
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of geminal bis- and tristriazoles: exploration of unconventional azide chemistry†
Hellmuth
Erhardt
a,
Fabian
Mohr
b and
Stefan F.
Kirsch
*a
aOrganic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany. E-mail: sfkirsch@uni-wuppertal.de
bInorganic Chemistry, Bergische Universität Wuppertal, Gaußstr. 20, 42119 Wuppertal, Germany
First published on 5th November 2015
Abstract
A range of geminal bis- and tristriazoles are presented. These rare and hardly studied compound classes were easily synthesized using ethyl 2,2-diazido-3-oxobutanoate as the common starting point. Firstly, CuAAC-reaction with an alkyne afforded the corresponding deacetylated bistriazoles. Upon further azidation yielding azidomethylenebistriazoles, a second CuAAC-functionalization then led to the creation of the geminal tristriazole compounds.
The generation of triazoles through the copper-catalyzed alkyne azide cycloaddition (CuAAC) reaction has become a major playground in many disciplines of science, ranging from chemical biology to material sciences.1 Contemporary research in the field of small molecules containing azide functionalities is also boosted,2 mainly due to their use as one reaction partner in the CuAAC, and several new methods for the simple azidation of compact chemical structures were reported over the last couple of years.3
We became involved into the question of how to azidate small molecules during our studies on oxidative functionalizations of 1,3-dicarbonyl compounds.4 In 2012, we demonstrated that 1,3-dicarbonyls can be azidated by use of IBX-SO3K,5 sodium azide and catalytic amounts of sodium iodide in aqueous DMSO at room temperature.6 With this protocol, we were capable of creating several geminal diazides.‡![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 A variant of this procedure also led to the synthesis of geminal triazides, highly energetic compounds that must be handled with caution.8 In the course of those projects, we realized that the di- and triazides can be converted into their corresponding geminal bis- and tristriazoles using cycloaddition reactions with alkynes.9 Besides our own reports, the two compound classes turned out to be somewhat neglected, and most previous reports on geminal bistriazoles focused on geminal bis(benzotriazol-1-yl)methyl derivatives, as shown, for example, in the detailed studies by Katritzky and co-workers.10,11 Even more surprising, our review of the literature revealed only one example for a geminal tristriazole, i.e. tris(benzotriazo-1-yl)methane.12
7 A variant of this procedure also led to the synthesis of geminal triazides, highly energetic compounds that must be handled with caution.8 In the course of those projects, we realized that the di- and triazides can be converted into their corresponding geminal bis- and tristriazoles using cycloaddition reactions with alkynes.9 Besides our own reports, the two compound classes turned out to be somewhat neglected, and most previous reports on geminal bistriazoles focused on geminal bis(benzotriazol-1-yl)methyl derivatives, as shown, for example, in the detailed studies by Katritzky and co-workers.10,11 Even more surprising, our review of the literature revealed only one example for a geminal tristriazole, i.e. tris(benzotriazo-1-yl)methane.12
In this communication, we describe a unique approach toward geminal tristriazoles that does neither rely on the hazardous geminal triazides8 as starting materials nor on the nucleophilic substitution of chloroform,12 a reaction that is highly limited in scope. We also show how azides (and triazoles) can dramatically alter the reactive behavior of adjacent functional groups and give rise to a whole new pool of unconventional disconnections that might become of interest for future reaction plannings.13
By pure serendipity, it was observed that CuAAC reaction with geminal diazide 1 under standard conditions does not result in the formation of the expected bistriazole 2a′. To our surprise, triazole formation was, with this particular substrate, always accompanied by deacetylation, and 2a [15N-NMR (DMSO) −13.3, −20.3, −134.4 ppm] was isolated as the only product when reaction time was prolonged (Scheme 1). The molecular structure of 2a was unequivocally confirmed by X-ray crystallography (Fig. 1); the two triazole rings are oriented in a perpendicular way. In contrast to 2a′, bistriazole 2a had a position with an additional hydrogen where a further functionalization could be anticipated. Indeed, the electron-withdrawing nature of the triazole groups activates this position, and azidation was possible by use of our standard azidation protocol with IBX-SO3K, sodium azide and catalytic amounts of sodium iodide.6 Now, the door was open to the synthesis of tristriazoles, and 3a [15N-NMR (DMSO) −17.6, −20.0, −127.2, −143.0, −152.8, −293.6 ppm] was transformed into the symmetric tristriazole 4a [15N-NMR (DMSO) −16.4, −19.3, −134.4 ppm] through standard cycloaddition. The sequence outlined in Scheme 1 was expected to allow for the synthesis of a broad range of novel bistriazoles (such as 2a), azidobistriazoles (such as 3a) and tristriazoles (such as 4a), depending only on the alkynes employed in the cycloaddition reactions.
