
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cooperative reaction chemistry derived from a borata-diene framework†
Jiangang
Yu
,
Gerald
Kehr
,
Constantin G.
Daniliuc‡
and
Gerhard
Erker
*
Organisch-Chemisches Institut der Universität Münster, Corrensstr. 40, 48149 Münster, Germany. E-mail: erker@uni-muenster.de
First published on 11th November 2015
Abstract
The bifunctional frustrated BH−/B hydridoborate nucleophile/borane Lewis acid pair 6 is prepared starting from the borata-diene 3 by a sequence of sequential protonation/hydride attachment, followed by hydroboration with Piers' borane [HB(C6F5)2]. The trans-1,2-bifunctional system 6 reduced carbon monoxide eventually to the aldehyde product 10.
Borohydrides are important reducing reagents. However, some of these systems show rather low hydride nucleophilicities, especially when additional strongly electron-withdrawing substituents are attached at the boron center.1 Lewis acid assistance is often helpful or even essential.2 We have now prepared a bifunctional –B(C6F5)2H−/–B(C6F5)2 borohydride/borane Lewis acid pair by selective construction starting from a readily available borata-diene precursor. The functionalities were separated similarly as it had previously been achieved in frustrated Lewis pair chemistry at rigid frameworks. The –B(C6F5)2H−/–B(C6F5)2 frustrated pair showed a high reaction potential, e.g. for the stoichiometric conversion of carbon monoxide.3 The preparation of the active hydridoborate/borane pair and some first reactions are described in this account.
Borata-alkenes represent mesomeric structures of α-boryl carbanions4 and consequently they can be prepared by deprotonation of the boranes with a suitable base.5 This reaction is, however, in most cases problematic since the majority of conventional bases would preferentially add to the borane Lewis acid site rather than deprotonate in the α-position. Therefore, a rather limited number of borata-alkenes were prepared in this straightforward way, isolated and amply characterized. The few structurally characterized examples show a pronounced B![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond character with boron–carbon bond lengths around 1.44 Å.6
C double bond character with boron–carbon bond lengths around 1.44 Å.6
We recently reported a base-free synthetic entry to the zwitterionic borata-diene compound 3 (see Scheme 1):7,8 hydroboration9 of the P-enyne 1 with Piers' borane [HB(C6F5)2] generated the geminal P/B Lewis pair 2 that underwent rapid intramolecular ring-closure to 3.7 Compound 3 served as the starting material for the construction of a reactive borohydride/borane pair. Treatment of 3 with triflic acid resulted in the protonation of the incipient carbanion site in the α-position to boron and attachment of the corresponding triflate anion to the boron atom.10 The resulting Brønsted acid addition product 4 was isolated in 67% yield. Its X-ray crystal structure analysis confirmed that a selective 1,2-addition of HOSO2CF3 had occurred across the BC double bond. Compound 4 contains a sp3-hybridized carbon atom C1 in the ring and has the adjacent C2–C3 double bond intact. The triflate anion is found to be attached at the boron atom B1 (for structural details and the spectroscopic characterization of compound 4 see the ESI†).
