
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemo- and regioselective reductive transposition of allylic alcohol derivatives via iridium or rhodium catalysis†
Rylan J.
Lundgren
 * and
Bryce N.
Thomas
* and
Bryce N.
Thomas
Department of Chemistry, University of Alberta, Edmonton, Alberta T6G 2G2, Canada. E-mail: rylan.lundgren@ualberta.ca
First published on 9th November 2015
Abstract
We report highly chemo- and regioselective reductive transpositions of methyl carbonates to furnish olefin products with complementary regioselectivity to that of established Pd-catalysis. These Rh- and Ir-catalysed transformations proceed under mild conditions and enable selective deoxygenation in the presence of functional groups that are susceptible to reduction by metal hydrides.
Deoxygenation reactions are important transformations in synthetic organic chemistry, finding applications in areas ranging from biomass conversion to the preparation of complex bioactive molecules.1,2 Mild, catalytic, chemoselective reductive deoxygenation of alcohols remains underdeveloped owing in large part to the difficulties associated with delivery of hydride equivalents to C–O sigma bonds in preference to C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, CO or C–X bonds.3 Thus classical methods that use stoichiometric additives such as the Barton–McCombie reaction4 or Mitsunobu reactions with diazene-precursors5,6 are still widely employed.
C, CO or C–X bonds.3 Thus classical methods that use stoichiometric additives such as the Barton–McCombie reaction4 or Mitsunobu reactions with diazene-precursors5,6 are still widely employed.
With specific regard to allylic substrates, Pd-based strategies have been developed to address some of the limitations associated with selective deoxygenation catalysis. For example, while deoxygenation of allylic alcohols via Mitsunobu reaction with diazene precursors NBSH or IPNBSH requires stoichiometric reagents such as diethyl azodicarboxylate (DEAD) (Fig. 1A),4 Movassaghi and co-workers