
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioinspired multi-block molecules
Takahiro
Muraoka
ab and
Kazushi
Kinbara
*a
aGraduate School of Bioscience and Biotechnology, Tokyo Institute of Technology, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: kkinbara@bio.titech.ac.jp
bPRESTO, Japan Science and Technology Agency, 4-1-8, Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 19th November 2015
Abstract
Multiblock motifs occur in proteins such as silk, elastin and ion channels. These motifs are advantageous to develop the characteristic mechanical properties through the formation of segregated intermolecular assemblies, and utilized to construct cylindrical channels in a membrane by folding and assembly with intra- or intermolecular interactions. As nature shows such elegant examples of multiblock molecules exerting sophisticated functions, synthetic multiblock molecules and copolymers have gained increasing attention as emerging structural motifs for realizing unique properties and functions. Recent notable examples of synthetic multiblock molecules and copolymers are highlighted, where linear molecules not only undergo folding elaborately but also form controlled and compartmentalized self-assemblies to realize characteristic functions in solution, the crystalline state, and membranous media.
 Takahiro Muraoka | Takahiro Muraoka is an Assistant Professor in Tokyo Institute of Technology since 2015. He studied Chemistry and Biotechnology at the University of Tokyo, where he earned PhD investigating molecular machines with interlocked movable units with Prof. Takuzo Aida in 2007. After working as a JSPS postdoctoral researcher in Prof. Samuel I. Stupp group at Northwestern University, he was promoted to an Assistant Professor in Tohoku University. Since 2013, he concurrently serves as a PRESTO researcher in Japan Science and Technology Agency. His present research efforts concentrate on the development of molecules functioning in aqueous media or liposomal membranes. |
 Kazushi Kinbara | Dr Kazushi Kinbara was born in 1967. He received a BS degree in Organic Chemistry from the University of Tokyo in 1991, and obtained a PhD in Organic Chemistry in 1996 under the direction of Professor Kazuhiko Saigo. He then began an academic career at the University of Tokyo, and had been involved until 2001 in the development of optical resolution upon crystallization. In 2001, he moved to Professor Takuzo Aida's group at School of Engineering, the University of Tokyo as a lecturer and associate professor. In 2008, he was promoted to Professor of the Institute of Multidisciplinary Research for Advanced Materials, Tohoku University. In 2015, he moved to Graduate School of Bioscience and Biotechnology, Tokyo Institute of Technology. His research interests include (1) development of biomimetic molecules, (2) supramolecular chemistry of macromolecules, and (3) protein engineering. |
Introduction
Repetitive motifs are commonly observed in functional biomacromolecules like proteins. Collagen, the main structural protein of connective tissues, is known to comprise repeating sequences of glycine–X–Y, where X and Y often correspond to proline and hydroxyproline, respectively.1,2 These repeating structure allows the molecules to fold into a triple helix so as to form robust elongated fibrils. Proteins including repeating domains, namely those with multiblock structures, also show characteristic functions. Elastin, a protein having an elastic property, is composed of two alternating domains; highly flexible domains rich in hydrophobic amino acids such as glycine and proline, and cross-linking domains rich in lysine and alanine.1–3 The multiblock alternating structure allows aggregation of the mobile hydrophobic domains, which is encouraged by the covalent cross-linking via lysine residues. Similar molecular structures are found in silk proteins possessing excellent mechanical properties.2,4 Silk proteins commonly contain well-oriented crystalline domains, whose typical sequences involve (glycine–alanine–glycine–alanine–glycine–serine)n and (alanine)n, and disordered domains rich in amino acids with bulky side groups. The crystalline domains form intra- and intermolecular assemblies surrounded by the buffering disordered domains, to realize the characteristic mechanical properties. In addition to those proteins with characteristic mechanical properties, some transmembrane proteins also possess multiblock molecular structures, which function as ion channels and signal transmitters.5 Conformational study of these transmembrane proteins revealed that they generally consist of alternating extramembranous and intramembranous domains, which contain hydrophilic and hydrophobic amino acid residues in abundance, respectively. The iterative multi-block molecular structure leads to a folded conformation with a multipass transmembrane (MTM) structure in a lipid bilayer, thereby controlling ion transportation and signal transduction.Such multiblock structures seen in nature have stimulated the design of functional synthetic polymers. Typical and successful examples are block copolymers that consist of covalently linked contiguous blocks of chemically heterogeneous and incompatible components. The simplest block copolymers comprising two components, namely AB-type diblock copolymers, have fascinated chemists especially in terms of the formation of nano- and micrometer scale regulated structures, such as cylindrical and spherical micelles, and lamellar structures. Such well-defined molecular and supramolecular architectures have been utilized for conductive materials, drug delivery carriers, and so on.6–12
One of the important applications of the AB-type diblock copolymers is the directed self-assembly (DSA) that combines a lithographical patterning of a substrate with the self-assembly of polymers to realize fine patterns with high resolution.13–16 Graphoepitaxy and chemical epitaxy methods are known as two major approaches for DSA. The graphoepitaxy method utilizes lamellar self-assembly of a block copolymer in a topographical prepattern of the substrate to increase the resolution by subdividing the prepattern. In the chemical epitaxy method, the self-assembly of a block copolymer is guided by a lithographically-defined chemical prepattern to improve the dimension control by reducing variations in the prepattern. Recent progress in DSA enables not only one-dimensional but also two-dimensional patterning.
Multiblock copolymers consisting of repeating (AB)n block pairs and sequential ABC⋯X, that are more resemblant to natural systems, have received increasing attention as an emerging class of functional molecules. Due to a wide structural diversity of the multiblock structures, various copolymers possessing elasticity, conductivity, and drug carrying capability with optimized properties have been developed so far.8,9,17–20 In addition to the numerous examples of these engineered multiblock copolymers, monodisperse small molecules with multiblock structures have also been attracting growing interest in the last decade. Successful recent examples of such small multiblock molecules demonstrate the folding and self-assembly of molecules in controlled manners so as to achieve the specific functions.
There are a number of available structural parameters in the design of multiblock molecules, such as hydrophilicity/hydrophobicity, difference in bulkiness, flexibility/rigidity, morphological difference (linear/cyclic structures), and possible interactions. By programming these parameters into the repeating (AB)n blocks or ABC⋯X block sequences, multimodal conformations and self-assembling structures with distinctive properties and functions can be realized elaborately. A systematic study by Patrickios and coworkers indicates the characteristic correlation of the number of blocks in an (AB)n-type multiblock copolymer with micellar structure in aqueous media.21 They have synthesized alternating multiblock copolymers consisting of two to six blocks of hydrophilic oligomeric 2-(dimethylamino)ethyl methacrylate (D) and oligomeric hydrophobic methyl methacrylate (M). It was revealed that the micelles formed by the diblock copolymer exhibits larger gyration and hydrodynamic radii (Rg and Rh, respectively) than those of the tri-, tetra-, and pentablock copolymers. Meanwhile, the Rg and Rh of the tri-, tetra-, and pentablock copolymers are comparable with one another. The micelles formed by the hexablock copolymer shows the largest Rg and Rh that are almost twice as large as those of the tri-, tetra-, and pentablock copolymers. In combination with the experimentally-evaluated numbers of molecules in the micelles and theoretical study, they proposed the micellar structures of these multiblock copolymers as shown in Fig. 1, representing different assembling modes with/without a folding process depending on the number of blocks. They suggested that tetra- and pentablock copolymers adopt a folded conformation, so that the maximum micellar radius is defined by nearly the same contour length, namely one hydrophilic block plus half of the hydrophobic block. The hexablock copolymer is suggested to favor a conformation with one fold per chain, leading to the larger and compartmentalized micellar structures. Such a “repeating-block effect” for folding and self-assembly is one of the characteristic features of multiblock molecules. An ABC⋯X-type sequential block design also has an advantage to covalently accumulate multiple components into one molecule with controlled positions to allow specific functions that could not be developed if any one of these components is lacking, or if these components are mixed as independent molecules.
 | ||
| Fig. 1 Multiblock copolymers with different repeating numbers of hydrophilic 2-(dimethylamino)ethyl methacrylate (D) and hydrophobic methyl methacrylate (M) segments, and the corresponding micelle-formation processes. For the tetra-, penta- and hexa-block copolymers, the folded conformations of a single copolymer chain encountered in the micelles are also shown. Calculated contour lengths of some segments, and experimentally estimated numbers of molecules in aggregates (Nagg), gyration radii (Rg) and hydrodynamic radii (Rh) are given at the bottom. Copyright 2009 from American Chemical Society ref. 21. | ||
In this Feature Article, we highlight varieties of emerging multiblock small molecules and copolymers by classifying them in terms of block motifs and folding/assembling manners, to describe the progresses in their functionalization and application.