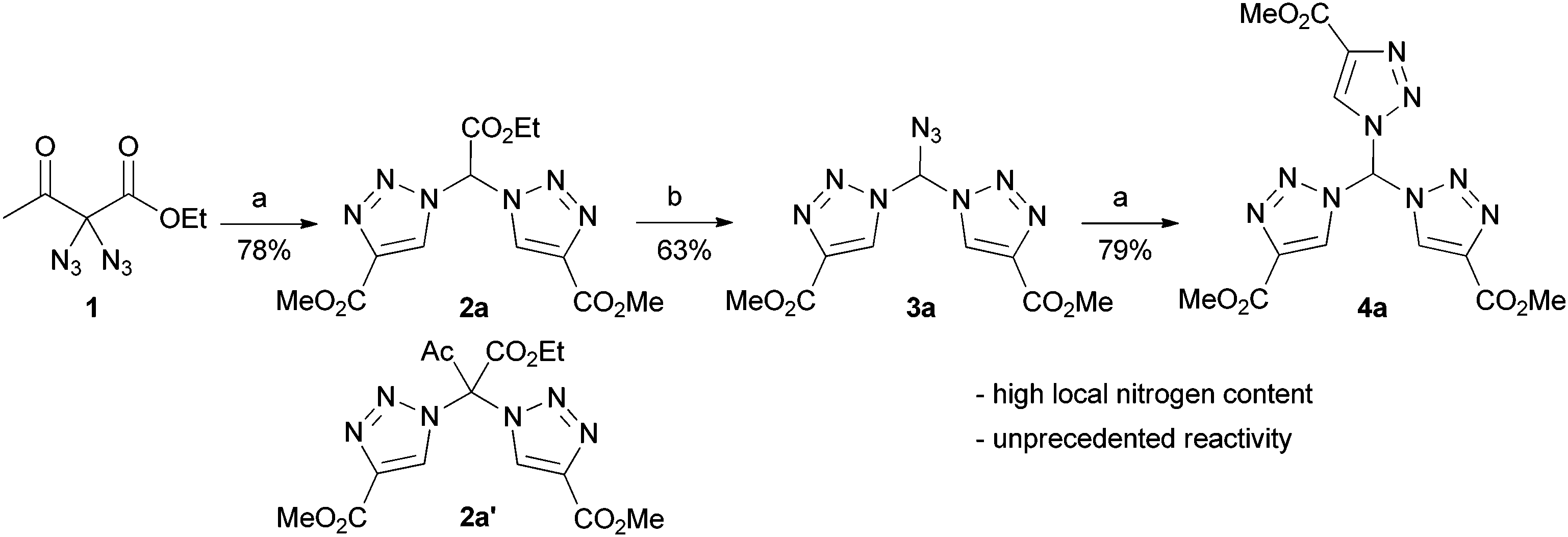 | ||
Scheme 1 Synthesis of tristriazole 4a by cycloaddition starting with diazide 1 and subsequent azidation. a: MeO2C–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH (2.2 eq.), CuSO4·5H2O (20 mol%), TBTA (3 mol%), Na ascorbate (42 mol%), rt, tBuOH/H2O. b: IBX-SO3K (4.0 eq), NaI (20 mol%), NaN3 (10.0 eq), rt, DMSO/H2O. CH (2.2 eq.), CuSO4·5H2O (20 mol%), TBTA (3 mol%), Na ascorbate (42 mol%), rt, tBuOH/H2O. b: IBX-SO3K (4.0 eq), NaI (20 mol%), NaN3 (10.0 eq), rt, DMSO/H2O. | ||
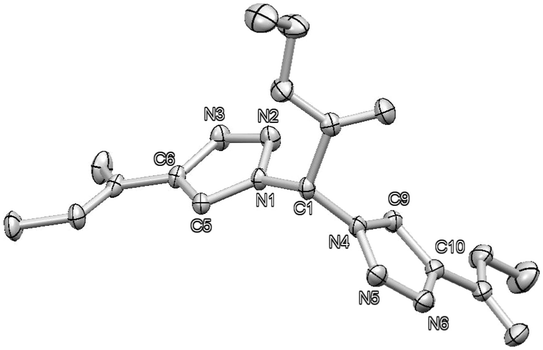 | ||
| Fig. 1 Crystal structure of 2a; ellipsoids are drawn at 50% probability. Hydrogen atoms are omitted for clarity.14 | ||
In every case, diazide 1 should be the starting material of choice due to its unique ability to undergo smooth deacetylation under the cycloaddition conditions. To this end, several bistriazoles 2 were generated through CuAAC reaction. As shown in Table 1, deacetylation of 1 took place in good yields when bistriazoles 2 were formed with arylalkynes. Aliphatic alkynes reacted with markedly reduced yields. Regarding the mechanism, we reasoned that the reaction proceeds via a retro-Claisen pathway in which water assumes the role of the nucleophile. When the reaction is stopped after 30 minutes, a mixture of acetylated and deacetylated bistriazoles was obtained in most cases. We note that the hydrogen atom attached at the alpha-carbon shows a tremendous downfield shift, ranging from 7.46 to 7.98 ppm in the 1H NMR spectrum. For a tetrahedral carbon (see Fig. 1), this downfield shift is quite astonishing.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | 2 | 3 | ||