reported an alternative IPNBSH-mediated reductive transposition using Pd-catalysis (Fig. 1B).7,8 The regiochemical outcome of the amination follows that expected for Pd-catalysed allylic substitution, generally featuring substrate steric control in the amination of a Pd-allyl species.9 Under these conditions, terminal olefin products are formed from both branched and linear allylic carbonates after sigmatropic elimination of dinitrogen from a linear monoalkyl diazene (Fig. 1B-1),10 while both formal SN2 and SN2′ displacement are observed with internal branched substrates (Fig. 1B-2).7 Similar to Pd-catalysed allylic reductions employing formate,11,12 generation of the alternative olefin regioisomers is not possible; thus complete regiocontrol of catalytic reductive transposition of allylic alcohol derivatives remains a significant unmet challenge. Furthermore, catalytic and chemoselective diazene-mediated deoxygenation in the presence of other reducible functional groups has not been demonstrated broadly. Herein we report a strategy to address these deficits by employing Ir- and Rh-catalysis (Fig. 1C). Under mild conditions, highly chemo- and regioselective reductive transposition is observed for allylic methyl carbonates. This new method can be considered a direct, catalytic alternative to stoichiometric Mitsunobu protocols for deoxygenation of allylic alcohols embedded within functionalised molecules.13
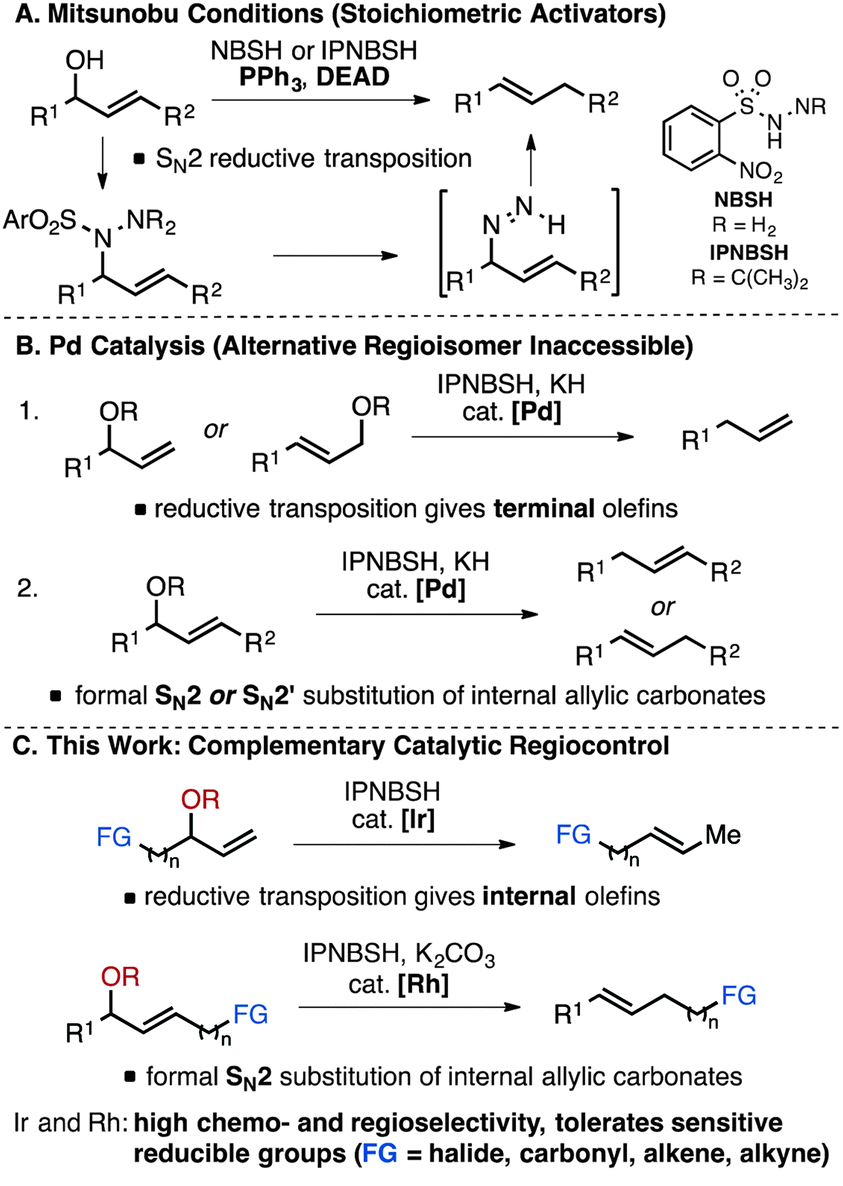 | ||
| Fig. 1 Overview of diazene-mediated reductive transposition of allylic alcohol derivatives. | ||
Conditions were optimized such that reactive functionalities, such as aliphatic chlorides are tolerated. Table 1 highlights how simple modifications to the conditions have a significant effect on the selectivity of the transformation when employing bulky diazene precursors.14,15a Under optimized conditions employing 2.5 mol% [Ir(COD)Cl]2, the desired branched N-alkyl N-sulfonyl hydrazone product formed in 91% yield at room temperature with no