Multiblock molecules foldable by intramolecular interactions
(AB)n-type linear and macrocyclic multiblock motifs have been utilized to develop foldamers by controlling intramolecular interactions.Huc and coworkers developed a multi-block molecule 1 (Fig. 2a) and its oligomeric isomers consisting of rigid aromatic units favorable for stacking, which are connected with hairpin-turn units.22,23 As the hairpin-turn unit, 4,6-dinitro-1,3-phenylenediamine was used because the intramolecular hydrogen bonds between the amine and nitro groups are advantageous for aligning the aromatic components in the parallel orientation. Disubstitution with methyl groups at the ortho-positions to the amino group effectively restricts rotation around the amino–phenyl bond, so as to bring the aromatic component in an orientation perpendicular to the dinitrophenyl unit, thereby forming π–π face-to-face stacking. With these alternatingly repeated components, 1 folds into a refined ladder-like conformation. Indeed, crystallographic analysis of 1 displays a five-stranded sheet-like conformation forming π–π stacking among the aromatic components (Fig. 2b). It is of importance that the multi-turn β-sheet structure is also formed in an organic medium.
 | ||
| Fig. 2 (a) Molecular structure of (AB)n-type multiblock molecule 1. (b) Side view of the crystal structure of 1. Hydrogen atoms and included solvent molecules are omitted for clarity. Copyright 2014 from American Chemical Society ref. 22. | ||
Stepwise folding of multiblock copolymers has been demonstrated by Meijer and coworkers. They developed ABA-type block copolymer 2 that forms orthogonal interactions in A and B segments, respectively, in a stepwise manner (Fig. 3).24,25 The block copolymer 2 incorporates o-nitrobenzyl-protected 2-ureidopyrimidinone (UPy) and benzene-1,3-5-tricarboxamide (BTA) units in A and B blocks, respectively. Temperature reduction prompts the self-assembly of the BTA moieties into helical aggregates, and UPy groups dimerize by hydrogen-bonding after light-triggered deprotection of the o-nitrobenzyl group intramolecularly under dilute conditions. Since these thermo-/photo-induced self-assembly steps take place independently with each other, two-step sequential association of the two self-assembling moieties leads to single-chain folding of the polymer, and the formation of compartmentalized nanometer-sized particles.
 | ||
| Fig. 3 (a) Folding of 2 into a single-chain nanoparticle by orthogonal assemblies at the BTA and UPy groups in response to cooling and light, respectively. (b) Molecular structure of multiblock copolymer 2. (c and d) Interactions in BTA and UPy groups, respectively. Copyright 2013 from American Chemical Society ref. 24. | ||
Multiblock molecules forming intermolecular interactions for functional self-assemblies
ABC-type multiblock molecules
Pioneering examples of small multiblock molecules that self-assemble in solution to form nano- or macroscopic refined structures include rod-coil molecules, where rigid rod-like segments are covalently attached to flexible coil chains. Stupp and coworkers have reported a series of ABC-type triblock rod-coil molecules consisting of an oligo(biphenyl carboxylate) or oligo(phenylene vinylene) rod segment and coil segments of oligostyryl and oligoisoprene, which form one- or two-dimensional ribbon-like supramolecular nanostructures.26–29 The chemical structure of the rod segments influences the morphology of nanostructures, and lowering the rod-to-coil volume ratio results in suppression of the nanostructure formation. Triblock rod-coil molecule 3 forms mushroom-shaped supramolecular assembly, which further organizes into unidirectionally stacked films (Fig. 4). This material is polar, exhibits a nonlinear optical property, and provides an adhesive-tape-like character with nonadhesive-hydrophobic and adhesive-hydrophilic surfaces. Subsequently, the group has developed a different type of triblock molecules, dendron rod-coils consisting of coil-like, rod-like and dendritic segments.30–33 The dendron rod-coils form one-dimensional aggregation through non-covalent interactions, possibly due to the frustration to form two-dimensional assemblies caused by the bulky dendron segment. A dendron rod-coil molecule containing a tetrathiophene unit affords conductive films composed of one-dimensionally aligned π–π stacking by an electric field.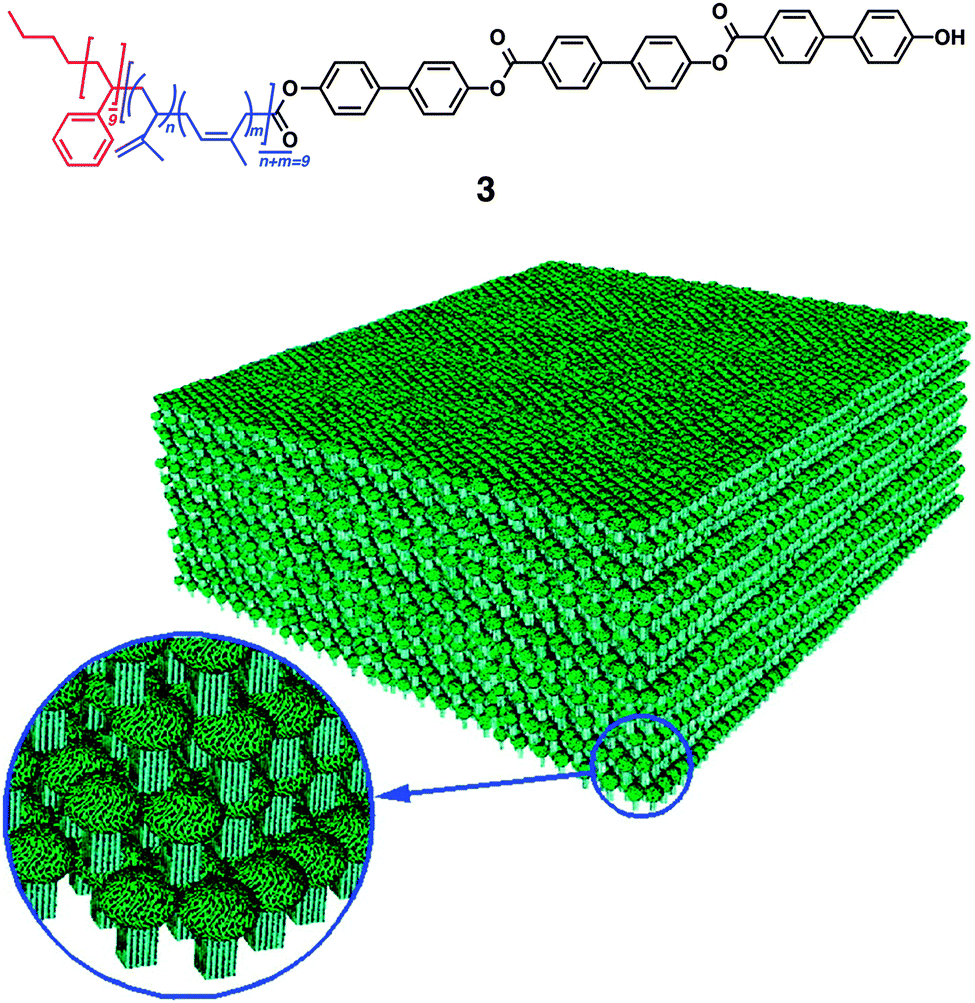 | ||
| Fig. 4 Multi-block rod–coil molecule 3 forming mushroom-shaped assemblies that organizes into a macroscopic film with asymmetric surfaces. Copyright 1997 from AAAS ref. 27. | ||
Lee and coworkers reported diverse supramolecular organization of rod-coil molecules comprising a biphenyl unit as the rod segment, possessing an oligo(propylene oxide) chain at one end (AB-type diblock molecules) or both ends (ABA-type triblock molecules).34–40 The AB-type rod-coil molecules form thermotropic liquid crystalline (LC) assemblies, while the ABA-type molecules form not only LC assemblies but also unusual crystalline assemblies with a body-centered lattice. More recently, the group reported the formation of water-miscible toroids by co-assembly of laterally grafted tri- and di-block amphiphiles 4, 5 and 6 composed of oligophenylene (B), oligo(ethylene glycol) (A), and paraffinic (only for 4 and 5) blocks (C, Fig. 5).41,42 At a low content of 6, a mixture of 4 (or 5) and 6 forms a ribbon-like structure. Increase in the content of 6 (>80 mol%) leads to the formation of toroids, likely due to the increased volume fraction of the hydrophilic portion to induce a curved nano-assembly. The toroid consists of a single layer of the amphiphiles, in which the rod segments align perpendicularly to the ring plane. Transmission electron microscopy reveals that the interior and exterior of toroids are hydrophobic and hydrophilic, respectively. This allows complexation of the toroids with fullerene in the hydrophobic cavity, resulting in the formation of tubular supramolecular nano-assemblies.