| Yieldb (%) | δ (ppm) | Yieldb (%) | δ (ppm) | ||
| a Conditions: a: alkyne (2.2 eq.), CuSO4·5H2O (20 mol%), TBTA (3 mol%), Na ascorbate (42 mol%), rt, tBuOH/H2O. b: IBX-SO3K (4.0 eq.), NaI (20 mol%), NaN3 (10.0 eq.), 60 °C, DMSO/H2O. b Isolated yields. c Determined by 1H NMR analysis of the product in CDCl3 at 400 MHz or 600 MHz. d Reaction at room temperature. e 2.0 equiv. of IBX-SO3K were used. | |||||
| 1 | CO2Me | 2a (78) | 7.78 | 3a (63)d | 8.77 |
| 2 | CO2Bn | 2b (19) | 7.98 | 3b (24)d,e | 8.56 |
| 3 | c-C3H5 | 2c (34) | 7.46 | 3c (26) | 8.06 |
| 4 | 4-BrC6H4 | 2d (76) | 7.72 | 3d (19) | 8.28 |
| 5 | 4-nPrC6H4 | 2e (91) | 7.68 | 3e (36) | 8.27 |
| 6 | C6H5 | 2f (74) | 7.71 | 3f (35) | 8.30 |
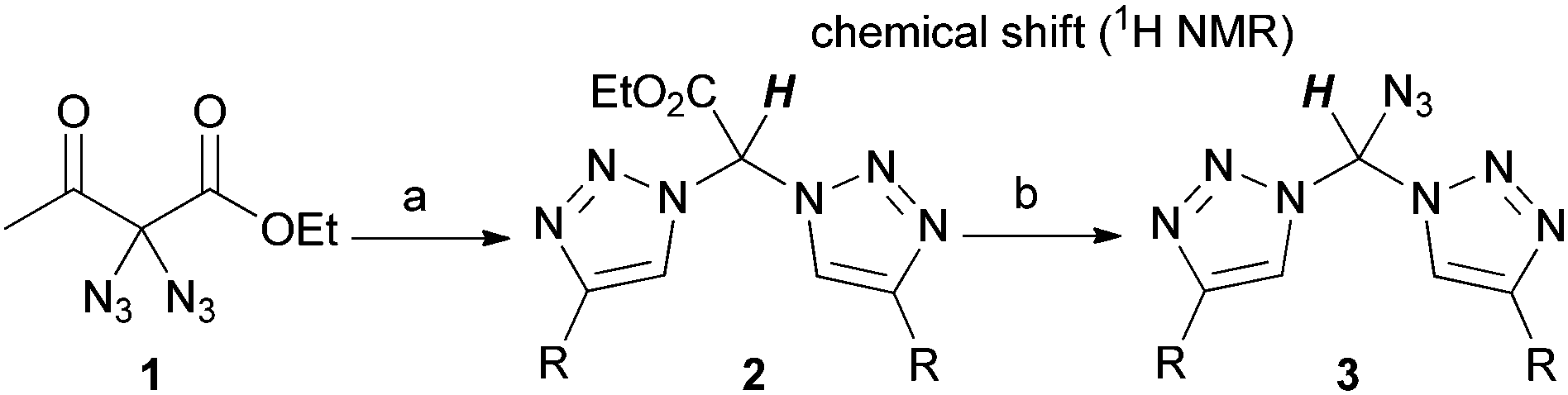
The results of the azidation are also listed in Table 1. In general, elevated reaction temperatures of 60 °C were required in order to obtain acceptable yields for the azidobistriazoles 3 (although traces of product formation were found as well at room temperature). The azidation proved to be scalable: azidobistriazole 3a, for example, was isolated with 63% yield in a preparative amount of 870 mg. In all cases, the formation of the azidomethylenebistriazoles was accompanied by the loss of the ester moieties. Despite their high local nitrogen content, compounds of type 3, however, never exhibited explosive behaviour and showed no degradation when stored under ambient conditions. We also point out that azidobistriazoles 3 are not stable under basic conditions; with LiOH in aqueous THF for example, they immediately engage in a strongly exothermic reaction under vigorous evolution of gas.