detectable amount of the linear allylic isomer. Rh- and Ru-based catalysts proved ineffective under these conditions (Table 1, entries 2 and 3). In solvents other than MeCN product yields were significantly lower and formation of the undesired byproducts was observed. The hydrazine reagent NBSH provided suboptimal yields (10%, Table 1, entry 6). Methyl carbonate is the preferred leaving group, as use of alternative alkyl carbonates or a phosphate ester resulted in lower yields.15b Finally, in situ hydrolysis and sigmatropic rearrangement of the allylic sulfonyl hydrazone at room temperature yielded the desired internal olefin in 71% isolated yield (eqn (1)).15c Of note, experiments under similar conditions using ammonium formate as the reducing agent resulted in unselective consumption of the substrate.
 | (1) |
|
|
|||
|---|---|---|---|
| Entry | Change from the standard conditions | Conv. | Yield (%) |
| 0.05 mmol scale, 24 h, conversions and yields determined by 1H NMR using Bn2O as an internal standard. | |||
| 1 | None | >98 | 91 |
| 2 | [Rh(COD)Cl]2 instead of [Ir(COD)Cl]2 | 8 | <2 |
| 3 | RuCp*(MeCN)3PF6 instead of [Ir(COD)Cl]2 | 94 | 10 |
| 4 | THF instead of MeCN | 74 | 15 |
| 5 | CH2Cl2 instead of MeCN | 61 | 12 |
| 6 | NBSH instead of IPNBSH | 23 | 10 |
| 7 | CO2t-Bu instead of CO2Me | 64 | 44 |

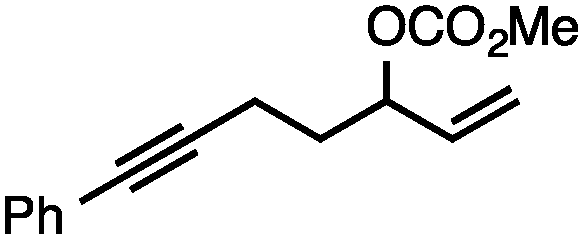
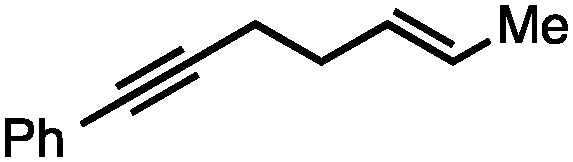
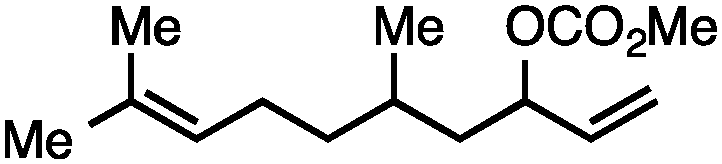
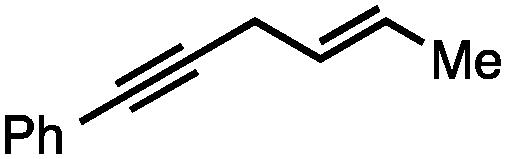
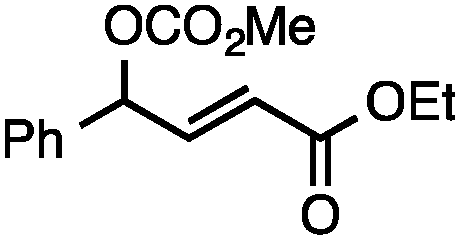
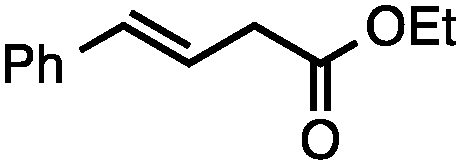
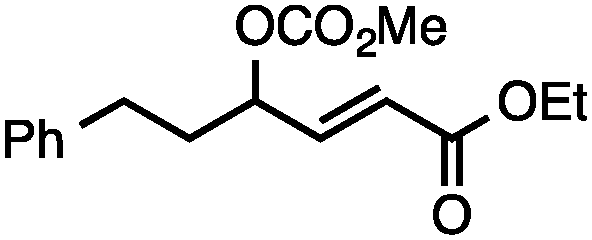
Both simple and functionalised alkyl-substituted allylic carbonates can be converted to the corresponding internal olefins in moderate to excellent yields with very high regioselectivities (Table 2).16 The reaction is tolerant of substitution β to the carbonate (Table 2, entries 2, 3 and 7), as well as oxygen, nitrogen, and halogen functional groups (Table 2, entries 2–5).15d For substrates containing pendant