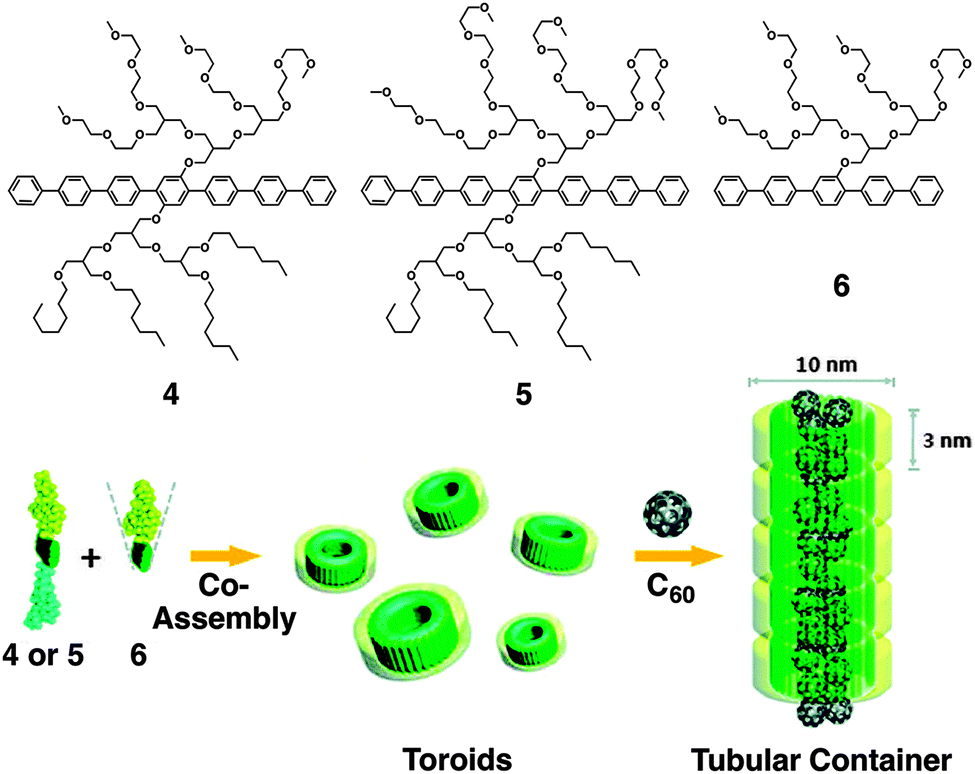 | ||
| Fig. 5 Laterally grafted multi-block amphiphiles 4, 5 and 6. Schematic illustration of the co-assembly of 4 or 5 and 6 into toroids and C60-triggered formation of tubular containers. Copyright 2009 from American Chemical Society ref. 41. | ||
Kataoka and coworkers developed smart drug carriers by deliberate multiblock motifs with an ABC-type sequence like copolymer 7 (Fig. 6),43 consisting of branched PEG, poly(L-glutamic acid) (PLGA) and cholesterol. In an aqueous medium at neutral pH, (1,2-diaminocyclohexane)platinum(II) (DACHPt), an anticancer drug, decreases the intramolecular electrostatic repulsion between the carboxylic moieties in a PLGA block, thereby inducing α-helix formation. In addition to the self-assembling nature of cholesterol, cylindrical α-helices have an advantage for lateral packing, so that 100 nm polymersomes are formed with narrow unimodal size distribution. While DACHPt is accumulated in the membrane, the polymersomes can encapsulate neutral hydrophilic substances such as fluorescent dextran into the inner aqueous cavity. A selective dual delivery of DACHPt and the encapsulated substance to a tumor tissue in mice is successfully demonstrated, thanks to the biocompatibility of the polymer and enhanced permeability and retention (EPR) effect.
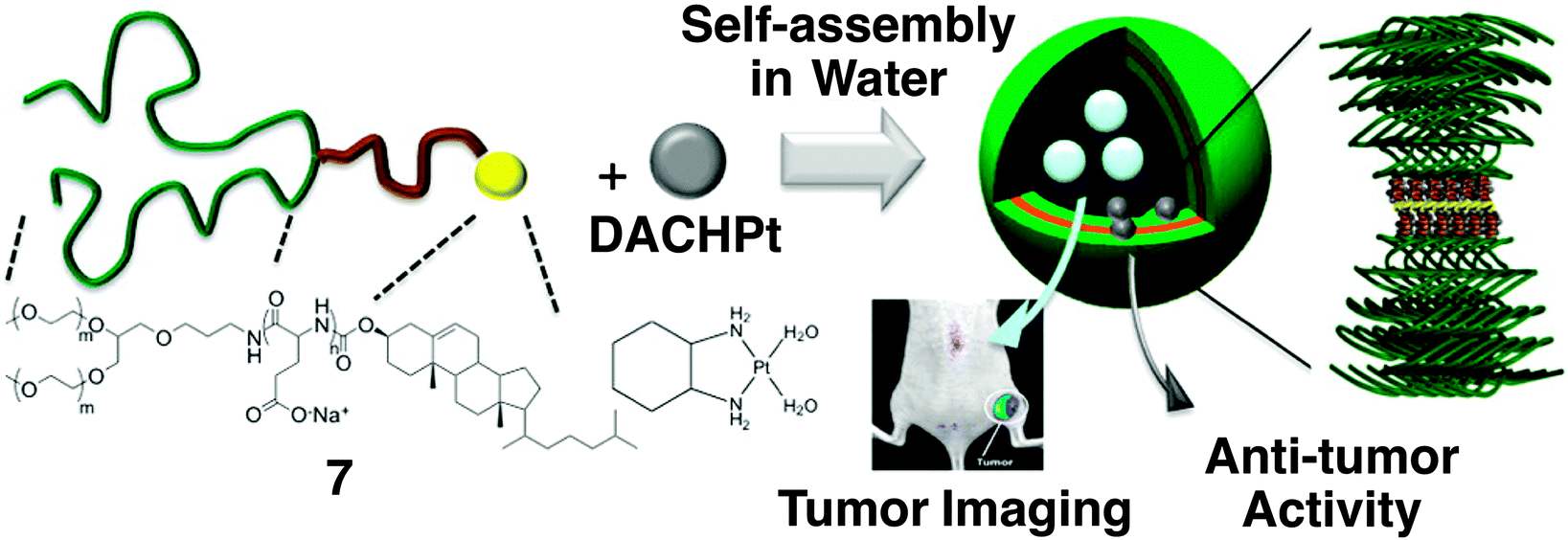 | ||
| Fig. 6 Molecular structures of triblock copolymer 7 consisting of PEG, PLGA and cholesterol blocks, and anti-cancer drug DACHPt, which form a polymersome. A proposed self-assembling structure is illustrated, displaying the formation of a metal complex between a Pt atom in DACHPt and the carboxylic moiety of the PLGA block. Copyright 2012 from American Chemical Society ref. 43. | ||
Hammond and Swager’s group also demonstrated effective in vivo tumor targeting with a multiblock copolymer.44 They designed an ABCBA-type pentablock copolymer including conjugated polymer units that provides highly fluorescent nanoparticles. Namely, the C block is a far-red emitting conjugated polymer, while A is a PEG chain giving water solubility, the stealth effect, and anti-fouling properties. B is introduced as reactive functionality enabling attachment of the ligands and therapeutics for tumor targeting. Indeed, folic acid is appended as the ligand to target folate receptors expressed on many gynecologic cancer cell membranes. In aqueous media, the pentablock copolymer forms 80–200 nm nanoparticles suitable for biological applications. The nanoparticles show prolonged persistence in mice, and in vivo fluorescence imaging of tumor was demonstrated with minimal cytotoxicity.
Thus, elaborate combination and design of three (in principle, even more) components in ABC-type multiblock molecules can realize a variety of hierarchical supramolecular assemblies functioning at nanometer to macroscopic scales.
(AB)n-type multiblock molecules
An (AB)n-type multiblock structure has been used to control not only folding by intramolecular interactions but also finely segregated self-assembling by intermolecular interactions. As described above, nature takes advantage of the (AB)n-type multiblock structure to develop distinguishing mechanical properties such as elasticity and stiffness, as were found in elastin and silk.1–4 The mechanical properties of elastin furnish the tissues with required strength, flexibility and resilience. Tropoelastin, the soluble precursor of elastin, consists of an alternating alignment of flexible segment rich in nonpolar amino acids and alanin-rich crosslinking segment containing a small amount of lysine.45 Kiick and Jia have developed a synthetic mimic of elastin by replacing the flexible segments with PEG that conjugates with the cross-linking domains with an alanine–lysine–(alanine)3–lysine–alanine sequence. The block copolymer is soluble in water, which readily forms a hydrogel upon covalent cross-linking with hexamethylene diisocyanate. The hydrogel is elastic, and the compression modulus is similar to that of a commercial polyurethane elastomer. Interestingly, the hydrogel hardly shows cytotoxicity, although the soluble precursor block copolymer exhibits some toxicity. The mechanical properties and biocompatibility of the block copolymer is suitable as a cell growth scaffold for normal growth and proliferation of a fibroblast.Development of conductive materials is another effective direction for the application of multiblock molecules. An efficient proton conductive material with high thermal stability was developed by Miyatake, Watanabe and coworkers utilizing multiblock (AB)n-type copolymers containing poly(arylene ether sulfone) block appending protogenic sulfonated group (A) and robust hydrophobic polybenzimidazole (PBI) block (B) in an alternating arrangement.46 For efficient proton conduction, the formation of continuous ionic transporting pathways is required. The transporting pathways can likely be constructed by well-developed phase separation between the hydrophilic and hydrophobic blocks of the multiblock copolymer. As expected, the designed poly-sulfone and PBI multiblock copolymer gives separated phases, where the hydrophilic sulfonic acid groups aggregate to form dispersed ionic clusters. The membrane of the multiblock copolymer shows significantly higher conductivity than that of randomly sulfonated PBIs.