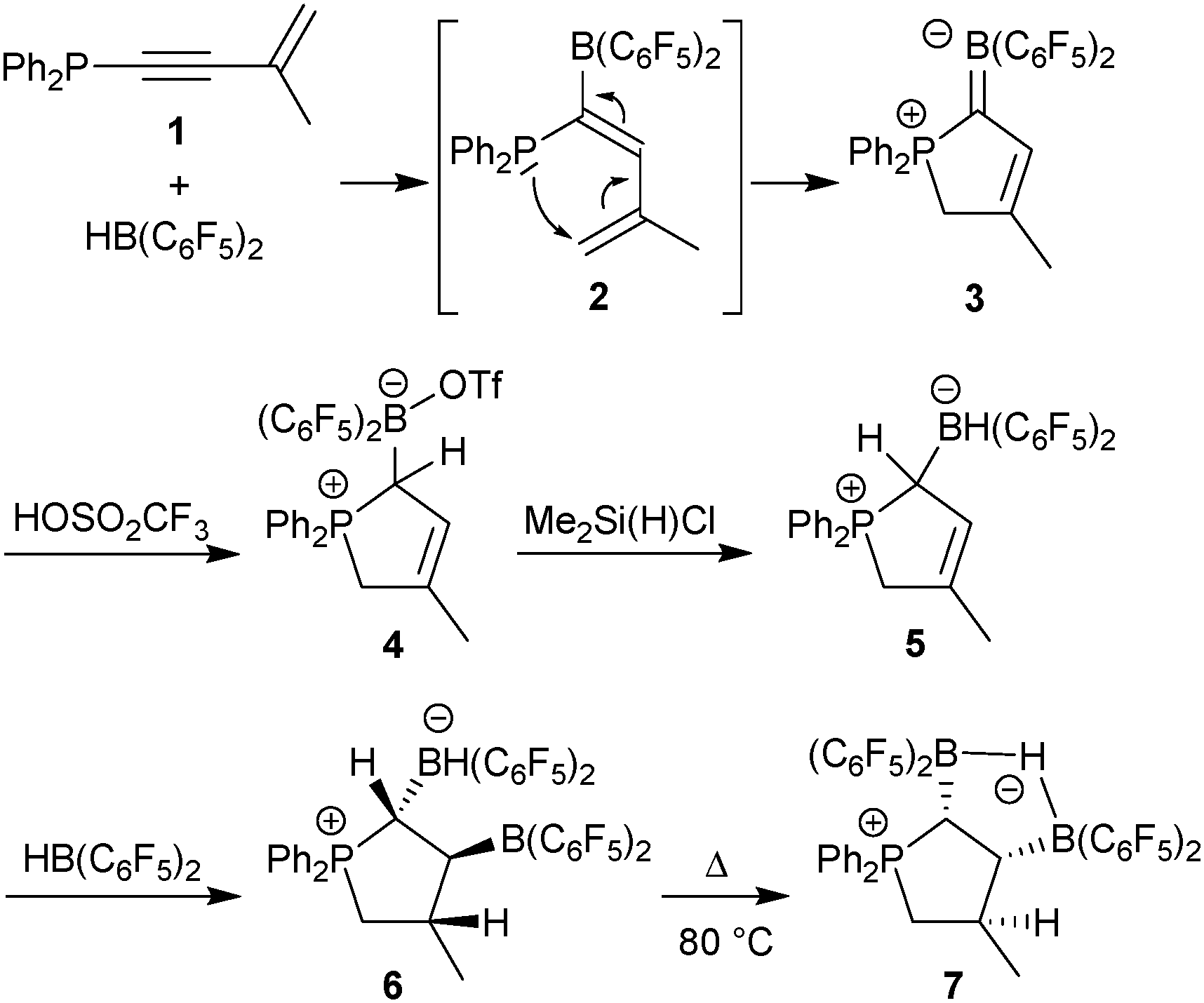 | ||
| Scheme 1 | ||
We then exchanged the OTf group at boron for hydride. This was effected by treatment of compound 4 with excess chlorodimethylsilane (30 min, 60 °C, dichloromethane).10 The borohydride product 5 was isolated as a colorless solid in 73% yield. It showed the broad [B]–H 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 1H NMR quartet at δ 2.78 ppm in [D2]-dichloromethane solution with a corresponding 11B NMR signal at δ −22.1 ppm (1JBH ∼ 94 Hz) [31P NMR: δ 51.1 ppm]. The X-ray crystal structure analysis of compound 5 shows the hydridoborate substituent attached at the ring carbon atom C1 (∑B1CCC = 331.9°). The substituent features a staggered conformation at the connecting C1–B1 vector with an anti-periplanar orientation of the H–C1–B1–H moiety. The five-membered phosphonium ring contains a distal carbon–carbon double bond (C2C3) (see Fig. 1).