unsaturation in the form of an alkyne, alkene or α,β-unsaturated ester, no over-reduction is observed allowing for facile deoxygenation of polyunsaturated carbonates (Table 2, entries 6–8).17 In a particularly striking example of chemoselective deoxygenation, methyl carbonate reduction proceeds smoothly in the presence of an allylic acetate group (Table 2, entry 9).18
|
|
|||
|---|---|---|---|
| Entry | Substrate | Product | Yield (%) |
Yields are of isolated material. Regioisomer ratios are ≥95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5, E/Z ratios are ≥92:8 in all cases. See ESI for details.a 91:9 regioisomer ratio.b 5 mol% [Ir(COD)Cl]2.c Allylic acetate E/Z = 85:15 in starting material.d Allylic acetate E/Z = 85:15. :5, E/Z ratios are ≥92:8 in all cases. See ESI for details.a 91:9 regioisomer ratio.b 5 mol% [Ir(COD)Cl]2.c Allylic acetate E/Z = 85:15 in starting material.d Allylic acetate E/Z = 85:15. |
|||
| 1 |
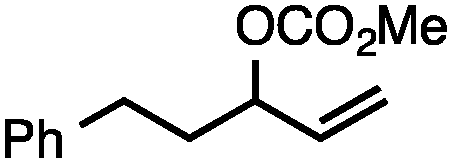
|

|
84 |
| 2 |
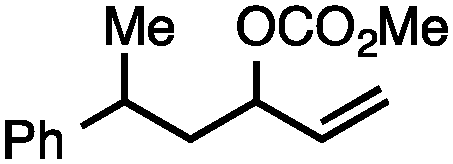
|
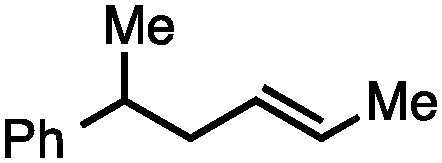
|
68a |
| 3b |
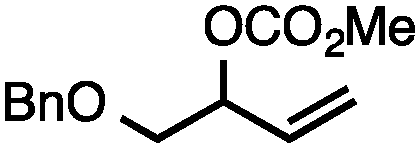
|
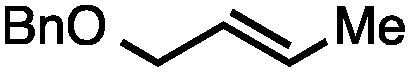
|
71 |
| 4 |
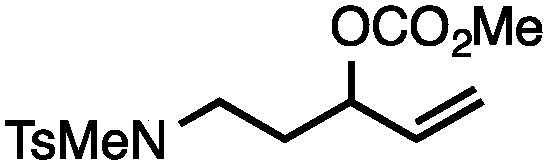
|

|
88 |
| 5 |
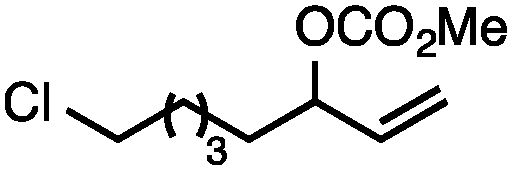
|
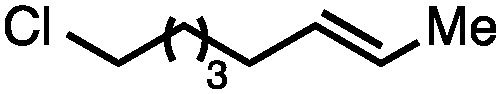
|
71 |
| 6 |
|
|
74 |
| 7 |
|

|
57 |
| 8 |
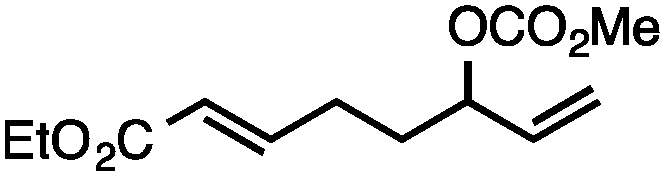
|
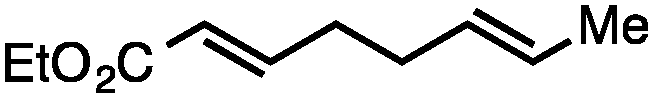
|
75 |
| 9c |
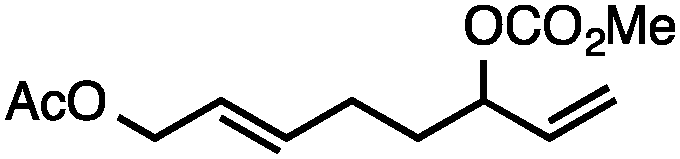
|
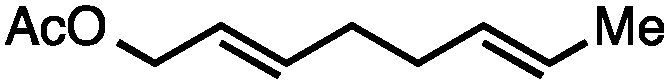
|
65d |
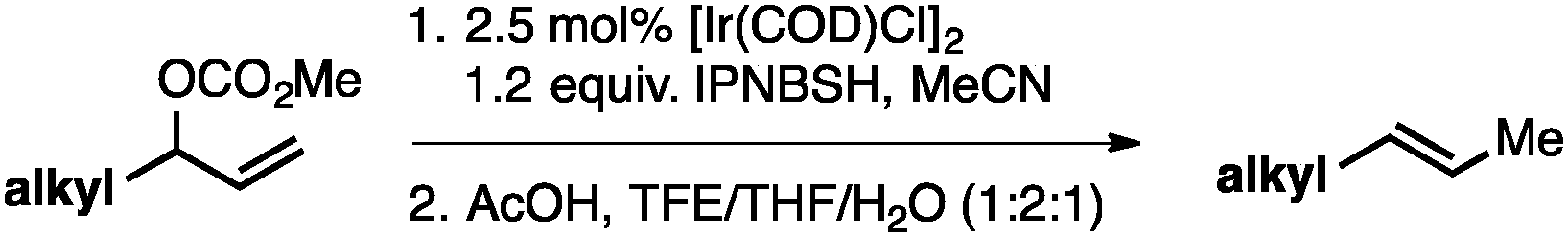
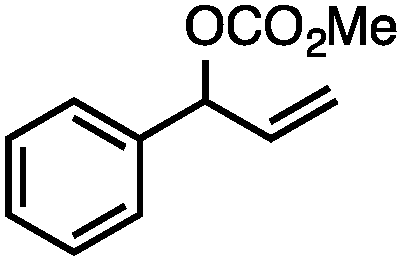
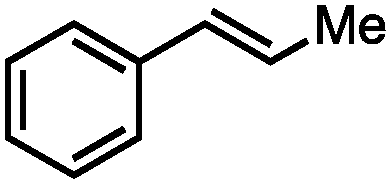
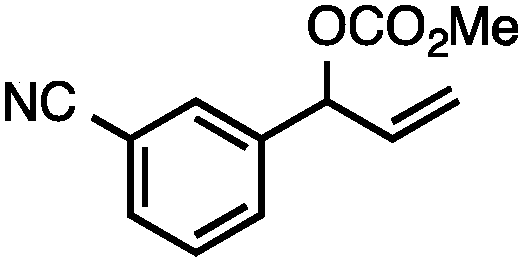
Without change to the standard conditions, aryl-substituted allylic carbonates are suitable substrates, allowing for the synthesis of functionalised β-methyl styrenes (Table 3). Electron-rich and electron-poor aryl-substituted carbonates can be deoxygenated under mild conditions. Potentially reactive functional groups that are prone to reduction under radical or metal hydride treatment, such as an aryl bromide and chloride, an allylic ether, ester, nitrile, ketone, and an aryl boronic ester, are tolerated highlighting the excellent chemoselectivity of the reduction.