Sommerdijk and coworkers developed ABABA-type amphiphilic multiblock copolymers 8 composed of hydrophobic polydisperse poly(methylphenylsilane) (PMS) and hydrophilic nearly monodisperse poly(ethylene glycol) (PEG) segments in the A and B blocks, respectively.47 The major oligomer was (PMPS–PEG)2–PMPS, as schematically shown in Fig. 7. The multiblock copolymer forms micellar-fibrils, vesicles and helical aggregates, upon changing the water content in tetrahydrofuran (THF), which influences the packing and conformation of the PMPS blocks. Below 40% water content in THF, 7 is molecularly dissolved. Between 40 and 80% water content, TEM visualizes the formation of micellar fibrils with a high aspect ratio. The fibrils are constructed by the assembly of the PMPS blocks as a core that are surrounded by PEG blocks. At the water content higher than 80%, helical aggregates, including both left- and right-handed screw senses, are formed. Upon dialysis of the THF/water mixture in water, the formation of vesicles is visualized by TEM. A spectroscopic study indicates that by increasing the water content, PMPS blocks form more highly ordered assemblies. In water, the polysilane blocks adopt a more extended transoid conformation, suggesting that the PMPS blocks form a parallel alignment in the interior of the membranes, which are separated from water by PEG blocks.
 | ||
| Fig. 7 Molecular structure of (AB)n-type multiblock copolymer 8 and its schematic drawing. Copyright 2000 from American Chemical Society ref. 47. | ||
It is known that the amino acid sequence of MTM proteins possesses an (AB)n-type alternating multi-block structure consisting of the domains rich in hydrophobic and hydrophilic amino acids.5 The hydrophobic domains span the plasma membrane, which are connected by rather flexible hydrophilic domains, to form an MTM architecture acting as an ion channel and a signal mediator complex for a signal transduction. One of the typical structures of transmembrane ion channel is a β-barrel structure, where the membrane-spanning domains form a cylindrical β-sheet to construct a channel. Inspired by this architecture, Matile and coworkers have developed pioneering self-assembling motifs of synthetic supramolecular ion channels,48–61 including ABA-type triblock molecules with β-sheet-forming short peptides grafted on a membrane spanning oligo-phenylene core. Non-planarity of the rod scaffold caused by biphenyl torsions and intermolecular β-sheet formation by the peptide units typically promote the formation of a tetrameric cylindrical self-assembly in a bilayer membrane to construct β-barrel-like channels. Taking advantage of this synthetic β-barrel pore, the group has developed a ligand-gating system.62–65 Graft-type triblock molecule 9 bears electron-deficient naphthalenediimide (NDI) acceptors as side chains of the p-octiphenyl rod (Fig. 8). Because of the slipped NDI stacks, 9 yields supramolecular π-helices with a closed pore. Upon complexation with a ligand 10, dialkoxynaphthalene (DAN) donor, a conformational change takes place to untwist the helices so as to open the pore. On the basis of this supramolecular system, photoproduction of a proton gradient across a lipid bilayer was also demonstrated.66p-Octiphenyl rod 9 bearing NDI is introduced in the phospholipid bilayer of vesicles. The vesicles encapsulate quinone as an electron acceptor and ethylenediaminetetraacetic acid (EDTA) is added in the outer phase as a hole acceptor. Ultrafast and relatively long-lived charge separation occurs at the π-architecture quantitatively, thereby promoting external oxidation of EDTA and reduction of quinone at the external and internal phases of the vesicle, respectively. This photosynthetic activity eventually induces the proton gradient. Addition of DAN donor 10 as a ligand allows ligand-gated helix-barrel transition to open the channel, and thus the light-induced proton gradient is canceled.
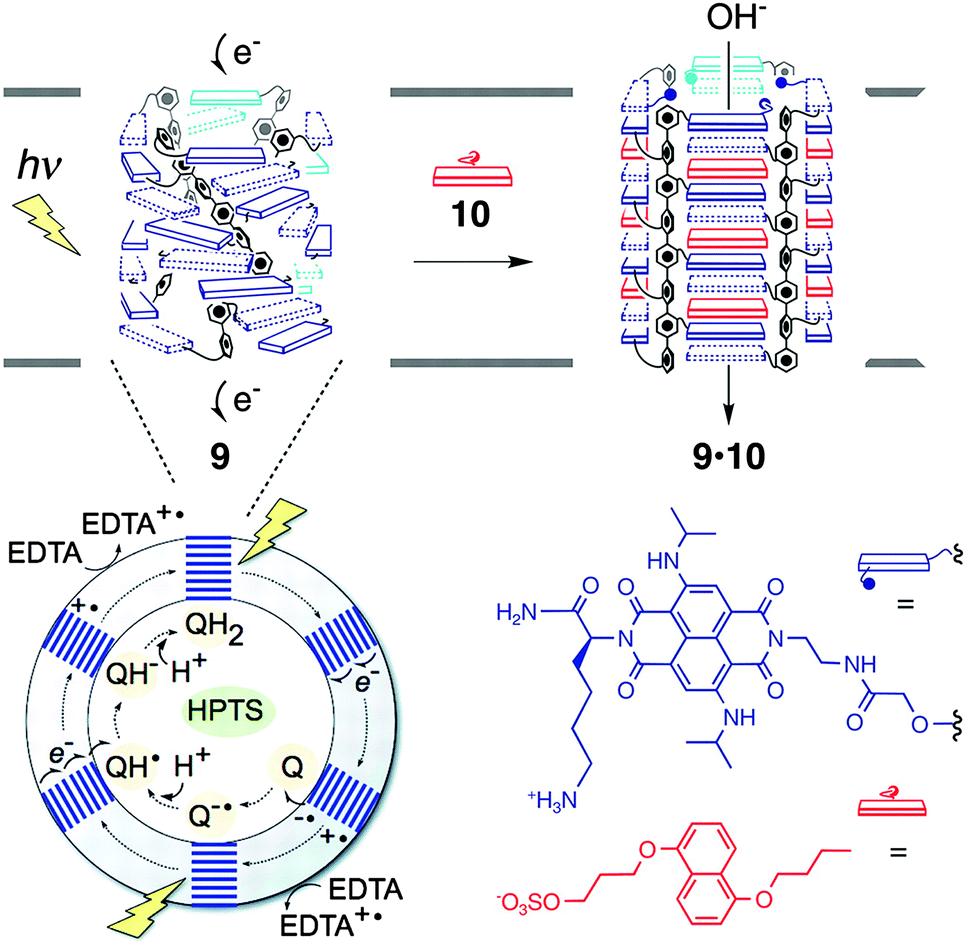 | ||
| Fig. 8 Supramolecular photosystem 9 consisting of a tetrameric assembly of the multiblock monomer in a closed state, which turns into an open state (9·10 complex) by ligand (10) gating. The phospholipid vesicle including 9 encapsulates HPTS for fluorometric detection of pH increase after photo-induced charge separation of 9, which fosters external EDTA oxidation and internal reduction of quinones (Q). Copyright 2006 from AAAS ref. 66. | ||
A different type of ion transporter with multi-block structure was developed by Gokel.67 Three crown ethers are simply connected by alkyl chains to form a heptablock amphiphile, which is successfully incorporated into a bilayer membrane. It is suggested that the inner crown locates inside the membrane and the two terminal rings are at the bilayer surface. Using this heptablock amphiphile, cation transportation across the membrane was demonstrated, where the three crown ethers likely provide donor relays to allow hopping of the cation for the transportation across a membrane.
Simpler and smaller multiblock amphiphiles have been recently developed by our group, which show hysteretic thermal responses in aqueous media.68–70 Amphiphiles 11 and 12 have an (AB)n-type alternating pentablock structure consisting of hydrophilic tetraethylene glycol chains and hydrophobic aromatic groups; phenyl and 4-methoxyphenyl groups in 11 and 12, respectively (Fig. 9a). These pentablock amphiphiles are miscible in water at room temperature, while the aqueous solutions turn into cloudy suspensions upon temperature elevation. Transmittance change at 800 nm displays a steep drop during the temperature elevation (Fig. 9b). The transition temperature depends on the concentration, where a solution at a higher concentration shows the transition at a lower temperature. Interestingly, the transition curves monitored by transmittance show a hysteretic feature during the heating and cooling processes (except 5.0 mM), where the transition curves in the cooling process are more gentle than those in the heating process. Dynamic light scattering (DLS) and phase-contrast microscopy (PCM) visualize that 11 and 12 form micrometer-scale aggregates by heating. As shown in Fig. 9c, PCM visualizes that 11 at 20 mM in water forms aggregates at 30.0 °C, and the quantity of the aggregates increases by temperature elevation. In the cooling process, the assembly and fusion of the aggregates are observed [indicated by white arrows with a square bracket in Fig. 9c at 38.5 °C (cooling)]. Such “inter-aggregate” interactions causing micrometer-scale assemblies are observed only in the cooling process, likely resulting in the hysteretic transmittance changes. The concentration dependence and the assembly of aggregates in the cooling process are considered to be unique features of non-polymeric small molecules showing cloud points in water. The variable-temperature nuclear magnetic resonance (NMR) studies suggest that increased hydrophobicity at the tetraethylene glycol chains due to the gauche-to-anti conformational changes of the ethylene moieties and aromatic interactions likely prompt the aggregation of 11 and 12 upon heating. Importantly, the thermo-driven micrometer-scale aggregation formation of 11 and 12 is applicable for selective extraction of peptides based on the content of hydrophobic and aromatic residues.