:1:1:1 1H NMR quartet at δ 2.78 ppm in [D2]-dichloromethane solution with a corresponding 11B NMR signal at δ −22.1 ppm (1JBH ∼ 94 Hz) [31P NMR: δ 51.1 ppm]. The X-ray crystal structure analysis of compound 5 shows the hydridoborate substituent attached at the ring carbon atom C1 (∑B1CCC = 331.9°). The substituent features a staggered conformation at the connecting C1–B1 vector with an anti-periplanar orientation of the H–C1–B1–H moiety. The five-membered phosphonium ring contains a distal carbon–carbon double bond (C2C3) (see Fig. 1).
 | ||
| Fig. 1 Molecular structure of the phosphonium/hydridoborate compound 5 (thermal ellipsoids are shown with 15% probability). Selected bond lengths (Å) and angles (deg): C1–B1 1.668(4), C1–P1 1.789(3), C1–C2 1.523(3), C2–C3 1.319(4), C3–C4 1.503(4), C4–P1 1.804(3), C1–P1–C4 97.2(1), B1–C1–C2 110.2(2). | ||
Compound 5 can formally be regarded as the 1,2-dihydrogen addition product to the borata-alkene 3. However, it was not obtained by means of a real hydrogenation process, but rather formed in a conventional way by adding a proton to the basic borata-alkene α-carbon followed by hydride attachment at boron. Compound 5 underwent hydroboration with Piers' borane [HB(C6F5)2] to give 6 (isolated in 63% yield).
The 1H NMR spectrum showed the C3–CH3 doublet at δ 1.34 ppm (3JHH = 6.3 Hz) which indicated regioselective formation of the anti-Markovnikov [B]–H addition product. The newly attached B(C6F5)2 group shows a 11B NMR resonance (δ 65.1 ppm) typical of a planar-tricoordinated boron Lewis acid. Consequently, this group shows only a single set of 19F NMR resonances of its pair of homotopic C6F5 groups with a typical large p,m-signal shift difference of Δδm,p = 12.0 ppm. The B(H)(C6F5)211B NMR resonance occurs at δ −16.3 ppm and this signal is split into a doublet (1JBH ∼ 77 Hz) that indicates the presence of an unbridged terminal [B]–H hydride.
The X-ray crystal structure analysis (see Fig. 2) has confirmed the formation of the anti-Markovnikov HB(C6F5)2-addition product 6, featuring the pair of boron containing substituents trans-vicinally attached at the carbon atoms C1 and C2 at the five-membered heterocyclic phosphonium core. The Lewis acidic B(C6F5)2 substituent is attached at C2 (∑B2CCC = 359.1°) whereas the B(H)(C6F5)2 group (∑B1CCC = 332.4°) is found to be bonded to C1. This group has retained the anti-periplanar arrangement of the H–C1–B1–H unit. Carbon atom C3 is now sp3-hybridized. C3–H is oriented cis- to the vicinal C2–B2 vector as expected from the hydroboration reaction.
 | ||
| Fig. 2 Molecular structure of compound 6 (thermal ellipsoids are shown with 15% probability). Selected bond lengths (Å) and angles (deg): C1–B1 1.675(4), C1–P1 1.806(3), C1–C2 1.597(3), C2–C3 1.550(4), C3–C4 1.530(4), C4–P1 1.788(3), C1–P1–C4 97.8(1), B1–C1–C2 110.0(2). | ||
The trans-vicinal arrangement of the borane and boryl substituents in compound 6 was probably determined by steric hindrance between these bulky groups during the hydroboration reaction. Compound 6 was formed under kinetic control. This was shown by thermolizing it subsequently. Keeping compound 6 for 3 days at 80 °C resulted in the formation of its cis-isomer 7 which was eventually isolated in 81% yield. We assume that it was formed by a dehydroboration/hydroboration sequence11 and thus we conclude that compound 7 is the thermodynamic hydroboration product under these conditions.