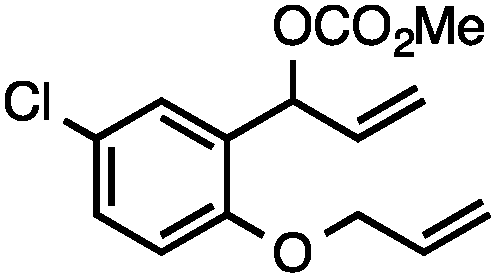
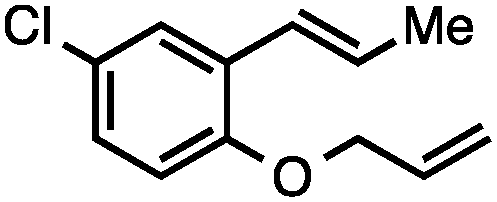
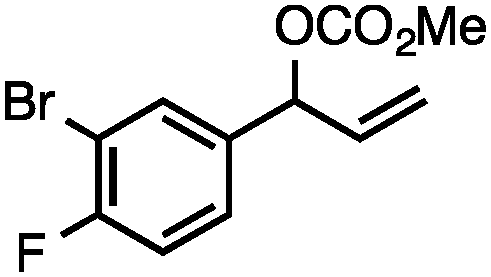
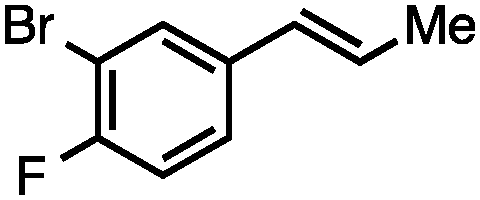
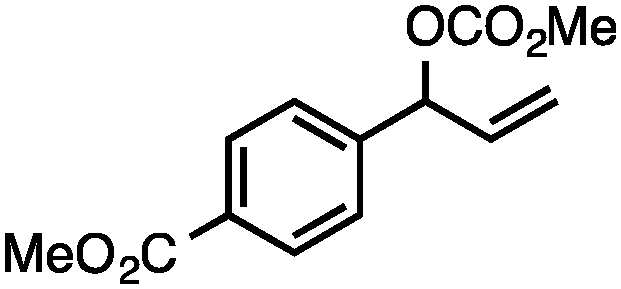
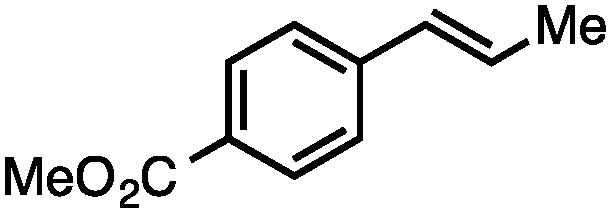
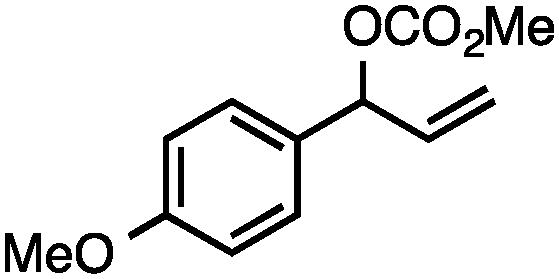
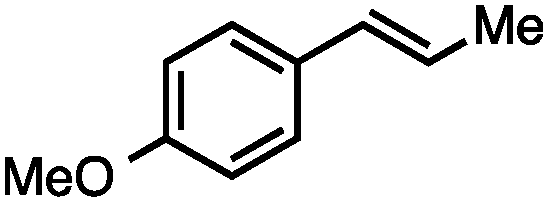
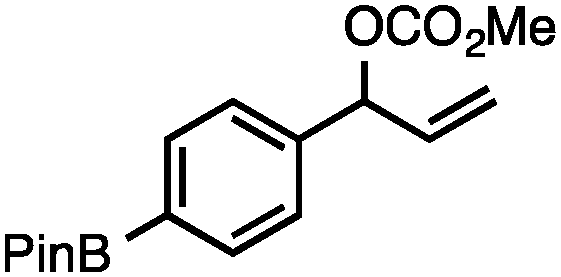
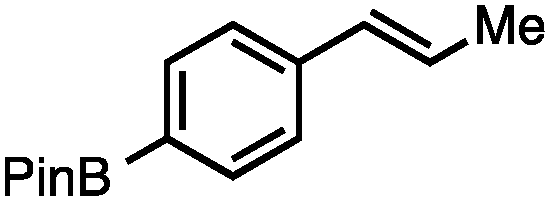
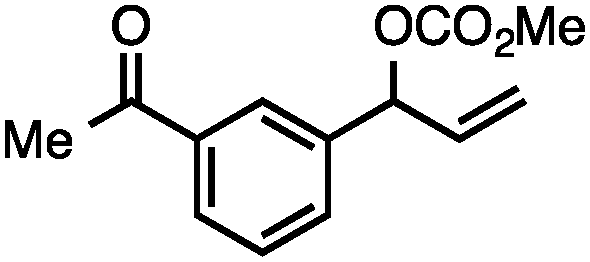
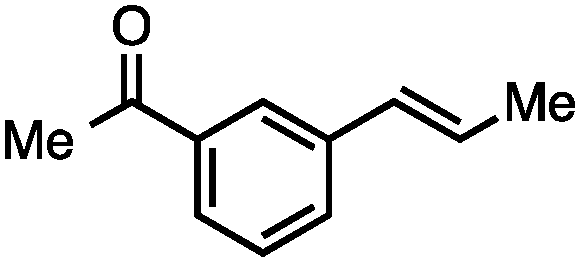
Allylic carbonates with an internal alkene were resistant to amination with IPNBSH under the standard Ir-catalysed conditions described above. Subsequent optimization, however, revealed that the use of catalytic mixtures of [Rh(COD)Cl]2 and P(OPh)3 with K2CO3 led to good yields and excellent regioselectivities (Table 4).15e,f,19 Aryl, alkenyl, alkynyl, and ethereal allylic methyl carbonates can be deoxygenated under these Rh-catalysed conditions, providing a simple and mild strategy for the preparation of sensitive skipped dienes and enynes (Table 4, entries 2 and 4). Allylic carbonates substituted with electron-withdrawing groups, such as an ester or ketone, also undergo amination with high formal SN2-selectivity, and upon reductive transposition, γ-unsaturated carbonyl compounds can be obtained (Table 4 entries 5–8). The reaction tolerates sterically demanding carbonates, such as an α-branched substrate (Table 4, entry 7). Collectively, these results demonstrate an attractive means to convert easily accessible conjugated systems into more valuable 1,4-polyunsaturated compounds that are otherwise difficult to prepare. In keeping with the observation of remarkably high formal SN2 amination selectivity, alkyl-, heteroaryl-, and alkenyl-substituted primary allylic carbonates generate terminal olefin products under the standard Rh-catalysed reaction conditions (Table 4, entries 9–11).