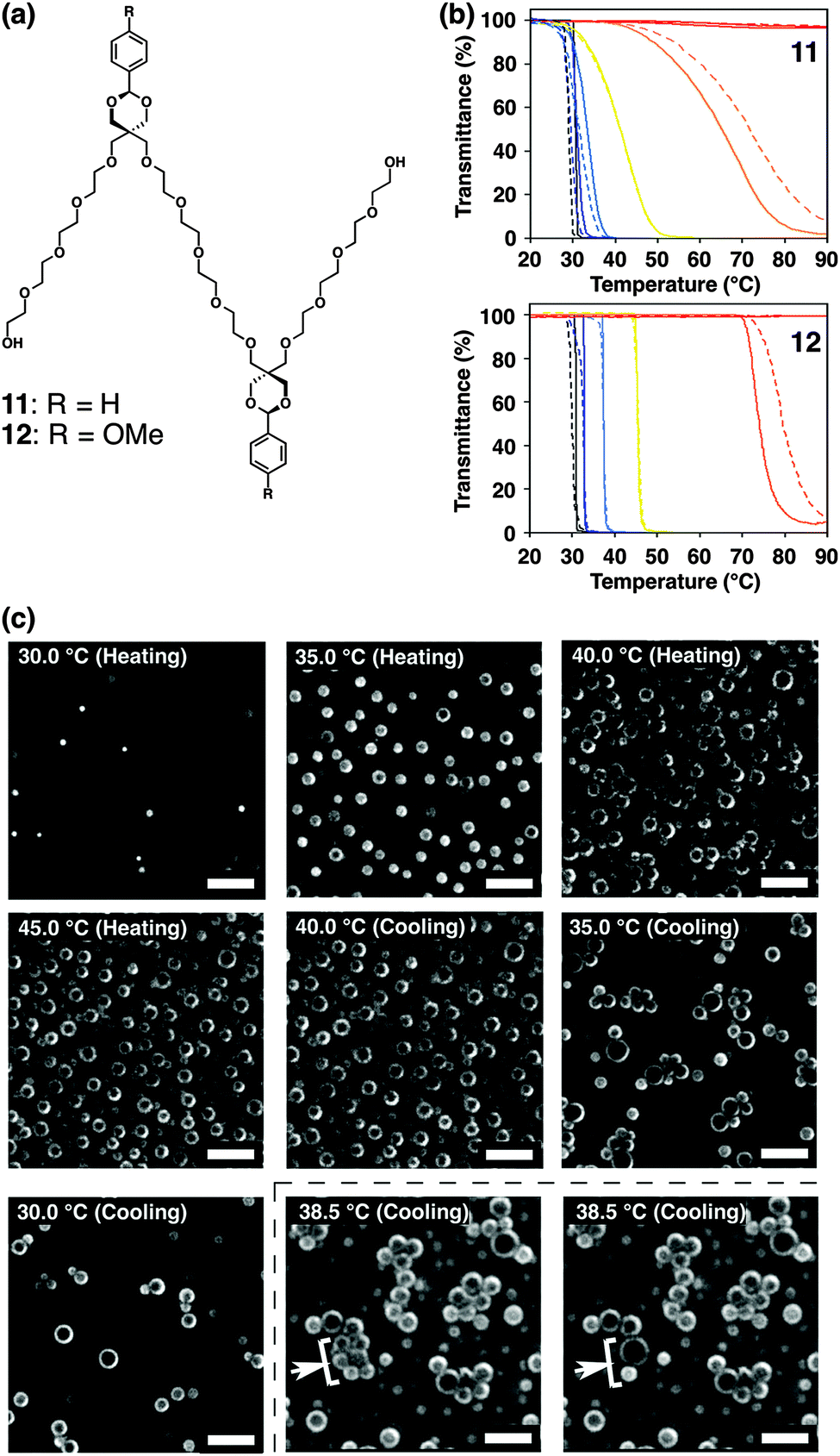 | ||
| Fig. 9 (a) Molecular structures of pentablock molecules 11 and 12. (b) Transmittance changes of (upper) 11 and (lower) 12 at 1.5 (red), 3.0 (orange), 5.0 (yellow), 10 (pale blue), 15 (blue), and 20 mM (black) in water upon temperature elevation (solid lines) and reduction (broken lines) at the rate of 1.0 °C min−1. (c) Phase-contrast micrographs of 11 in water (20 mM) upon temperature elevation and reduction (20–45 °C) at the rate of 1.0 °C min−1. The white arrows and square brackets at 38.5 °C in the cooling step indicate the fusing spherical objects. Scale bars: 20 μm ref. 68. | ||
Foldable multiblock molecules forming higher-order structures by self-assembly
Recently, we have developed synthetic mimics of α-helical MTM ion channels.71,72 The molecular structure of α-helical MTM proteins is iterative multi blocks between extramembranous hydrophilic domains and membrane-spanning rod domains. As a synthetic mimic of this structure, (AB)n-type multi-block amphiphiles 13 and 14 consisting of hydrophilic and flexible oligoethyelene glycols conjugated with hydrophobic, rigid, and fluorescent aromatic 1,4-bis(4-phenyl ethynyl) benzene (BPEB) units (Fig. 10a) were prepared. Fluorescent microscopy and a fluorescence depth quenching study indicate that 13 and 14 are embedded in a DOPC liposomal membrane, with the BPEB units penetrating the hydrophobic layer. Intermolecular assembly of the BPEB unit of 13 in the membrane is indicated by red-shift of the fluorescence upon increase in the concentration. Meanwhile, 14 shows significantly red-shifted fluorescence even at a low concentration and the wavelength of the maximal fluorescence hardly changes upon concentration enhancement, thus suggesting the intramolecular stacking of the BPEB units in 14. Namely, 14 likely adopts an MTM-like conformation in the lipid bilayer. Interestingly, 14 forms a tetrameric assembly (Fig. 10b) acting as a supramolecular ion channel, although 13 hardly shows ion transportation capability even at high concentration. It is suggested that the panel-like shape of 14 formed by folding into an MTM-like conformation is advantageous to construct the supramolecular ion channel. | ||
| Fig. 10 (a) Molecular structures of (AB)n-type multiblock amphiphiles 13 and 14 with different repeating numbers (one and four). (b) Schematic illustration of dynamic supramolecular ion channel formation of 14 by tetrameric assembly ref. 72. | ||
Changing the rigid linear hydrophobic segment into a flexible component leads to the formation of a supramolecular half-channel.73 Amphiphile 15 consists of five blocks, where the central hydrophobic unit bears a flexible chiral aliphatic part with aromatic units (Fig. 11a). At the junction point between the aromatic and octaethylene glycol units, phosphate groups are inserted as ligand-binding sites capturing aromatic amines through electrostatic, hydrogen-bonding and aromatic interactions. At the both ends of the molecule, hydrophobic triisopropylsilyl (TIPS) groups are appended. Microscopic observation, spectroscopic study and Langmuir–Blodgett assay indicate that the central and terminal hydrophobic parts are embedded in a bilayer membrane so that the molecule adopts a “M-shape” conformation with intramolecular aromatic stacking. In contrast to membrane-spanning foldamer 14, membrane-half-spanning foldamer 15 hardly shows the capability of ion transportation by itself. Interestingly, however, 15 embedded in a membrane interacts with 2-phenethyl amine (PA), thereby changing the conformation around the aromatic units so as to initiate the ion transportation. It is indicated that trimeric assembly of 15 forms a half channel with PA in a lipid monolayer of the membrane, that stacks with another half channel in the opposite-side monolayer, to form an active ion channel (Fig. 11b). Addition of β-cyclodextrin to dissociate PA from 15 stops the ion transportation, thus reversible ligand-gated ion transportation is successfully demonstrated. Of importance, amphiphile 16, adopting “V-shape” conformation due to the hydrophilic termini with no TIPS groups, hardly shows ion transportation capability either in the absence or presence of the ligand. This suggests the importance of the terminal TIPS groups, likely acting as anchors to assist ion-channel formation.
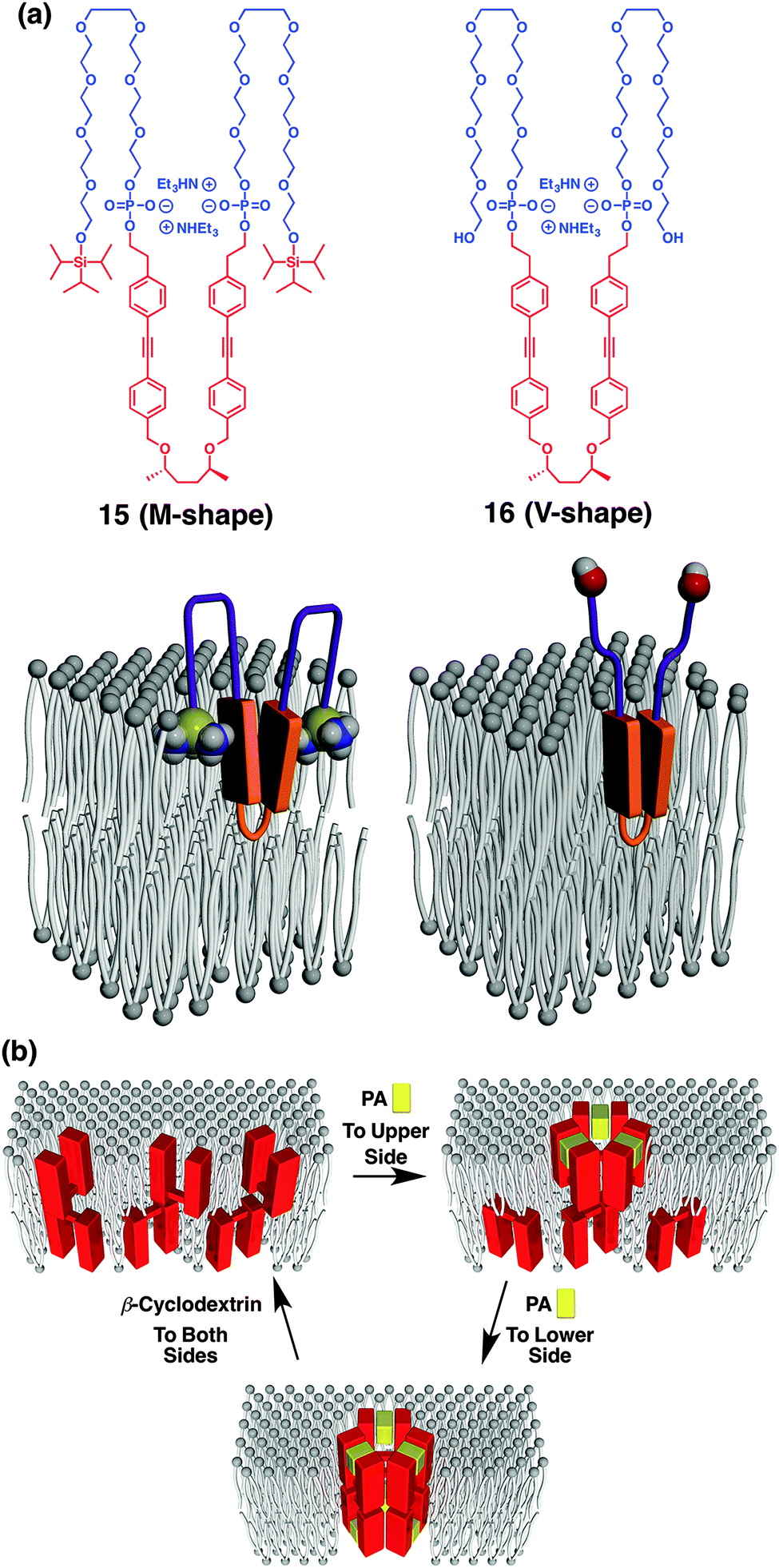 | ||
| Fig. 11 (a) Molecular structures of (AB)n-type multiblock amphiphiles 15 and 16 with different terminal structures, and models of 15 and 16 in a membrane adopting M-shape and V-shape conformations, respectively. (b) Schematic illustration of step-wise supramolecular ion channel formation by ligand (PA) addition to upper and lower sides of the membrane, and its reversible disassembly upon ligand-removal by β-cyclodextrin. PA, 2-phenethylamine ref. 73. | ||
In the course of our study to develop synthetic mimics of α-helical MTM ion channels, we serendipitously found that macrocyclic multi-block amphiphiles with alternative oligoethyelene glycol and aromatic units show dynamic thermal and photochemical responses. A single crystal of macrocyclic multi-block amphiphile 17 exhibits reversible thermal phase-transition without loss of the single crystallinity, leading to macroscopic mechanical motion.7417 consists of alternately aligned tetraethylene glycol chains and aromatic groups such as BPEB and azobenzene units (Fig. 12a). Differential scanning calorimetry displays a thermal transition of single crystalline 17 at 333 K below its melting point (422 K). A thermal transition is observed at 329 K in the cooling process from 370 K indicating a reversible transition. When a needle-shape single crystal of 17 is heated on a hot stage, bending and straightening motions are observed (Fig. 12b). By using a relatively large single crystal, the range of the motion reaches a millimeter-scale. The similar macroscopic motion occurs in the cooling process as well. X-ray crystallographic analyses clearly visualize the mechanism of the crystalline motion at the molecular level (Fig. 12c), where 107-fold amplification of the range of the motions, namely, from the angstrom-scale conformational changes to the millimeter-scale macroscopic motions are achieved in this case. The space group of the low-temperature phase crystal belongs to Pc. The asymmetric unit includes a pair of molecules (A and B), which stacks along the a-axis. The azobenzene and BPEB units align alternately along the a-axis, forming intra- and intermolecular π–π interactions. Importantly, the