Compound 7 shows a broad [B]–H signal in the 1H NMR spectrum at δ 2.12 ppm and a pair of broad 11B NMR resonances at δ 9.4 and 5.5 ppm, respectively. It shows the typical 1H NMR –C3H–CH3 methyl doublet at δ 0.99 ppm (3JHH = 6.5 Hz) and the three 1,2-cis-3-trans CH resonances at δ 4.44 (C1–H), 2.78 (C2–H) and 3.11 ppm (C3–H) (C4H2: δ 3.17 and 2.35 ppm). The structure of compound 7 was confirmed by X-ray diffraction. It shows a cis-1,2-arrangement of the H-bridged –B(C6F5)2(μH) B(C6F5)2-unit at the ring carbon atoms C1 and C2 (see Fig. 3). The methyl group at C3 is cis-oriented to the C2–B2 vector. Both the boron atoms feature markedly distorted tetrahedral coordination geometries (∑B1CCC = 346.9°, ∑B2CCC = 345.8°).12
 | ||
| Fig. 3 Molecular structure of compound 7 (thermal ellipsoids are shown with 15% probability). Selected bond lengths (Å) and angles (deg): C1–B1 1.649(3), C1–P1 1.809(2), C1–C2 1.568(3), C2–C3 1.541(3), C3–C4 1.540(3), C4–P1 1.812(2), C1–P1–C4 98.4(1), B1–C1–C2 104.0(2). | ||
Compound 7 represents just an example of a rather conventional hydride bridged B–H–B type compound. In contrast, compound 6 is remarkable in that it contains an unquenched pair of an active borohydride nucleophile and a strongly Lewis acidic boryl electrophile attached at adjacent carbon atoms at the central five-membered heterocyclic core structure. It is due to these specific structural features of the phosphoniacyclopentane framework that these two units are very effectively hindered from annihilating their typical reactivities by “neutralizing” bridging μ-hydride formation. In a way this situation may be regarded as a new form of a frustrated Lewis pair (FLP), hereby having a pair of a hydride nucleophile and a borane Lewis acid being effectively hindered from mutual quenching by the specific nature of the framework that they are attached to. This opens the chance to find cooperative reaction with added substrates.
Reaction of the bifunctional compound 6 with benzaldehyde (r.t., 1 h) gave the reduction product 9 (isolated in 77% yield after crystallization from pentane) (see Scheme 2).13 The X-ray crystal structure analysis showed that hydride had been added to the carbonyl carbon atom to form the [B]-benzyl alcoholate product. The alcoholate oxygen is rather symmetrically bonded to both boron atoms. The oxygen atom shows a trigonal planar coordination geometry (∑O1CBB = 356.9°). The boryl substituents are found to be trans-1,2-attached at the ring carbon atoms C1 and C3 (see the ESI† for structural details and the spectroscopic characterization of compound 9).
 | ||
| Scheme 2 | ||
Both isomers 6 and 7 were exposed to carbon monoxide. While the conventional hydride-bridged cis-isomer 7 was inert toward CO under our typical reaction conditions, BH−/B FLP 6 reduced carbon monoxide under mild reaction conditions. We exposed compound 6 in dichloromethane to carbon monoxide (2.5 bar) under close to ambient conditions (r.t., 30 min). After a short reaction time, workup gave product 10 in 63% yield (see Scheme 3). The product was characterized by C,H elemental analysis, by NMR spectroscopy and X-ray diffraction (see Fig. 4).