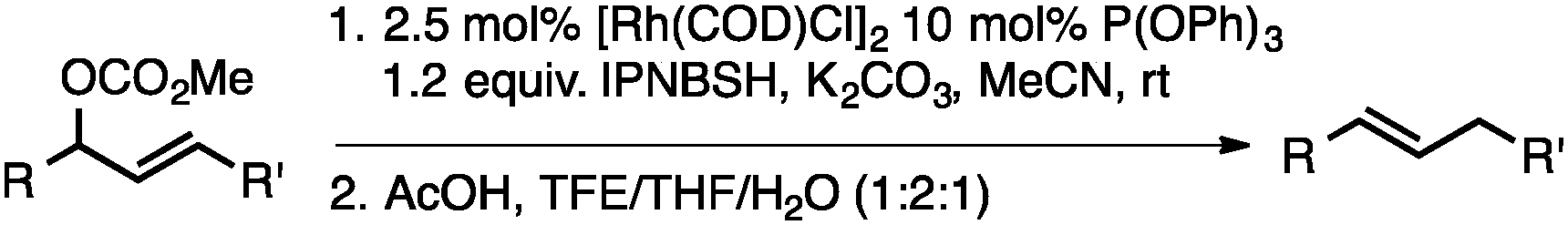
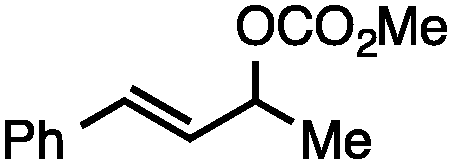
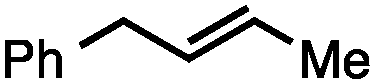
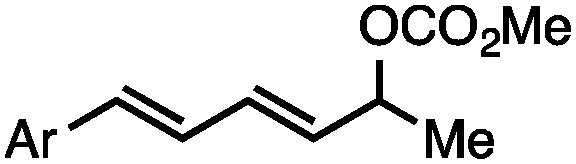
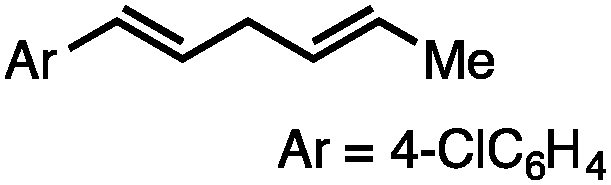
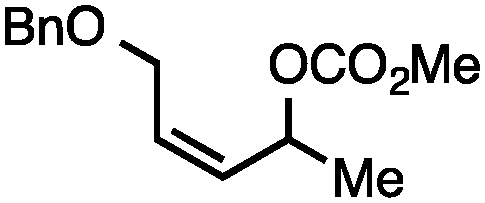
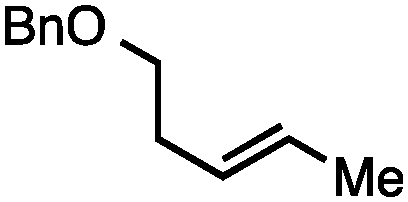
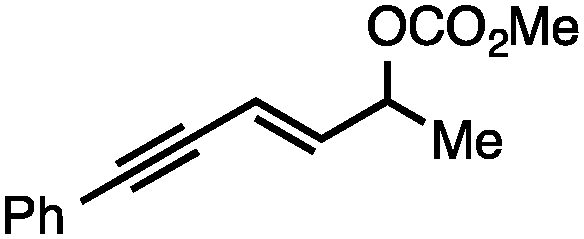
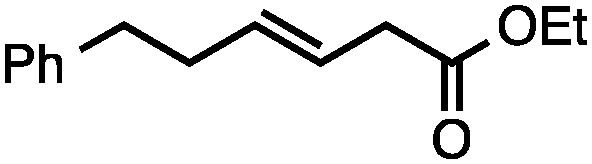
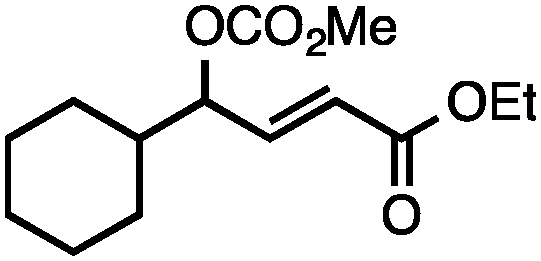
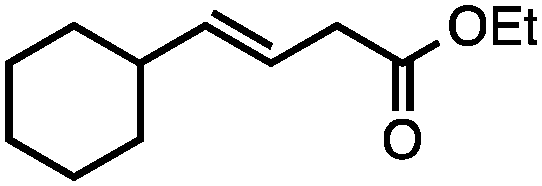
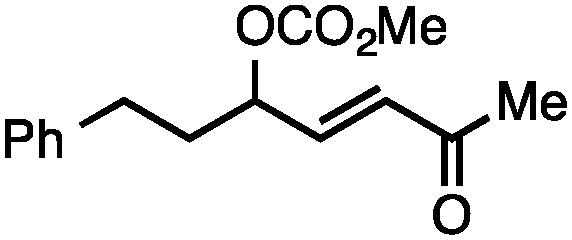
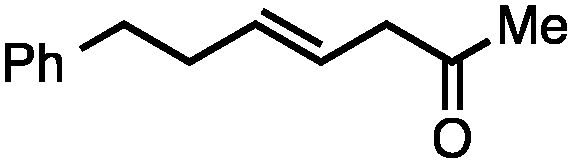
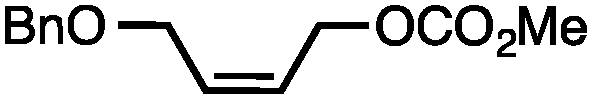

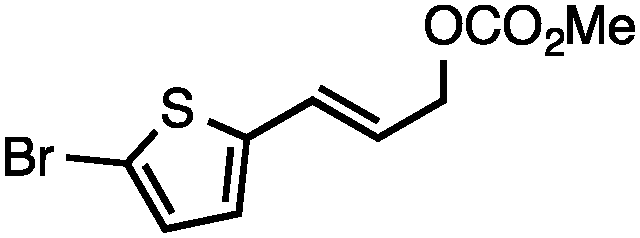
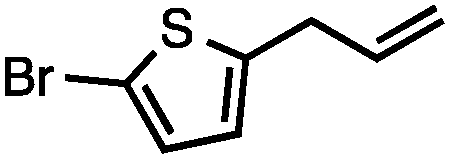
Both of the methods reported herein proceed well on larger scales, as demonstrated by the gram-scale syntheses of a halogenated β-methyl styrene via Ir-catalysis (eqn (2)) and a γ-unsaturated ester via Rh-catalysis (eqn (3)).15g
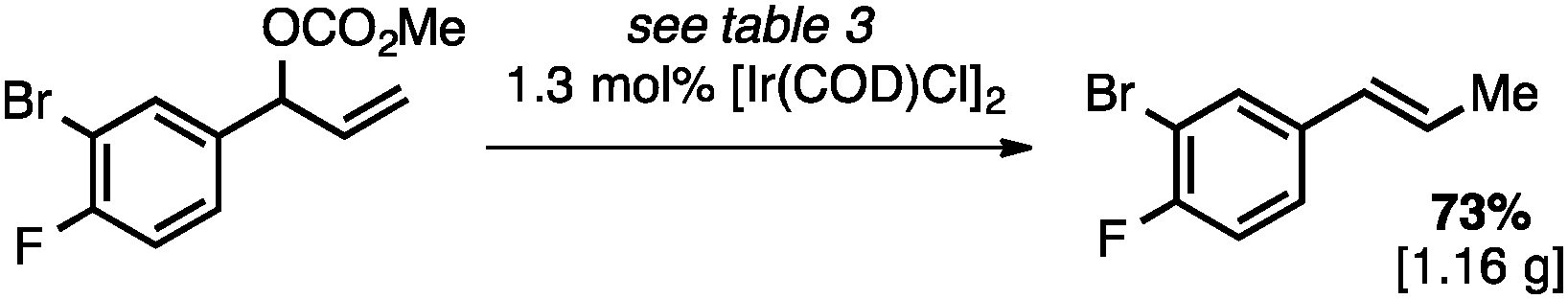 | (2) |
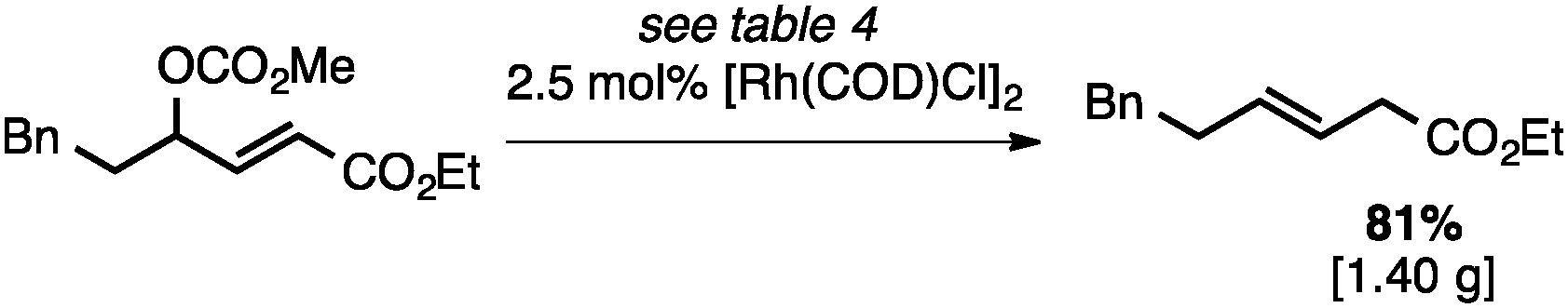 | (3) |
In summary, we have developed new catalytic strategies for the mild and selective reductive transposition of allylic alcohol derivatives employing Ir- or Rh-based catalysts. The deoxygenation process tolerates a wide range of functional groups that are susceptible to radical or hydride reduction and provides complementary regioselectivity to that of Pd-catalysed methodologies. The ability of this method to be used in place of stoichiometric Mitsunobu-type deoxygenation processes should result in widespread appeal.
We thank NSERC Canada (Discovery Grant, Research Tools and Infrastructure Grant), the Canadian Foundation for Innovation, the University of Alberta, and faculty within the Department of Chemistry for generous donations of equipment and chemicals. Chris Godwin is acknowledged for assistance with substrate synthesis.
Notes and references