longest axis of the needle-shape single crystal is parallel to the a-axis. The space group of single crystal at the high-temperature phase remains unchanged, but interestingly, C45 in the molecule, a carbon atom in the tetraethylene glycol chain in proximity to the triazole ring, flips during this thermal transition. Other carbon atoms such as C48 and C57 also exhibit flipping. In contrast, the azobenzene and BPEB units are almost unchanged in terms of the geometry and conformations, which allows for the retention of π–π stacking. The conformational changes cause changes in the alignment of the molecules. In the thermal transition from the low- to the high-temperature phases, the largest change in the length is observed for the a-axis, which expands by 2.6%. This change in the lattice parameters likely generates a mechanical force to induce the bending motion. In the heating process on a hot stage, the thermal structural transformation should start from the molecules located at the heated surface of the single crystal to cause internal strain. Since the a-axis corresponds to the long-axis of the single crystal, the expansion along the a-axis at the heated surface likely prompts curving of the needle crystal along the long-axis to raise a tip of the crystal from the hot stage. The internal strain is gradually released, when the heat reaches to the top side, to complete the phase transition in the whole crystal, thereby straightening the crystal again. Thus, both bending and straightening occur in a single heating process. The deformation of a crystal with maintaining its single crystallinity is likely attained by the characteristic crystal structure originated from the multiblock molecular structure, including the well-stacked aromatic columns separated by rather flexible tetraethylene glycol domains. This packing feature is similar to the structure of elastin composed of separated covalently cross-linked domain and flexible region.
 | ||
| Fig. 12 (a) Molecular structure of macrocyclic multiblock molecule 17. (b) Snapshots of a macroscopic motion of a 15 mm long single crystal of 17 on a hot stage upon heating and cooling processes between 326.0 and 338.0 K. One end of the crystal is adhered by glue. Scale bars: 5.0 mm. (c) Merged X-ray crystal structures of 17 in the crystals of the low-temperature (blue, 273 K) and high-temperature phases (red, 343 K) viewed down the a-axis ref. 74. | ||
Macrocyclic multi-block amphiphile 18, that consists of the same molecular components as 17 in a larger macrocycle, was revealed to cause light-triggered giant vesicle formation (Fig. 13).75,76 In the process of a gentle hydration method using a mixture of DOPC and 18, shapeless particles are prepared in aqueous medium (Fig. 13a). Upon irradiation of these particles with UV light (330–385 nm), micrometer-size vesicles are readily formed from their surfaces, as displayed by phase-contrast microscopy (Fig. 13b–f). The size of the vesicles continuously increases during irradiation, and eventually, the vesicles launch from the particles. Exposure of the film to sonication in the initial stage of hydration is effective to encourage the light-triggered vesicle formation. Importantly, neither heating nor IR-laser irradiation induce the vesicle formation, suggesting that light is the essential trigger. Taking advantage of the benefit to use light, single vesicle formation with micrometer-scale spatiotemporal control is demonstrated. Irradiation of the particle with 405 nm laser focused with 1 μm diameter readily triggers the formation of a single vesicle, which eventually detaches from the particle. Transmission electron microscopy visualizes that the nonirradiated particles are enveloped with multilayer membranes, which likely turn into the vesicles. Control experiments indicate that the BPEB units are the essential components for the light-triggered vesicle formation and the azobenzene unit likely encourages the formation through photoisomerization. In addition, the phase of the phospholipid membrane also influences the vesicle formation. Fluid phase membranes such as DOPC or a mixture of DSPC and cholesterol at 20 °C afford the vesicles upon photo-irradiation in the presence of 17, while DSPC at 20 °C in a solid phase hardly shows the vesicle formation.
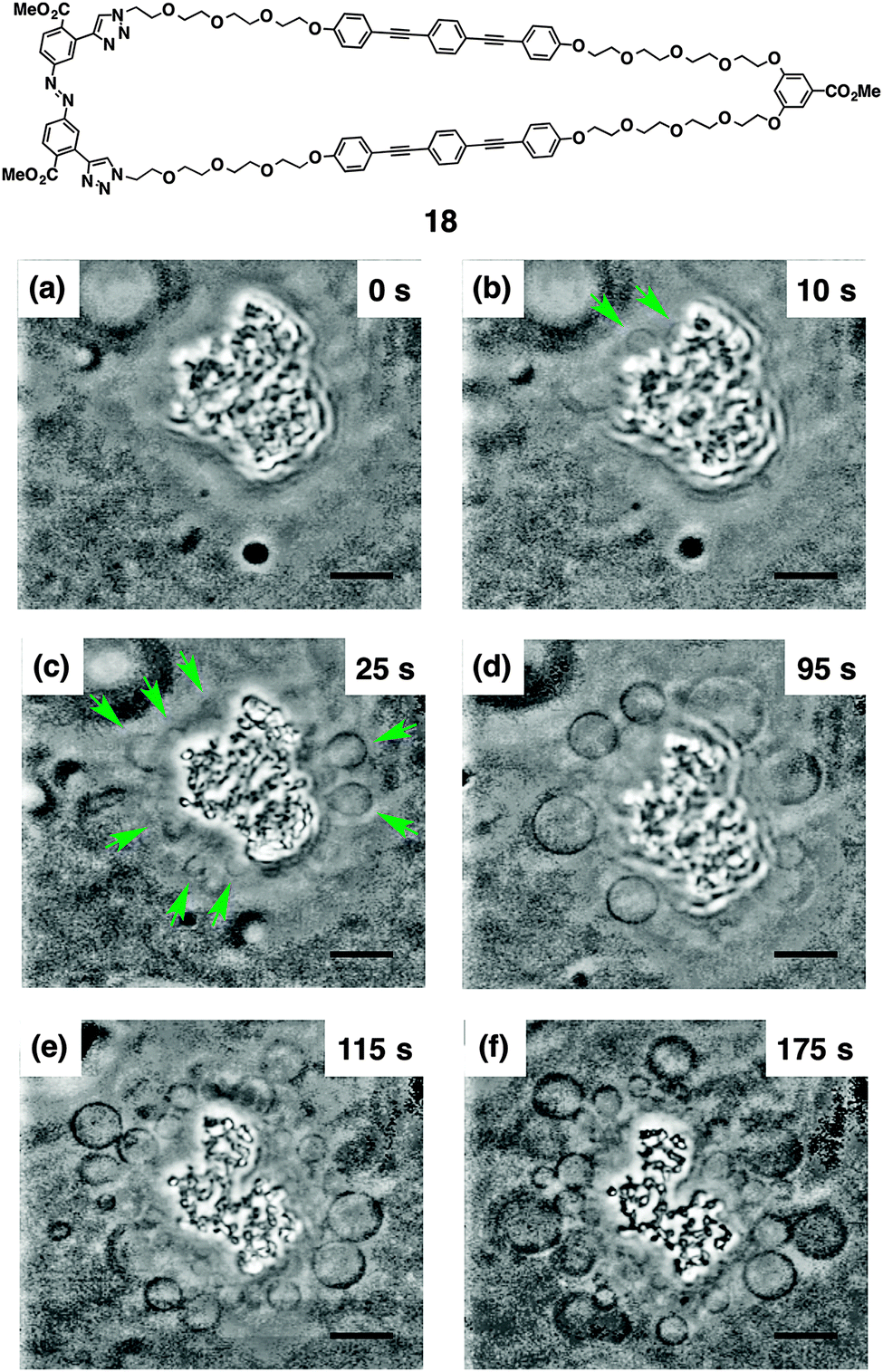 | ||
| Fig. 13 Molecular structure of 18. Time-course phase-contrast microscopic observation of the light-triggered μm-size vesicle formation from a shapeless particle composed of DOPC and 18 in an aqueous solution of glucose and sucrose ([DOPC] = 200 μM, [18] = 40 μM, [glucose] = [sucrose] = 100 mM). Snapshots were taken after irradiation (330–385 nm) at 20 °C for (a) 0, (b) 10, (c) 25, (d) 95, (e) 115, and (f) 175 s, respectively. The green arrows indicate the generated μm-size vesicles. Scale bars, 10 μm ref. 75. | ||
Conclusions and perspective
Several proteins take advantage of the multi-block motifs to encourage folding and self-assembly by intramolecular and intermolecular interactions, thereby acquiring specific mechanical properties and constructing channels for substance transportation. Inspired by such natural molecular architectures, synthetic multi-block molecules and polymers, which have the capability of folding and self-assembly in solution, membranous media and crystalline state in well-controlled manners, have been developed. In the self-assembling systems, there are two distinct molecular-design concepts. The ABC-type sequential structure allows the conjugation of triple and more block components; elaborate combination of the multi-blocks with an adequate sequence leads to development of multi-dimensional self-assemblies such as tubular, film, and spherical structures, yielding versatile microscopic and macroscopic functions. The (AB)n-type structure consisting of iterative dual orthogonal blocks provides segregated macroscopic assembly of the two blocks useful for the development of elastic and conductive materials. Discrete architectures have also been constructed by the self-assembly of (AB)n-type multi-block molecules, leading to the realization of functional supramolecular channels. In such systems, combination of folding and self-assembly, as seen in the hierarchical secondary and tertiary structures in proteins, has been successfully demonstrated, realizing the formation of dynamic supramolecular ion channels. The structural simplicity and diversity of monodisperse multi-block molecules are attractive due to their capability of folding and