 | ||
| Scheme 3 | ||
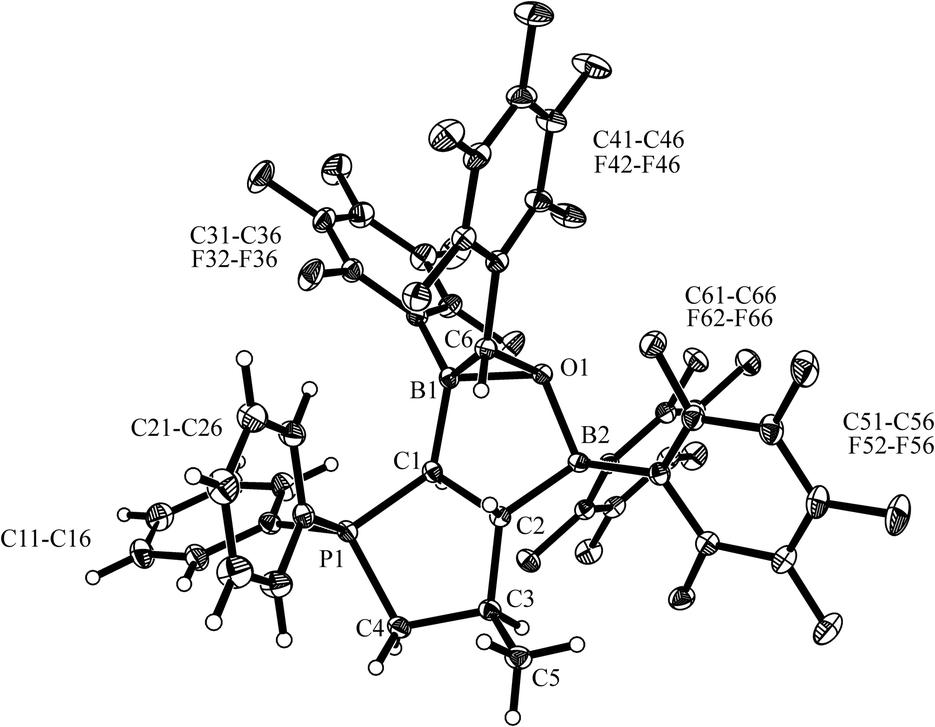 | ||
| Fig. 4 A projection of the carbonylation product 10 (thermal ellipsoids are shown with 15% probability). Selected bond lengths (Å) and angles (deg): B1–O1 1.577(2), B2–O1 1.593(2), C1–B1 1.612(3), C1–P1 1.783(2), C1–C2 1.555(2), C2–C3 1.552(2), C2–B2 1.633(3), C3–C4 1.562(3), C4–P1 1.816(2), C6–O1 1.501(2), C1–P1–C4 95.8(1), B1–C1–C2 105.8(1), O1–B1–C6 57.1(1), B1–C6–O1 61.9(1), B1–O1–C6 61.0(1). | ||
The X-ray crystal structure analysis (single crystals were obtained from dichloromethane/pentane) showed that CO had become reduced at the BH−/B framework of 6 and coupled with a –C6F5 group to give a pentafluorobenzaldehyde moiety. However, this reduction product is bonded in a special manner to the framework of 6: the newly introduced oxygen atom is found to bridge between the pair of boron atoms and the aldehyde carbon atom (which is derived from the CO molecule) is bonded to boron atom B1. So the C6F5–CHO unit is found η2-bonded to B13 and κO-bonded to B2. This situation generates a rather special stereochemical situation by introducing a pair of new chirality centers, namely at the former CO carbon atom (C6) and its adjacent boron atom (B1). Together with the three carbon chiral centers of the framework (C1, C2, C3) this makes a total of five stereogenic centers present in compound 10. Three of these (at the framework) are determined in their relative stereochemistry by the starting material 6 (which was formed as a single diastereomer), but in principle the pair of newly introduced stereocenters could have led to the formation of 10 as a mixture of four diastereomers. However, we find only a single diastereomer of 10 being formed in the carbonylation reaction of 6. It is characterized by a syn-arrangement of the C6 group to the C5-methyl substituent and an exo-orientation of the C6-pentafluorophenyl substituent.
In solution (CD2Cl2) we have observed the 1H/13C NMR signals of the CO derived –CH–Ar group at δ 4.29 ppm/δ 60.8 ppm. Compound 10 shows the 19F NMR resonances of four different C6F5 substituents (for details see the ESI†) and we have recorded the heteronuclear magnetic resonances of the pair of boron atoms (11B: δ 4.2 and −3.5 ppm) and the phosphorus atom (31P: δ 38.9 ppm).
It has long been known that boranes can form carbonyl adducts upon exposure to CO. The borane carbonyl H3B–CO14 and Piers' borane carbonyl (C6F5)2HB–CO15 are typical examples. Therefore, we were led to assume a reaction pathway for the formation of compound 10 that involved the reactive borane carbonyl intermediate 11. Subsequent reduction by the adjacent borohydride reagent to the formyl stage followed by nucleophilic C6F5 attack would then generate the C6F5–CHO subunit.16 Thermodynamically driven rearrangement would then give the final product 10 (see Scheme 3).