Azidomethylenebistriazoles 3 were then converted into the geminal tristriazoles 4 as summarized in Table 2. The overall sequence allows for the preparation of homosubstituted tristriazoles (R = R′) and tristriazoles with two different substituents (R ≠ R′). Again, we found extraordinary downfield shifts in the 1H NMR spectra for the central tetrahedral hydrogen. This observation points to a quite acidic character, a feature that will become the theme of future studies from our group.
|
|
|||||
|---|---|---|---|---|---|
| Entry | R | R′ | # | Yieldb (%) | δ (ppm) |
| a Conditions: a: alkyne (3.0 eq.), CuSO4·5H2O (40 mol%), Na ascorbate (84 mol%), rt, tBuOH/H2O. b Isolated yields. c Determined by 1H NMR analysis of the product in CDCl3 at 400 MHz or 600 MHz. d Determined by 1H NMR analysis of the product in d6-DMSO at 400 MHz or 600 MHz. | |||||
| 1 | CO2Me | CO2Me | 4a | 79 | 10.34d |
| 2 | CO2Bn | CO2Bn | 4b | 60 | 9.62c |
| 4 | c-C3H5 | c-C3H5 | 4c | 69 | 9.17c |
| 5 | 4-nPrC6H4 | 4-nPrC6H4 | 4d | 75 | 9.54c |
| 6 | C6H5 | C6H5 | 4e | 77 | 9.52c |
| 7 | CO2Me | CO2Bn | 4f | 64 | 10.32d |
| 8 | CO2Me | c-C3H5 | 4g | 64 | 10.26d |
| 9 | CO2Me | C6H5 | 4h | 82 | 10.39d |
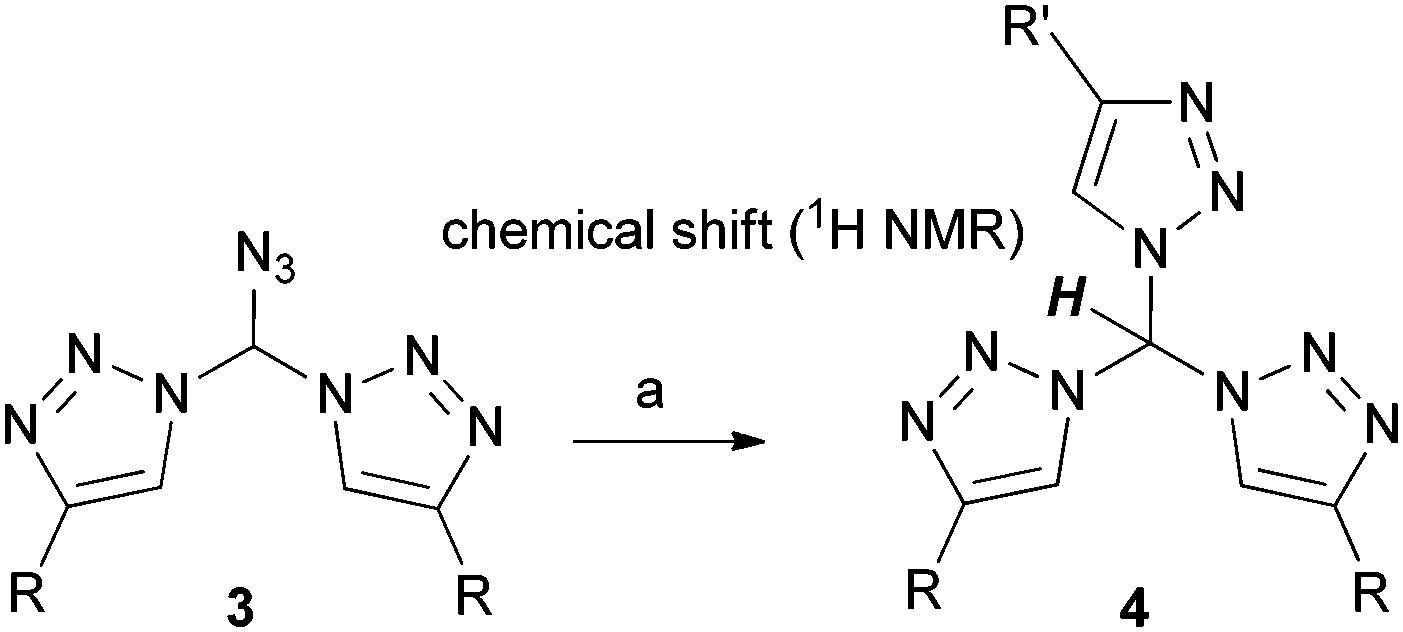
Single crystal analysis of compound 4b was possible and is depicted in Fig. 2. The Cα–N bonds have a length of 1.449 Å/1.453 Å and are, therefore, in perfect agreement with an average C–N single bond. Two triazole rings are pointing in the same direction, whereas one ring has the opposite orientation. The central carbon shows an almost ideal tetrahedral geometry.