self-assembling with segregated structures, endowing with sophisticated functions.As overviewed in this article, the multi-block structures have been adopted in the monodisperse small molecular systems as well as in the polydisperse macromolecular systems. Although sophisticated functions have been demonstrated in both cases by controlling folding and assembly, the monodisperse systems are likely advantageous to form closed discrete supramolecular structures such as supramolecular ion channels and toroidal assemblies with a uniform size as demonstrated by 4–6, 9, 14, 15. The most significant advancement of the multi-block systems, which could not be achieved by the small mono-domain molecules, is the capability to form higher-order structures with folded conformations. The sheet-like intramolecularly-stacked architecture and the MTM structure formed by 1, 14 and 15 are the typical examples, which cannot be constructed by small mono-domain molecules. A more complex system has been demonstrated by 2, accomplishing the multi-step stimuli-controlled folding with orthogonal assemblies. A monodisperse system utilizing this strategy would have a great potential for the development of sophisticated systems like proteins.
It would be also meaningful to compare the characteristics of the behaviors of those multiblock molecules in different media. In solution, the high mobility allows molecular-scale dynamic controls such as stepwise folding, stimuli-induced assembling/disassembling, and guest encapsulation in a self-assembled host. In membrane, although the molecular motions are restricted in the two-dimensional media, the dynamic processes such as self-assembly and stimuli-responsive conformational change are possible to allow for achievement of a dynamic channel function, as demonstrated by elaborately designed monodisperse multi-block molecules. Meanwhile, in crystalline or bulk states, the conformation and alignment of molecules is mostly fixed, so that anisotropic macroscopic properties can be developed. Importantly, a dynamic response such as the conformational change is possible in some cases, which results in the macroscopic morphological change of crystals. This would lead to the development of the stimuli-responsive crystalline materials with mechanical functions.
What comes next? Proteins can be regarded as ultimate foldamers/assemblies composed of monodisperse “multi-domain” macromolecules. The domains and their controlled assembly allow hierarchical construction of the whole structure, through the formation of the secondary, tertiary and quaternary structures. As a consequence, the structures of the proteins have diversity in the morphology, and even a single protein displays a structural diversity due to the conformational flexibility responsive to the external stimuli, leading to a wide variety of sophisticated functions. In order to approach such diverse but well-controlled structure formation of proteins by synthetic molecules, the molecular design concept based on the multi-block architecture likely gives a promising direction. Existing synthetic foldamers constructed by multi-block molecules correspond to the domains of proteins forming the secondary structures. Covalent integration of the synthetic foldamers into a single molecule affords a multi-domain structure, and subsequent assembly of the folded domains produces a higher-order structure corresponding to the tertiary structure. Finally, the assembly of such folded molecules would give the quaternary structure. Such a multi-domain design strategy allowing the controlled multi-step folding gives a potential future direction for multi-block molecules inspired by nature.
Acknowledgements
This work was partially supported by Grant-in-Aid for Scientific Research on Innovative Areas “Spying Minority in Biological Phenomena (No. 3306)” (23115003 to KK), Grant-in-Aid for Scientific Research on Innovative Areas “π-System Figuration (No. 2601) (26102001 to TM), Grant-in-Aid for Scientific Research C (TM) from MEXT, JST PRESTO program “Molecular Technology and Creation of New Functions” (TM), and the Mitsubishi Foundation (KK).Notes and references
- D. R. Eyre, M. A. Paz and P. M. Gallop, Annu. Rev. Biochem., 1984, 53, 717–748 CrossRef CAS PubMed.
- R. D. B. Fraser and T. P. MacRae, Conformation in Fibrous Proteins and Related Synthetic Polypeptides, Academic Press, New York, 1973 Search PubMed.
- D. W. Urry, J. Protein Chem., 1988, 7, 1–34 CrossRef CAS PubMed.
- Silk Polymers: Materials Science and Biotechnology, ed. D. Kaplan, W. W. Adams, B. Farmer and C. Viney, ACS Symposium Series 544, American Chemical Society, Washington, DC, 1994 Search PubMed.
- B. Alberts, A. Johnson, J. Lewis, D. Morgan, M. Raff, K. Roberts and P. Walter, Mol. Biol. Cell, Garland Science, New York, 6th edn, 2014 Search PubMed.
- R. E. Zeebe, J. C. Zachos, K. Caldeira and T. Tyrrell, Science, 2008, 321, 50–51 CrossRef PubMed.
- J. S. Lee and J. Feijen, J. Controlled Release, 2012, 161, 473–483 CrossRef CAS PubMed.
- J.-F. Gohy and Y. Zhao, Chem. Soc. Rev., 2013, 42, 7117–7129 RSC.
- Z. Ge and S. Liu, Chem. Soc. Rev., 2013, 42, 7289–7325 RSC.
- M. J. Robb, S.-Y. Ku and C. J. Hawker, Adv. Mater., 2013, 25, 5686–5700 CrossRef CAS PubMed.
- B.-K. Cho, RSC Adv., 2014, 4, 395–405 RSC.
- N. Yamashita, S. Watanabe, K. Nagai, M. Komura, T. Iyoda, K. Aida, Y. Tada and H. Yoshida, J. Mater. Chem. C, 2015, 3, 2837–2847 RSC.
- J. Y. Cheng, C. T. Rettner, D. P. Sanders, H.-C. Kim and W. D. Hinsberg, Adv. Mater., 2008, 20, 3155–3158 CrossRef CAS.
- J. Xu, S. Park, S. Wang, T. P. Russell, B. M. Ocko and A. Checco, Adv. Mater., 2010, 22, 2268–2272 CrossRef CAS PubMed.
- H. Yi, X.-Y. Bao, J. Zhang, C. Bencher, L.-W. Chang, X. Chen, R. Tiberio, J. Conway, H. Dai, Y. Chen, S. Mitra and H.-S. P. Wong, Adv. Mater., 2012, 24, 3107–3114 CrossRef CAS PubMed.
- G. S. Doerk, J. Y. Cheng, G. Singh, C. T. Rettner, J. W. Pitera, S. Balakrishnan, N. Arellano and D. P. Sanders, Nat. Commun., 2014, 5, 5805 CrossRef CAS PubMed.
- O. Rathore and D. Y. Sogah, J. Am. Chem. Soc., 2001, 123, 5231–5239 CrossRef CAS PubMed.
- L. Ayres, M. R. J. Vos, P. J. H. M. Adams, I. O. Shklyarevskiy and J. C. M. van Hest, Macromolecules, 2003, 36, 5967–5973 CrossRef CAS.
- H. Yu, S. Asaoka, A. Shishido, T. Iyoda and T. Ikeda, Small, 2007, 5, 768–771 CrossRef PubMed.
- N. Li and M. D. Guiver, Macromolecules, 2014, 47, 2175–2198 CrossRef CAS.
- N. A. Hadjiantoniou, A. I. Triftaridou, D. Kafouris, M. Gradzielski and C. S. Patrickios, Macromolecules, 2009, 42, 5492–5498 CrossRef CAS.