- For a review see: J. M. Herrmann and B. König, Eur. J. Org. Chem., 2013, 7017–7027 CrossRef CAS.
- (a) For select recent examples of new deoxygenation methods see: L. L. Adduci, T. A Bender, J. A. Dabrowski and M. R. Gagné, Nat. Chem., 2015, 7, 576–581 CrossRef CAS PubMed; (b) H. Dang, N. Cox and G. Lalic, Angew. Chem., Int. Ed., 2014, 53, 752–756 CrossRef CAS PubMed; (c) L. L. Adduci, M. P. McLaughlin, T. A. Bender, J. J. Becker and M. R. Gagné, Angew. Chem., Int. Ed., 2014, 53, 1646–1649 CrossRef CAS PubMed; (d) M. Shiramizu and F. D. Toste, Angew. Chem., Int. Ed., 2013, 52, 12905–12909 CrossRef CAS PubMed; (e) J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997–2009 CrossRef CAS PubMed.
- (a) For reviews that discuss such problems in modern organic chemistry see: J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954–10990 CrossRef CAS PubMed; (b) N. A. Afagh and A. K. Yudin, Angew. Chem., Int. Ed., 2010, 49, 262–310 CrossRef CAS PubMed.
- For a review see: (a) S. W. McCombie, W. B. Motherwell and M. J. Tozer, The Barton-McCombie Reaction, Org. React., John Wiley & Sons, Inc., 2012 Search PubMed; (b) for recent applications in complex molecule synthesis see: H. Sugimura, S. Sato, K. Tokudome and T. Yamada, Org. Lett., 2014, 16, 3384–3387 CrossRef PubMed; (c) M. Zhang, N. Liu and W. Tang, J. Am. Chem. Soc., 2013, 135, 12434–12438 CrossRef CAS PubMed; (d) B. S. Fowler, K. M. Laemmerhold and S. J. Miller, J. Am. Chem. Soc., 2012, 134, 9755–9761 CrossRef CAS PubMed; (e) M. Bian, Z. Wang, X. Xiong, Y. Sun, C. Matera, K. C. Nicolaou and A. Li, J. Am. Chem. Soc., 2012, 134, 8078–8081 CrossRef CAS PubMed.
- (a) For diazene mediated alcohol reduction with sulfonyl hydrazine reagents see: M. Movassaghi and O. K. Ahmad, J. Org. Chem., 2007, 72, 1838–1841 CrossRef CAS PubMed; (b) A. G. Myers, M. Movassaghi and B. Zheng, J. Am. Chem. Soc., 1997, 119, 8572–8573 CrossRef CAS; (c) A. G. Myers and B. Zheng, J. Am. Chem. Soc., 1996, 118, 4492–4493 CrossRef CAS; (d) A. G. Myers and B. Zheng, Tetrahedron Lett., 1996, 37, 4841–4844 CrossRef CAS.