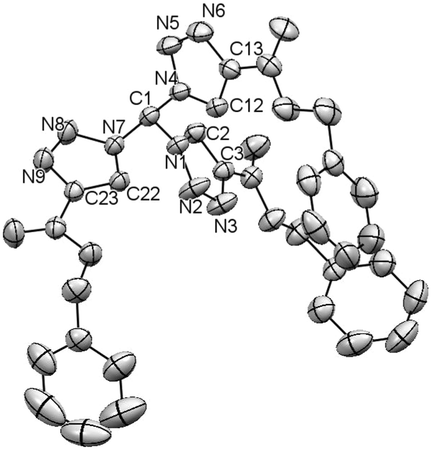 | ||
| Fig. 2 Crystal structure of 4b; ellipsoids are drawn at 50% probability level. Hydrogen atoms are omitted for clarity.15 | ||
To underline the possibilities with our leading sequence toward tristriazoles, we briefly looked into two variations: firstly, an excess of 3a was connected with tripropargyalmine under formation of tristriazole 5 (Fig. 3). The six ester groups in the periphery of the nitrogen rich system allow for further functionalization, thus the overall molecule 5 provides a starting point for dendritic growth. Secondly, we were able to generate tristriazole 6 with three different triazole units attached to the central carbon atom. As shown in Scheme 2, the synthetic strategy was slightly altered, and diazide 1 was combined first under thermal cycloaddition conditions with tosyl acetylene, an appropriately electron-deficient alkyne. Although double cycloaddition was preferred, it was possible to isolate monosubstituted triazole 7 in 32% yield. Of note, a single cycloaddition to azidotriazoles such as 7 was never possible when employing copper-catalyzed CuAAC conditions. In those cases, the exclusive formation of bistriazoles 2 was observed indicating that the second cycloaddition proceeds with a markedly increased rate. It was then possible to convert azide 7 into bistriazole 8 using copper-catalyzed conditions; the