Compound 6 contains an interesting construction principle. It contains a pair of antagonistic17 boron bound functional groups. The strongly Lewis acidic B(C6F5)2 moiety is attached at carbon atom C2. It has the anionic hydridoborate moiety attached in close vicinity at its adjacent carbon atom C1. Nevertheless, these two divergent functionalities do not hinder each other's typical reactivity (e.g. by hydride bridging) because of the rigid separation caused by the typical stereochemical features of the five-membered heterocyclic framework. This allows cooperative reactions with added reagents which show a remote resemblance of the behavior of frustrated Lewis pairs. In our case the carbonyl reagents, activated by attachment at the Lewis acid site, got converted by nucleophilic hydride attack from the adjacent hydridoborate. The bifunctional BH−/B pair has conveniently become available by a straightforward synthetic route derived from a readily available borata-alkene starting material. Our study indicates that the general principles that have made frustrated Lewis pair chemistry successful are beginning to stretch out to related fields in the vicinity of FLP chemistry and enlarge its scope.18
Financial support from the European Research Council is gratefully acknowledged.
Notes and references
- Z. M. Heiden and A. P. Lathem, Organometallics, 2015, 34, 1818 CrossRef CAS.
- D. W. Stephan and G. Erker, Top. Curr. Chem., 2013, 332, 85 CrossRef CAS PubMed.
- See for comparison: M. Sajid, L.-M. Elmer, C. Rosorius, C. G. Daniliuc, S. Grimme, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 2243 CrossRef CAS PubMed.
- P. Moquist, G.-Q. Chen, C. Mück-Lichtenfeld, K. Bussmann, C. G. Daniliuc, G. Kehr and G. Erker, Chem. Sci., 2015, 6, 816 RSC.
- (a) T. Tomioka, Y. Takahashi, T. G. Vaughan and T. Yanase, Org. Lett., 2010, 12, 2171 CrossRef CAS PubMed; (b) T. Tomioka, R. Sankranti, T. G. Vaughan, T. Maejima and T. Yanase, J. Org. Chem., 2011, 76, 8053 CrossRef CAS PubMed; (c) T. Tomioka, Y. Takahashi and T. Maejima, Org. Biomol. Chem., 2012, 10, 5113 RSC.
- (a) M. W. Rathke and R. Kow, J. Am. Chem. Soc., 1972, 94, 6854 CrossRef CAS; (b) J. J. Eisch, in Adv. Organomet. Chem., ed. A. S. F. Gordon and W. Robert, Academic Press, 1996, vol. 39, pp. 355 Search PubMed; (c) P. P. Power, Chem. Rev., 1999, 99, 3463 CrossRef CAS PubMed; (d) M. Yamashita and K. Nozaki, Bull. Chem. Soc. Jpn., 2008, 81, 1377 CrossRef CAS; (e) C. Fedorchuk, M. Copsey and T. Chivers, Coord. Chem. Rev., 2007, 251, 897 CrossRef CAS.
- J. Yu, G. Kehr, C. G. Daniliuc and G. Erker, Eur. J. Inorg. Chem., 2013, 3312 CrossRef CAS.
- J. Möbus, G. Kehr, C. G. Daniliuc, R. Fröhlich and G. Erker, Dalton Trans., 2014, 43, 632 RSC.
- (a) D. J. Parks, R. E. von H. Spence and W. E. Piers, Angew. Chem., Int. Ed. Engl., 1995, 34, 809 CrossRef CAS; (b) D. J. Parks, W. E. Piers and G. P. A. Yap, Organometallics, 1998, 17, 5492 CrossRef CAS.
- See for comparison: (a) J. Yu, G. Kehr, C. G. Daniliuc, C. Bannwarth, S. Grimme and G. Erker, Org. Biomol. Chem., 2015, 13, 5783 RSC ; See also: ; (b) G. C. Welch, R. R. San Juan, J. D. Masuda and D. W. Stephan, Science, 2006, 314, 1124–1126 CrossRef CAS PubMed.
- (a) H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1966, 88, 1433 CrossRef CAS; (b) L. D. Field and S. P. Gallagher, Tetrahedron Lett., 1985, 26, 6125 CrossRef CAS; (c) C. Chen, M. Harhausen, R. Liedtke, K. Bussmann, A. Fukazawa, S. Yamaguchi, J. L. Petersen, C. G. Daniliuc, R. Fröhlich, G. Kehr and G. Erker, Angew. Chem., Int. Ed., 2013, 52, 5992 CrossRef CAS PubMed.
- For examples of chelate bis-borane chemistry: (a) S. P. Lewis, N. J. Taylor, W. E. Piers and S. Collins, J. Am. Chem. Soc., 2003, 125, 14686 CrossRef CAS PubMed; (b) P. A. Chase, L. D. Henderson, W. E. Piers, M. Parvez, W. Clegg and M. R. J. Elsegood, Organometallics, 2006, 25, 349 CrossRef CAS; (c) H. Zhao and F. P. Gabbaï, Organometallics, 2012, 31, 2327 CrossRef CAS; (d) A. Hübner, A. M. Diehl, M. Bolte, H.-W. Lerner and M. Wagner, Organometallics, 2013, 32, 6827 CrossRef.
- (a) D. J. Parks, W. E. Piers, M. Parvez, R. Atencio and M. J. Zaworotko, Organometallics, 1998, 17, 1369 CrossRef CAS; (b) D. J. Morrison and W. E. Piers, Org. Lett., 2003, 5, 2857 CrossRef CAS PubMed; (c) P. Spies, G. Erker, G. Kehr, K. Bergander, R. Fröhlich, S. Grimme and D. W. Stephan, Chem. Commun., 2007, 5072 RSC . See for comparison: ; (d) T. Mahdi and D. W. Stephan, J. Am. Chem. Soc., 2014, 136, 15809 CrossRef CAS PubMed; (e) D. J. Scott, M. J. Fuchter and A. E. Ashley, J. Am. Chem. Soc., 2014, 136, 15813 CrossRef CAS PubMed.
- (a) A. B. Burg and H. I. Schlesinger, J. Am. Chem. Soc., 1937, 59, 780 CrossRef CAS; (b) S. H. Bauer, J. Am. Chem. Soc., 1937, 59, 1804 CrossRef CAS; (c) R. D. Cowan, J. Chem. Phys., 1950, 18, 1101 CrossRef CAS; (d) W. Gordy, H. Ring and A. B. Burg, Phys. Rev., 1950, 78, 512 CrossRef CAS; (e) G. W. Bethke and M. K. Wilson, J. Chem. Phys., 1957, 26, 1118 CrossRef CAS . See for comparison: Q. Zhang, W.-L. Li, C.-Q. Xu, M. Chen, M. Zhou, J. Li, D. M. Andrada and G. Frenking, Angew. Chem., Int. Ed., 2015, 54, 11078 Search PubMed.
- M. Sajid, G. Kehr, C. G. Daniliuc and G. Erker, Angew. Chem., Int. Ed., 2014, 53, 1118 CrossRef CAS PubMed.
- See for comparison: (a) A. Berkefeld, W. E. Piers, M. Parvez, L. Castro, L. Maron and O. Eisenstein, J. Am. Chem. Soc., 2012, 134, 10843 CrossRef CAS PubMed; (b) R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 4974 CrossRef CAS PubMed.
- W. Tochtermann, Angew. Chem., Int. Ed. Engl., 1966, 5, 351 CrossRef CAS.
- D. W. Stephan and G. Erker, Angew. Chem., Int. Ed., 2015, 54, 6400 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental, analytical and structural details. CCDC 1418752–1418757. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5cc07954c |
| ‡ X-Ray crystal structure analysis. |
| This journal is © The Royal Society of Chemistry 2016 |