- (a) For selected applications of reductions employing sulfonyl hydrazine reagents in complex molecule synthesis see: D. Yang and G. C. Micalizio, J. Am. Chem. Soc., 2012, 134, 15237–15240 CrossRef CAS PubMed; (b) M. Shan, E. U. Sharif and G. A. O'Doherty, Angew. Chem., Int. Ed., 2010, 49, 9492–9495 CrossRef CAS PubMed; (c) M. Movassaghi, G. Piizzi, D. S. Siegel and G. Piersanti, Angew. Chem., Int. Ed., 2006, 45, 5859–5863 CrossRef CAS PubMed; (d) M. G. Charest, C. D. Lerner, J. D. Brubaker, D. R. Siegel and A. G. Myers, Science, 2005, 308, 395–398 CrossRef CAS PubMed.
- M. Movassaghi and O. K. Ahmad, Angew. Chem., Int. Ed., 2008, 47, 8909–8912 CrossRef CAS PubMed.
- (a) For acid catalysed substitution reactions of activated propargylic substrates with sulfonyl hydrazines to generate allenes see: Z. Liu, P. Q. Liao and X. Bi, Chem. – Eur. J., 2014, 20, 17277–17281 CrossRef CAS PubMed; (b) D. A. Mundal, K. E. Lutz and R. J. Thomson, J. Am. Chem. Soc., 2012, 134, 5782–5785 CrossRef CAS PubMed.
- For a detailed discussion of the topic see: B. S. Kim, M. M. Hussain, P. O. Norrby and P. J. Walsh, Chem. Sci., 2014, 5, 1241–1250 RSC and references therein.
- A. Jabbari, E. J. Sorensen and K. N. Houk, Org. Lett., 2006, 8, 3105–3107 CrossRef CAS PubMed.
- (a) For a review see: J. Tsuji and T. Mandai, Synthesis, 1996, 1–24 CrossRef CAS; (b) strong hydride donors such as DIBAL-H or SmI2 can afford the opposite regioisomers, but these reagents would exhibit minimal chemoselectivity in the presence of reducible groups. For more recent examples of Pd-catalyzed formate reduction see: A. Chau, J.-F. Paquin and M. Lautens, J. Org. Chem., 2005, 71, 1924–1933 CrossRef PubMed; (c) T. Konno, T. Takehanna, M. Mishima and T. Ishihara, J. Org. Chem., 2006, 71, 3545–3550 CrossRef CAS PubMed; (d) in very rare cases, select substrates can be converted to the internal alkene, see: H. Cheng, C. Sun and D. Hou, J. Org. Chem., 2007, 72, 2674–2677 CrossRef CAS PubMed.
- For the phosphine-catalysed reduction of allylic bromides with LiAlH(OtBu)3 to terminal olefins see: K. D. Reichl, N. L. Dunn, N. J. Fastuca and A. T. Radosevich, J. Am. Chem. Soc., 2015, 137, 5292–5295 CrossRef CAS PubMed.
- For an unselective Ir-catalysed deoxygenation process (alkenes are also reduced) employing hydrazine at 160 °C, see: J. L. Huang, X. J. Dai and C. J. Li, Eur. J. Org. Chem., 2013, 6496–6500 CrossRef CAS.
- (a) For early examples of Ir- and Rh-catalysed allylic amination see: P. A. Evans, J. E. Robinson and J. D. Nelson, J. Am. Chem. Soc., 1999, 121, 6761–6762 CrossRef CAS; (b) R. Takeuchi, N. Ue, K. Tanabe, K. Yamashita and N. Shiga, J. Am. Chem. Soc., 2001, 123, 9525–9534 CrossRef CAS PubMed; (c) For Ir-catalysed allylation of hydrazines and hydrazones see: R. Matunas, A. J. Lai and C. Lee, Tetrahedron, 2005, 61, 6298–6308 CrossRef CAS.
- Notes:
(a) the stable, crystalline reagents NBSH and IPNBSH are commercially available, or readily synthesized on decagram scale;
(b) for additional optimization data see the ESI†;
(c) hydrolysis was performed by removal of MeCN prior to addition of THF/TFE/H2O (2:1:1) and AcOH, direct addition of TFE/H2O without THF resulted in ∼10% lower yields;
(d) electron-rich aryl methyl carbonates are prone to rearrangement to the linear isomer, pyridine and quinoline substrates are aminated effectively but undergo reduction with low yield and regioselectivity, cyclic allylic methyl carbonates are not viable substrates, as is generally observed in Ir- and Rh-catalysed allylic functionalization;
(e) see ESI† for details on optimization of internal allylic substrates;
(f) these conditions are effective for terminal allylic carbonates, however the reactions proceed with slightly diminished branched/linear selectivity compared to the use of [Ir(COD)Cl]2;
(g) no glovebox is required for these reactions, see the ESI†.
- Aside from being useful chemical building blocks, methyl-substituted olefins are found in numerous bioactive molecules, such as cyclosporines, corallopyronins and penibruguieramide A.
- Free diazene generated from sulfonyl hydrazines can reduce olefins, for examples see: (a) B. J. Marsh and D. R. Carbery, J. Org. Chem., 2009, 74, 3186–3188 CrossRef CAS PubMed; (b) M. H. Haukaas and G. A. O'Doherty, Org. Lett., 2002, 4, 1771–1774 CrossRef CAS PubMed.
- Secondary allylic acetates are less reactive substrates. Under the standard Ir-catalysed conditions with the acetate version of the substrate in Table 3, entry 1, 27% (>20:1 b/l) product is observed, compared to >95% for the allylic methyl carbonate substrate, (the isolated yield of olefin is lower due to volatility of the product). Under the standard Rh-catalysed conditions 34% (20:80 b/l) product is observed.
- For the Rh-catalyzed allylation of sulfonamides see: P. A. Evans, J. E. Robinson and K. K. Moffett, Org. Lett., 2001, 3, 3269–3271 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic procedures and characterization data. See DOI: 10.1039/c5cc07993d |
| This journal is © The Royal Society of Chemistry 2016 |