subsequent azidation gave rise to azide 9 where two different triazole moieties were incorporated. Finally, copper-catalyzed cycloaddition with phenylacetylene led to the desired geminal tristriazole 6, a chiral (but racemic) compound containing three different triazole units.
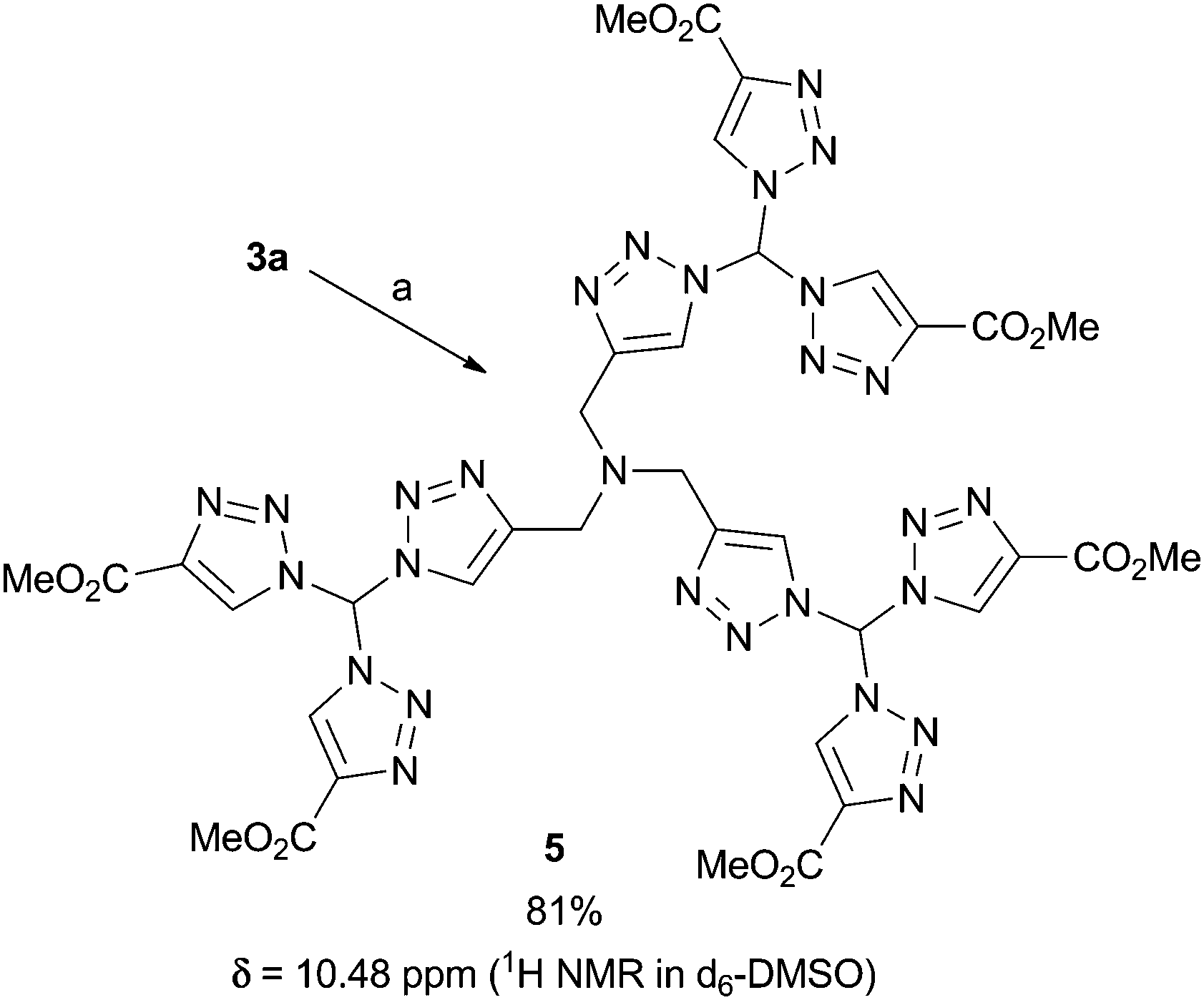 | ||
| Fig. 3 Synthesis of the first evolution dendritic structure 5. Conditions: a: tripropargyl amine (0.2 eq.), CuSO4·5H2O (40 mol%), Na ascorbate (84 mol%), rt, tBuOH/H2O. | ||
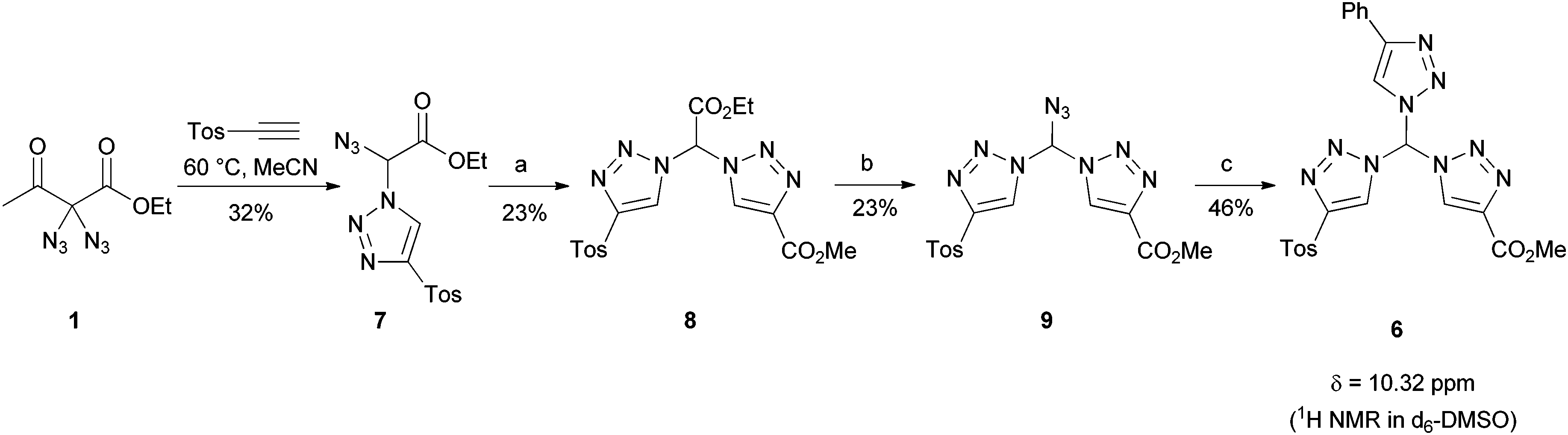 | ||
| Scheme 2 A strategy for the synthesis of geminal tristriazoles with three different substituents. a: MeO2C–CCH (1.1 eq.), CuSO4·5H2O (20 mol%), TBTA (1 mol%), Na ascorbate (42 mol%), rt, tBuOH/MeCN/H2O. b: IBX-SO3K (4.0 eq.), NaI (20 mol%), NaN3 (10.0 eq.), rt, DMSO/H2O. c: Ph–CCH (4.0 eq.), CuSO4·5H2O (40 mol%), Na ascorbate (84 mol%), rt, tBuOH/H2O. | ||
In conclusion, we have described unconventional variants of the now classical CuAAC reaction and an exceptional azidation under oxidative conditions that combine to a straightforward sequence for the synthesis of a diverse array of geminal bis- and tristriazoles, most of which are not accessible with established methods. Several compounds with an unprecedented substitution pattern were presented; synthetic applications and studies on their photochemical properties will be reported in due course.
The supporting work from Yasemin Özkaya (BUW) and Phillip Biallas (BUW) is gratefully acknowledged. We thank Dr A. Kotthaus (BUW) for helpful discussions and Michael Dlugosch (Australian National University) for initial studies.
Notes and references
- (a) P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905 CrossRef CAS PubMed; (b) G. C. Tron, T. Pirali, R. A. Billington, P. L. Canonico, G. Sorba and A. A. Genazzani, Med. Res. Rev., 2008, 28, 278 CrossRef CAS PubMed; (c) T. Michinobu, Macromol. Chem. Phys., 2015, 216, 1387 CrossRef CAS; (d) J. E. Moses and A. D. Moorhouse, Chem. Soc. Rev., 2007, 36, 1249 RSC; (e) C. P. R. Hackenberger and D. Schwarzer, Angew. Chem., Int. Ed., 2008, 47, 10030 CrossRef CAS PubMed; (f) H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004 CrossRef CAS; (g) G. Franc and A. K. Kakkar, Chem. Soc. Rev., 2010, 39, 1536 RSC.
- (a) S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188 CrossRef PubMed; (b) D. K. Kölmel, N. Jung and S. Bräse, Aust. J. Chem., 2014, 67, 328 CrossRef.
- (a) Q.-H. Deng, T. Bleith, H. Wadepohl and L. H. Gade, J. Am. Chem. Soc., 2013, 135, 5356 CrossRef CAS PubMed; (b) S. D. Sarkar and A. Studer, Org. Lett., 2010, 12, 1992 CrossRef PubMed; (c) X. Huang, T. M. Bergsten and J. T. Groves, J. Am. Chem. Soc., 2015, 137, 5300 CrossRef CAS PubMed; (d) M. V. Vita and J. Waser, Org. Lett., 2013, 15, 3246 CrossRef CAS PubMed; (e) W. Song, S. I. Kozhushkov and L. Ackermann, Angew. Chem., Int. Ed., 2013, 52, 6576 CrossRef CAS PubMed; (f) A. Sharma and J. F. Hartwig, Nature, 2015, 517, 600 CrossRef CAS PubMed; (g) M. J. Galligan, R. Akula and H. Ibrahim, Org. Lett., 2014, 16, 600 CrossRef CAS PubMed; (h) J. Barluenga, M. Tomás-Gamasa and C. Valdés, Angew. Chem., Int. Ed., 2012, 51, 5950 CrossRef CAS PubMed; (i) K. Banert, C. Berndt, S. Firdous, M. Hagedorn, Y.-H. Joo, T. Rüffer and H. Lang, Angew. Chem., Int. Ed., 2010, 49, 10206 CrossRef CAS PubMed.
- (a) A. Duschek and S. F. Kirsch, Chem. – Eur. J., 2009, 15, 10713 CrossRef CAS PubMed; (b) S. F. Kirsch, J. Org. Chem., 2005, 70, 10210 CrossRef CAS PubMed.
- A. Bredenkamp, F. Mohr and S. F. Kirsch, Synthesis, 2015, 1937 CAS.
- T. Harschneck, S. Hummel, S. F. Kirsch and P. Klahn, Chem. – Eur. J., 2012, 18, 1187 CrossRef CAS PubMed.
- (a) M. O. Forster, H. E. Fierz and W. P. Joshua, J. Chem. Soc., 1908, 93, 1070 RSC; (b) K. Nishiyama and T. Yamaguchi, Synthesis, 1988, 106 CrossRef CAS; (c) A. Hassner and J. Keogh, J. Org. Chem., 1986, 51, 2767 CrossRef CAS; (d) A. Hassner, M. Stern, H. E. Gottlieb and F. Frolow, J. Org. Chem., 1990, 55, 2304 CrossRef CAS; (e) D. A. Evans, T. C. Britton, J. A. Ellman and R. L. Dorow, J. Am. Chem. Soc., 1990, 112, 4011 CrossRef CAS; (f) D. A. Kamble, P. U. Karabal, P. V. Chouthaiwale and A. Sudalai, Tetrahedron Lett., 2012, 53, 4195 CrossRef CAS; (g) N. Okamoto, T. Sueda, H. Minami, Y. Miwa and R. Yanada, Org. Lett., 2015, 17, 1336 CrossRef CAS PubMed; (h) J.-c. Meng, V. V. Fokin and M. G. Finn, Tetrahedron Lett., 2005, 46, 4543 CrossRef CAS.
- P. Klahn, H. Erhardt, A. Kotthaus and S. F. Kirsch, Angew. Chem., Int. Ed., 2014, 53, 7913 CrossRef CAS PubMed.
- (a) P. Wu, A. K. Feldman, A. K. Nugent, C. J. Hawker, A. Scheel, B. Voit, J. Pyun, J. M. J. Fréchet, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2004, 43, 3928 CrossRef CAS PubMed; (b) J. Totobenazara and A. J. Burke, Tetrahedron Lett., 2015, 56, 2853 CrossRef CAS.
- (a) M. H. Mosslemin and A. E. Movahhed, E-J. Chem., 2012, 9, 301 CrossRef CAS; (b) Z.-Y. Zhong, R. Hong and X.-X. Wang, Tetrahedron Lett., 2012, 51, 6763 CrossRef; (c) G.-C. Kuang, H. A. Michaels, J. T. Simmons, R. J. Clark and L. Zhu, J. Org. Chem., 2010, 75, 6540 CrossRef CAS PubMed; (d) J. E. Hein, J. C. Tripp, L. B. Krasnova, K. B. Sharpless and V. V. Fokin, Angew. Chem., Int. Ed., 2009, 48, 8018 CrossRef CAS PubMed; (e) G. P. Savage and G. T. Wernert, Aust. J. Chem., 2005, 58, 877 CrossRef CAS; (f) R. Ying, L. Zhou, H. Liu, H. Mao, L. Xin and X. Wang, Chin. J. Chem., 2013, 31, 143 CrossRef; (g) Y.-S. Hong, H.-M. Kim, J.-G. Lee, Y.-T. Park and H.-S. Kim, J. Korean Chem. Soc., 1996, 40, 615 CAS; (h) P. Ballesteros, R. M. Claramunt, M. C. Lopez, J. Elguero and G. Gomez-Alarcon, Chem. Pharm. Bull., 1988, 36, 2036 CrossRef CAS PubMed.
- (a) A. R. Katritzky, S. Bobrov, H. Tao and K. Kirichenko, Tetrahedron, 2005, 61, 3305 CrossRef CAS; (b) A. R. Katritzky, S. Bobrov, K. Kirichenko and Y. Ji, J. Org. Chem., 2004, 69, 303 CrossRef CAS PubMed; (c) A. R. Katritzky, C. N. Fali and M. Qi, Tetrahedron Lett., 1998, 39, 2289 CrossRef CAS; (d) A. R. Katritzky and M. Qi, J. Org. Chem., 1997, 62, 4116 CrossRef CAS; (e) A. R. Katritzky, W. Kuzmierkiewicz, B. Rachwal, S. Rachwal and J. Thomson, J. Chem. Soc., Perkin Trans. 1, 1987, 811 RSC.
- (a) A. R. Katritzky, Z. Yang and J. N. Lam, Synthesis, 1990, 666 CrossRef CAS; (b) D. A. Androsov and D. C. Neckers, J. Org. Chem., 2007, 72, 1148 CrossRef CAS PubMed; (c) A. R. Katritzky and L. Xie, Tetrahedron Lett., 1996, 37, 347 CrossRef CAS.
- (a) K. Banert, Y.-H. Joo, T. Rüffer, B. Walfort and H. Lang, Angew. Chem., Int. Ed., 2007, 46, 1168 CrossRef CAS PubMed; (b) E. F. V. Scriven and K. Turnbull, Chem. Rev., 1988, 88, 297 CrossRef CAS; (c) B. Hu and S. G. DiMagno, Org. Biomol. Chem., 2015, 13, 3844 RSC.
- Structure was deposited with CCDC 1428562.
- Structure was deposited with CCDC 1428563.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1428562 and 1428563. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc08163g |
| ‡ Organic azides are always potentially explosive compounds and should be handled with appropriate care and safety equipment. |
| This journal is © The Royal Society of Chemistry 2016 |