32–42 CrossRef CAS.
- M. J. Bernaerts and J. De Ley, J. Gen. Microbiol., 1960, 22, 129–136 CrossRef CAS PubMed.
- E. Stoppok and K. Buchholz, Carbohydrate Biotechnology Protocols (Methods in Biotechnology), Humana Press, Totowa, NY, USA, 1999th edn, 1999 Search PubMed.
- V. Timme, R. Buczys, K. Buchholz and B. Germany, Starch - Stärke, 1998, 50, 29–32 CrossRef CAS.
- J. Anders, R. Buczys, E. Lampe, M. Walter, E. Yaacoub and K. Buchholz, Carbohydr. Res., 2006, 341, 322–331 CrossRef CAS PubMed.
- M. Pietsch, M. Walter and K. Buchholz, Carbohydr. Res., 1994, 254, 183–194 CrossRef CAS PubMed.
- V. Timme, R. Buczys and K. Buchholz, Chem. Ing. Tech., 2001, 73, 1357–1361 CrossRef CAS.
- M. Noll-Borchers and K. Buchholz, Biotechnol. Lett., 1993, 15, 139–144 CrossRef CAS.
- J. Walter, E. Stoppok and K. Buchholz, Chem. Ing. Tech., 1991, 63, 631–633 CrossRef CAS.
- E. Stoppok, J. Walter and K. Buchholz, Appl. Microbiol. Biotechnol., 1995, 43, 706–712 CAS.
| This journal is © The Royal Society of Chemistry 2016 |