- L. Sebaoun, V. Maurizot, T. Granier, B. Kauffmann and I. Huc, J. Am. Chem. Soc., 2014, 136, 2168–2174 CrossRef CAS PubMed.
- L. Sebaoun, B. Kauffmann, T. Delclos, V. Maurizot and I. Huc, Org. Lett., 2014, 16, 2326–2329 CrossRef CAS PubMed.
- N. Hosono, M. A. J. Gillissen, Y. Li, S. S. Sheiko, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2013, 135, 501–510 CrossRef CAS PubMed.
- N. Hosono, P. J. M. Stals, A. R. A. Palmans and E. W. Meijer, Chem. – Asian J., 2014, 9, 1099–1107 CrossRef CAS PubMed.
- S. I. Stupp, S. Son, H. C. Lin and L. S. Li, Science, 1993, 259, 59–63 CAS.
- S. I. Stupp, V. LeBonheur, K. Walker, L. S. Li, K. E. Huggins, M. Keser and A. Amstutz, Science, 1997, 276, 384–389 CrossRef CAS PubMed.
- G. N. Tew, L. Li and S. I. Stupp, J. Am. Chem. Soc., 1998, 120, 5601–5602 CrossRef CAS.
- G. N. Tew, M. U. Pralle and S. I. Stupp, J. Am. Chem. Soc., 1999, 121, 9852–9866 CrossRef CAS.
- E. R. Zubarev, M. U. Pralle, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2001, 123, 4105–4106 CrossRef CAS PubMed.
- E. R. Zubarev and S. I. Stupp, J. Am. Chem. Soc., 2002, 124, 5762–5773 CrossRef CAS PubMed.
- B. W. Messmore, J. F. Hulvat, E. D. Sone and S. I. Stupp, J. Am. Chem. Soc., 2004, 126, 14452–14458 CrossRef CAS PubMed.
- E. R. Zubarev, E. D. Sone and S. I. Stupp, Chem. – Eur. J., 2006, 12, 7313–7327 CrossRef CAS PubMed.
- M. Lee, N.-K. Oh and W.-C. Zin, Chem. Commun., 1996, 1787–1788 RSC.
- M. Lee, B.-K. Cho, H. Kim and W.-C. Zin, Angew. Chem., Int. Ed., 1998, 37, 638–640 CrossRef CAS.
- M. Lee, D.-W. Lee and B.-K. Cho, J. Am. Chem. Soc., 1998, 120, 13258–13259 CrossRef CAS.
- M. Lee and B.-K. Cho, Chem. Mater., 1998, 10, 1894–1903 CrossRef CAS.
- M. Lee, B.-K. Cho, Y.-G. Jang and W.-C. Zin, J. Am. Chem. Soc., 2000, 122, 7449–7455 CrossRef CAS.
- B.-K. Cho and M. Lee, J. Am. Chem. Soc., 2001, 123, 9677–9678 CrossRef CAS PubMed.
- M. Lee, B.-K. Cho, N.-K. Oh and W.-C. Zin, Macromolecules, 2001, 34, 1987–1995 CrossRef CAS.
- E. Lee, J.-K. Kim and M. Lee, J. Am. Chem. Soc., 2009, 131, 18242–18243 CrossRef CAS PubMed.
- Y. Kim, W. Li, S. Shin and M. Lee, Acc. Chem. Res., 2013, 46, 2888–2897 CrossRef CAS PubMed.
- K. Osada, H. Cabral, Y. Mochida, S. Lee, K. Nagata, T. Matsuura, M. Yamamoto, Y. Anraku, A. Kishimura, N. Nishiyama and K. Kataoka, J. Am. Chem. Soc., 2012, 134, 13172–13175 CrossRef CAS PubMed.
- E. Ahmed, S. W. Morton, P. T. Hammond and T. M. Swager, Adv. Mater., 2013, 25, 4504–4510 CrossRef CAS PubMed.
- S. E. Grieshaber, A. J. E. Farran, S. Lin-Gibson, K. L. Kiick and X. Jia, Macromolecules, 2009, 42, 2532–2541 CrossRef CAS PubMed.
- F. Ng, B. Bae, K. Miyatake and M. Watanabe, Chem. Commun., 2011, 47, 8895–8897 RSC.
- N. A. J. M. Sommerdijk, S. J. Holder, R. C. Hiorns, R. G. Jones and R. J. M. Nolte, Macromolecules, 2000, 33, 8289–8294 CrossRef CAS.
- G. Das, L. Ouali, M. Adrian, B. Baumeister, K. J. Wilkinson and S. Matile, Angew. Chem., Int. Ed., 2001, 40, 4657–4661 CrossRef CAS.
- N. Sakai and S. Matile, J. Am. Chem. Soc., 2002, 124, 1184–1185 CrossRef CAS PubMed.
- G. Das, P. Talukdar and S. Matile, Science, 2002, 298, 1600–1602 CrossRef CAS PubMed.
- N. Sakai, D. Houdebert and S. Matile, Chem. – Eur. J., 2003, 9, 223–232 CrossRef CAS PubMed.
- N. Sakai, N. Sordé and S. Matile, J. Am. Chem. Soc., 2003, 125, 7776–7777 CrossRef CAS PubMed.
- N. Sakai and S. Matile, Chem. Commun., 2003, 2514–2523 RSC.
- N. Sordé, G. Das and S. Matile, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 11964–11969 CrossRef PubMed.
- S. Litvinchuk, G. Bollot, J. Mareda, A. Som, D. Ronan, M. R. Shah, P. Perrottet, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 10067–10075 CrossRef CAS PubMed.
- V. Gorteau, F. Perret, G. Bollot, J. Mareda, A. N. Lazar, A. W. Coleman, D.-H. Tran, N. Sakai and S. Matile, J. Am. Chem. Soc., 2004, 126, 13592–13593 CrossRef CAS PubMed.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2005, 38, 79–87 CrossRef CAS PubMed.
- S. Litvinchuk, N. Sordé and S. Matile, J. Am. Chem. Soc., 2005, 127, 9316–9317 CrossRef CAS PubMed.
- S. Bhosale, A. L. Sisson, N. Sakai and S. Matile, Org. Biomol. Chem., 2006, 4, 3031–3039 CAS.
- S. Litvinchuk, H. Tanaka, T. Miyatake, D. Pasini, T. Tanaka, G. Bollot, J. Mareda and S. Matile, Nat. Mater., 2007, 6, 576–580 CrossRef CAS PubMed.
- N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2008, 41, 1354–1365 CrossRef CAS PubMed.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2005, 127, 6528–6529 CrossRef CAS PubMed.
- P. Talukdar, G. Bollot, J. Mareda, N. Sakai and S. Matile, Chem. – Eur. J., 2005, 11, 6525–6532 CrossRef CAS PubMed.
- H. Tanaka, S. Litvinchuk, D.-H. Tran, G. Bollot, J. Mareda, N. Sakai and S. Matile, J. Am. Chem. Soc., 2006, 128, 1600–16001 Search PubMed.
- H. Tanaka, G. Bollot, J. Mareda, S. Litvinchuk, D.-H. Tran, N. Sakai and S. Matile, Org. Biomol. Chem., 2007, 5, 1369–1380 CAS.
- S. Bhosale, A. L. Sisson, P. Talukdar, A. Fürstenberg, N. Banerji, E. Vauthey, G. Bollot, J. Mareda, C. Röger, F. Würthner, N. Sakai and S. Matile, Science, 2006, 313, 84–86 CrossRef CAS PubMed.
- A. Nakano, Q. Xie, J. V. Mallen, L. Echegoyen and G. W. Gokel, J. Am. Chem. Soc., 1990, 112, 1287–1289 CrossRef CAS.
- S. Kawasaki, T. Muraoka, H. Obara, T. Ishii, T. Hamada and K. Kinbara, Chem. – Asian J., 2014, 9, 2778–2788 CrossRef CAS PubMed.
- N. Sadhukhan, T. Muraoka, D. Abe, Y. Sasanuma, D. R. G. Subekti and K. Kinbara, Chem. Lett., 2014, 43, 1055–1057 CrossRef CAS.
- N. Sadhukhan, T. Muraoka, M. Ui, S. Nagatoishi, K. Tsumoto and K. Kinbara, Chem. Commun., 2015, 51, 8457–8460 RSC.
- T. Muraoka, T. Shima, T. Hamada, M. Morita, M. Takagi and K. Kinbara, Chem. Commun., 2011, 47, 194–196 RSC.
- T. Muraoka, T. Shima, T. Hamada, M. Morita, M. Takagi, K. V. Tabata, H. Noji and K. Kinbara, J. Am. Chem. Soc., 2012, 134, 19788–19794 CrossRef CAS PubMed.
- T. Muraoka, T. Endo, K. V. Tabata, H. Noji, S. Nagatoishi, K. Tsumoto, R. Li and K. Kinbara, J. Am. Chem. Soc., 2014, 136, 15584–15595 CrossRef CAS PubMed.
- T. Shima, T. Muraoka, N. Hoshino, T. Akutagawa, Y. Kobayashi and K. Kinbara, Angew. Chem., Int. Ed., 2014, 53, 7173–7178 CrossRef CAS PubMed.
- T. Shima, T. Muraoka, T. Hamada, M. Morita, M. Takagi, H. Fukuoka, Y. Inoue, T. Sagawa, A. Ishijima, Y. Omata, T. Yamashita and K. Kinbara, Langmuir, 2014, 30, 7289–7295 CrossRef CAS PubMed.
- T. Shima, T. Muraoka, K. V. Tabata, H. Noji and K. Kinbara, Pure Appl. Chem., 2014, 86, 1259–1267 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |