
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fast microwave treatments of single source alkoxides for nanostructured Li-ion battery electrodes†
Josefa Vidal
Laveda
a,
Vibhuti
Chandhok
a,
Claire A.
Murray
b,
Gary W.
Paterson
c and
Serena A.
Corr
*a
aSchool of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: serena.corr@glasgow.ac.uk; Tel: +44 (0)141 3302274
bDiamond Light Source, Didcot, Oxfordshire OX11 0DE, UK
cSchool of Physics and Astronomy, University of Glasgow, Glasgow G12 8QQ, UK
First published on 15th October 2015
Abstract
Microwave or ultrasonic treatment of metal alkoxides presents a fast, low cost route to both anode and cathode nanomaterials for Li-ion battery applications. Here, we demonstrate the formation of LiMPO4 (M = Fe, Mn) and Mn3O4 nanostructures via this simple route which exhibit excellent electrochemical performances. This approach opens up a new avenue for the targeted design of nanostructured materials, where co-location of the desired metals in a single starting material shortens reaction times and temperatures since there is a decrease in diffusional energy requirements usually needed for these reactions to proceed.
Control over the design of inorganic nanostructures of preferred crystal structure, particle size and morphology is highly desirable and challenges remain in achieving this in a reliable and reproducible manner. There are continuous research efforts to obtain electrode materials for use in Li-ion batteries with high energy densities to satisfy the increasing need for long lasting energy storage devices.1 Amongst the positive insertion electrodes, olivine-structured lithium transition metal phosphates such as LiFePO4 have been recognized as low cost, non-toxic, safe alternatives to layered LiCoO2 showing good thermal stability, high specific charge capacity (∼170 mA h g−1) and good cyclability.2 Crystalline LiMPO4 (M = Fe or Mn) has an orthorhombic unit cell in which O2− ions form strong covalent bonds with P5+ to form PO43− tetrahedral units stabilizing the entire network, assuring stable operation at high temperatures. Nanostructured insertion electrodes are of particular interest due to the potential improvements in cycling performance, as smaller sizes allow shorter path lengths for the Li-ions to diffuse, while the increased surface areas enhance electrode–electrolyte interactions.3 Also of interest is the possibility of partial substitution of Fe2+ with Mn2+ in the olivine structure to afford mixed metal LiFe1−xMnxPO4 phosphates with increased redox potential due to the higher Mn2+/3+ potentials compared to the Fe2+/3+ pair (3.45 V vs. Li+/Li0 for LiFePO4 and 4.10 V vs. Li+/Li0 for LiMnPO4).4 Therefore, there exist many opportunities to improve the behaviour and electrochemical performance of nanostructured electrode materials, beginning with careful consideration of the synthesis route employed. In the case of anode materials, metal oxides are finding increasing use as alternatives to graphite for negative electrodes in Li-ion batteries. Since transition metal oxides can react with more than one Li per transition metal atom, exceptionally high specific capacities are theoretically possible.5 However, the low electronic conductivity and large volume expansion associated with these materials often lead to poor cycling performance. Drastic volume variations during charging/discharging can cause disintegration of the electrode. Amongst several metal oxides, Mn3O4 is an attractive candidate for use as an anode material due to the high abundance of Mn, its environmental benignity and low cost.6 Furthermore, electrochemical studies of Mn3O4 nanostructures have shown that decreasing the particle size markedly reduces the polarization displayed by the electrodes and improves the reversibility of the cycling process and the rate capability. For example, Luo et al. demonstrated that a carbon nanotube–Mn3O4 composite displayed a reversible capacity of 1912.9 mA h g−1 for 10 nm particles compared to 367 mA h g−1 for 165 nm Mn3O4 particles.7 Increased capacities can also be realised through the use of conductive coatings such as reduced graphene oxide and the growth of porous or hollow morphologies.6,8,9,10
Often, routes to exotic morphologies or nanostructured particles can be time consuming and non-trivial. Fast, low temperature syntheses are therefore highly desirable for the preparation of high quality functional inorganic nanoparticles. Compared to conventional solid state reactions which often require high temperatures and long reaction times, syntheses employing microwave or ultrasonic irradiation can provide relatively low temperature routes to desired crystal structures that generally offer a significant reduction in reaction times and energy consumption.11–13 Our efforts have led us to develop a series of metalorganic precursors which provide an additional advantage for decreased reaction times and temperatures. Metal alkoxides have previously been employed as excellent starting materials for metal oxide thin films.14 The attractiveness of a heterometallic alkoxide as a starting material lies in their easy thermal decomposition and in the possibility of having, in a single compound, more than one of the desired metals with a known stoichiometry. Co-location of several metals in these precursors bypasses the need of diffusional mixing and could afford highly crystalline materials through a less energy consuming process. Here, we demonstrate the versatility of this approach for the clean preparation of both positive and negative electrodes for Li-ion batteries through relatively fast and low temperature microwave and ultrasound-assisted methods. Furthermore, electrochemical testing reveal the excellent performance of these nanostructures, establishing this synthetic approach as one to yield high performance electrode materials across the olivines and metal oxides.
Initially, the previously reported metal alkoxide [Fe(OtBu)2(THF)]2 and a new, similarly synthesized Mn alkoxide were employed as precursors for the preparation of olivine structured positive electrodes LiMPO4 (M = Fe and Mn) and the hausmannite structured negative electrode Mn3O4, according to Fig. 1. In the case of the olivine nanostructures, powder products were obtained after only ten minutes of microwave treatment. For the formation of Mn3O4, a hydrolysis route promoted by ultrasonic irradiation was used. Previous reports have demonstrated that hydrolysis of the metal alkoxide [Fe(OtBu)2(THF)]2 yields magnetite nanocrystals.15 In the current work, the Mn alkoxide analogue has been prepared for the first time and hydrolysed under ultrasonic treatment at room temperature to afford nanostructured Mn3O4. High resolution powder X-ray diffraction (XRD) measurements of the LiFePO4 and LiMnPO4 samples obtained at the I11 beamline at Diamond Light Source reveal phase-pure materials crystallised in the orthorhombic Pnma space group. The slight shifting of the peaks towards lower angles for LiMnPO4 is a signature of the larger cation size of Mn2+ in comparison to Fe2+. Powder XRD data were fit by Rietveld analysis, with excellent agreement to the Pnma model obtained.
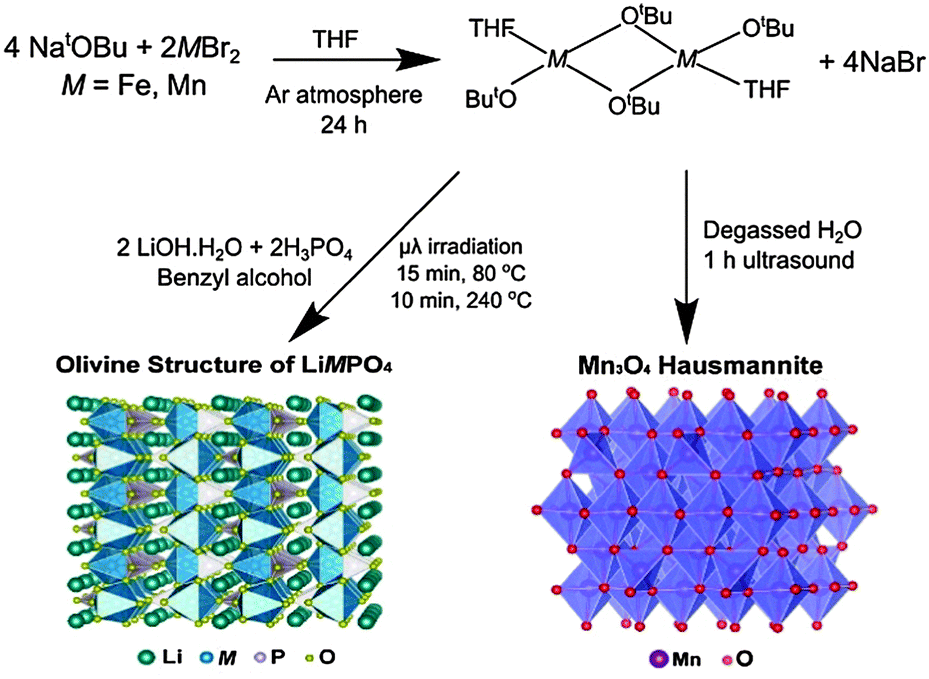 | ||
| Fig. 1 Reaction schematic for the synthesis of LiMPO4 (M = Fe or Mn) and Mn3O4 using metal alkoxide precursors. | ||
In order to establish this alkoxide route as applicable for the preparation of a range of LiFe1−xMnxPO4 materials, stoichiometric amounts of the individual Fe and Mn alkoxides were reacted following the same procedure to prepare LiFe0.5Mn0.5PO4. However, analysis of the resulting powder XRD pattern reveals a mixture of LiFe0.5Mn0.5PO4 and Fe3O4 magnetite is obtained (Fig. S1, ESI†). In an effort to avoid impurities, a bimetallic alkoxide with a suggested formula “[Fe0.5Mn0.5(OtBu)2(THF)]2” was prepared and used as a starting material. We suggest this formula according to our synthesis, but as yet we have not obtained a single crystal of sufficient quality to elucidate the crystal structure. Further efforts to characterise this compound are currently underway. However, we are confident that this route does not simply produce a mixture of the previous Fe- and Mn-alkoxides since the use of this proposed alkoxide to prepare LiFe0.5Mn0.5PO4 using the same conditions as Fig. 1 affords single phase products. Rietveld refinements of the high resolution powder XRD data shown in Fig. 2(b) indicates that phase pure LiFe0.5Mn0.5PO4 was obtained, with no evidence of any impurities. Table S1 (ESI†) summarises the calculated lattice parameters for the different olivines. In the case of the ultrasonic treatment of the “[Mn(OtBu)2(THF)]2” precursor, a brown powder was obtained which, when characterised with XRD, could be fit to a I41/amdZ space group, confirming phase pure Mn3O4 hausmannite (Fig. S2, ESI†). SEM images of the LiFePO4 sample revealed non-uniform platelets, with sizes ranging from 50 to 250 nm. Elongated particles were obtained for LiFe0.5Mn0.5PO4, with rod-like particles found for LiMnPO4. This highlights the strong influence of the Mn content on the resulting particle morphology and size. The fringes visible in TEM images of the LiFe1−xMnxPO4 (x = 0, 0.5 and 1) samples (Fig. S3, ESI†) reveal the samples are crystalline and lattice spacings are consistent with the planes indicated the images.16 The insets are Fourier transforms of the TEM images. Indexing of the patterns reveals the crystal long-axes, indicated by the green lines in the images, lie within a few degrees of the [101], [020] and [112] axes for LiMnPO4, LiFePO4 and LiFe0.5Mn0.5PO4, respectively. Fig. 3(d) reveals quasi-spherical Mn3O4 nanoparticles with a typical size of 80 nm.
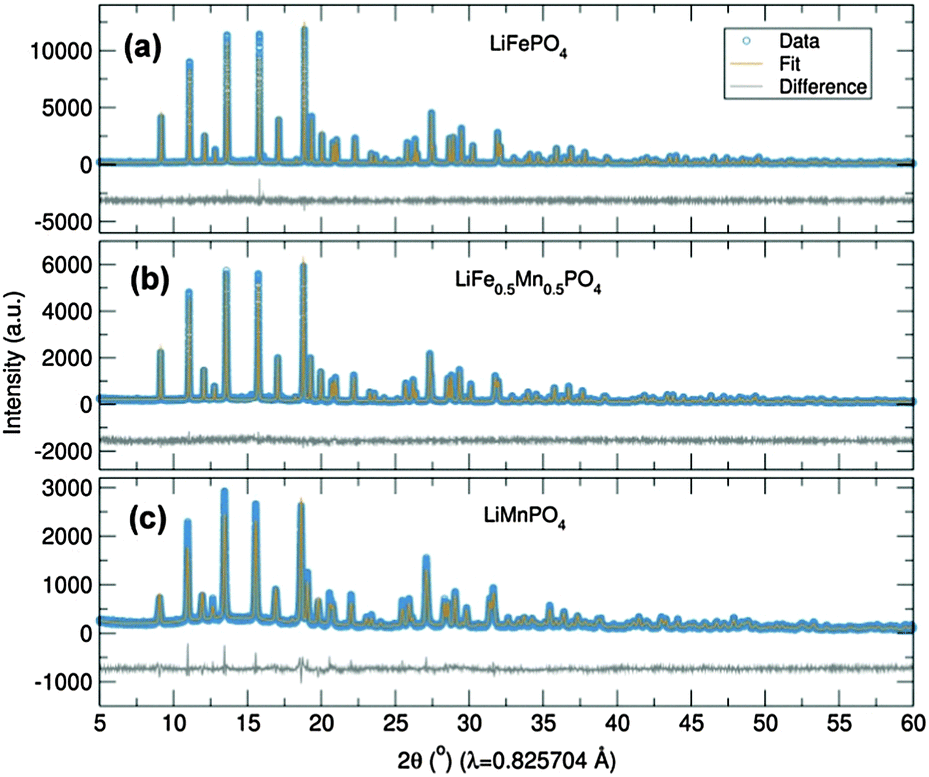 | ||
| Fig. 2 Rietveld analysis of XRD data of (a) LiFePO4, (b) LiFe0.5Mn0.5PO4 and (c) LiMnPO4 nanostructures prepared from [Fe(OtBu)2(THF)]2, “[Fe0.5Mn0.5(OtBu)2(THF)]2” and “[Mn(OtBu)2(THF)]2” alkoxides, respectively. | ||
 | ||
| Fig. 3 SEM images of (a) LiFePO4, (b) LiFe0.5Mn0.5PO4, (c) LiMnPO4 and (d) Mn3O4 nanostructures prepared from metal alkoxide precursors. | ||
Galvanostatic cycling at room temperature of the LiFePO4 sample was conducted over the voltage range of 2.2 to 4.0 V at a C/20 rate in a Swagelok cell-type. To enhance the electronic conductivity, a simple carbon coating using sucrose was performed, as poor electron mobility will limit the Li+ extraction/insertion. From the voltage-composition plot (Fig. S4, ESI†) it can be observed that approximately 0.6 Li+ were reversibly extracted during the cycling process. It is well established that the major LiFePO4 capacity is attributed to the Li+ insertion/extraction process from the LiFePO4 particles (LiFePO4 ⇌ FePO4 + Li+ + e−). The two main limiting factors reported for why Li-ions cannot be fully deinserted from the ordered-olivine structure are: limited Li+ phase-boundary diffusion and the low electronic conductivity during the charge/discharge processes. For example, Li+ diffusion can be blocked by ionic disorder, foreign phases or stacking faults, impeding the reversible LiFePO4/FePO4 phase transformation.17 The representative charge and discharge voltage profile for the LiFePO4 nanoparticles obtained through microwave-treatment of the alkoxide precursor shown in Fig. 4(a) indicates that charge and discharge capacities of approximately 150 mA h g−1 were obtained, close to the theoretical capacity of this material (170 mA h g−1). Furthermore, there is almost no capacity fading over at least 20 cycles [inset, Fig. 4(a)]. The rate behaviour of the C/LiFePO4 and the C/LiFe0.5Mn0.5PO4 phases was also investigated in order to examine the rate capabilities as a function of Mn content and particle morphology (see also Fig. S5, ESI†). Fig. 4(b) shows that the discharge capacities of the C/LiFePO4 and C/LiFe0.5Mn0.5PO4 phases decrease with higher C rates. This effect is more pronounced for the LiFePO4 sample. High C rates up to 10C delivered discharge capacities of 75 mA h g−1 and 50 mA h g−1 for the C/LiFe0.5Mn0.5PO4 and C/LiFePO4 phases, respectively.
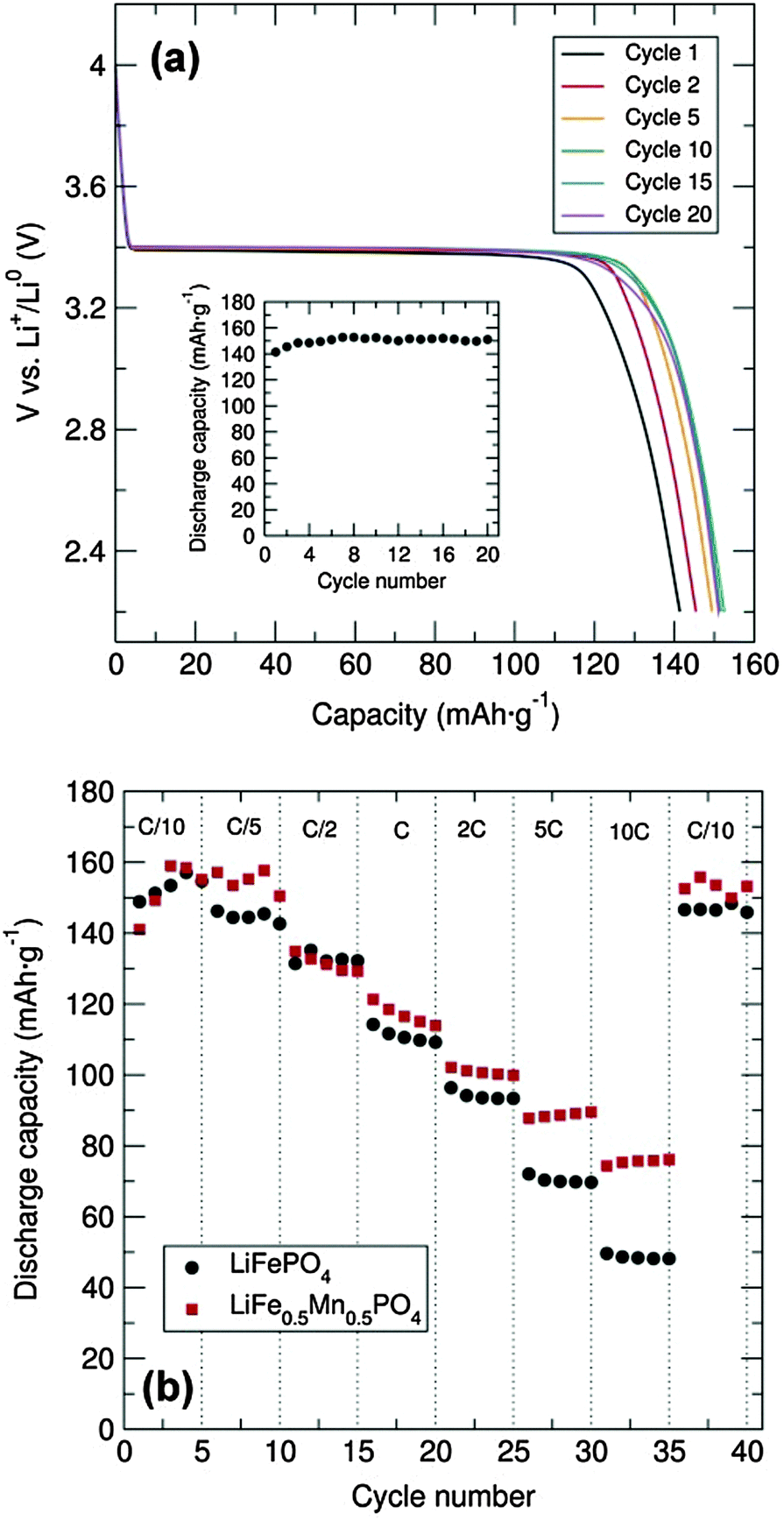 | ||
Fig. 4 (a) Cycling performance and capacity fading (inset) of C/LiFePO4 prepared from [Fe(OtBu)2(THF)]2 alkoxide precursor (mixed with C black and PTFE in 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30:10 weight%) between 2.2 and 4.0 V at C/20 rate. (b) Rate performance of C/LiFePO4 and C/LiFe0.5Mn0.5PO4 (mixed with C black and PTFE in 60:30:10) at different charge–discharge C rates. :30:10 weight%) between 2.2 and 4.0 V at C/20 rate. (b) Rate performance of C/LiFePO4 and C/LiFe0.5Mn0.5PO4 (mixed with C black and PTFE in 60:30:10) at different charge–discharge C rates. | ||
The C/LiFe0.5Mn0.5PO4 presented here showed an improved rate capability at 10C in comparison to the ∼55 mA h g−1 at 8C reported previously for LiFe0.5Mn0.5PO4 prepared via an electrospun synthetic procedure.18 Moreover, it must be noted that in both cases here our capacity was recovered when cycling back to C/10 rate after increasingly faster rates. These results suggest that partial substitution of Fe2+ by Mn2+ in the olivine structure affords a noticeable enhancement in the rate capability of these electrodes. The observed improvement in the electrochemical performance of the C/LiFe0.5Mn0.5PO4 material in comparison to C/LiFePO4 could also be attributed to the decrease in particle size. XRD characterization of the cycled materials in the discharge state showed that the extraction/insertion process did not affect the structure of the C/LiFePO4 and the C/LiFe0.5Mn0.5PO4 composites (Fig. S6 and S7, ESI†), confirming the good structural stability of these positive insertion electrodes upon cycling. Cyclic voltammetry (CV) measurements of the C/LiFePO4 and the C/LiFe0.5Mn0.5PO4 composites were performed at a scan rate of 0.1 mV s−1 between 2.5 and 4.5 V and demonstrated good electrochemical reversibility (Fig. S8, ESI†). Electrochemical measurements of C/Mn3O4 at a current density of 100 mA h g−1 over a 0.01–3.00 V range revealed an initial discharge capacity of ∼3200 mA h g−1 that sharply decreased to approximately 1300 mA g−1 at the second cycle (Fig. S9, ESI†). It must be remarked that this initial discharge capacity of the C/Mn3O4 composite was significantly larger than previous reports.19–22 The large capacity drop after the first cycle could be attributed to irreversible processes such as solid electrolyte interface formation and electrolyte decomposition. Fig. S7 (ESI†) shows a drastic decrease in the capacity with cycling but a discharge capacity of 460 mA h g−1 is still reached at the 20th cycle. Lack of Bragg diffraction peaks in the XRD patterns of the cycled C/Mn3O4 material suggests degradation, possibly due to significant volume changes (Fig. S10 and S11, ESI†). Studies are currently underway to try to alleviate this breakdown through surface coatings. CV shows good peak overlap after the first cycle, confirming the good cycling reversibility (Fig. S12, ESI†). An intense cathodic peak at around 0.3 V and an anodic peak at approximately 1.4 V were observed. The possible formation of a solid electrolyte interface and/or electrolyte decomposition during the first charge/discharge cycle is confirmed from the difference observed in the first discharge CV curve.
We have presented a low cost and green synthetic approach for the preparation of metal alkoxides to be used as precursors in the synthesis of phase pure olivine nanostructured LiFe1−xMnxPO4 (x = 0, 0.5 and 1) and Mn3O4 hausmannite nanocrystals through fast microwave and ultrasound-assisted routes, respectively. Moreover, we have demonstrated the versatility of these metal alkoxide precursors for the synthesis of both cathode and anode materials for Li-ion batteries. Electrochemical testing of LiFePO4 and LiFe0.5Mn0.5PO4 samples revealed excellent cyclability with charge/discharge capacities close to theoretical values (∼155 mA h g−1vs. 170 mA h g−1 at C/20 rate). These positive insertion electrodes also displayed rate capabilities comparable to the best performing olivine-structured metal phosphates in the literature, suggesting the suitability of the metal alkoxide precursors presented here in the generation of high quality olivine mixed-metal phosphates via a fast microwave-assisted synthesis. Further improvements in the carbon coating of our metal oxide nanoparticles synthesised by a straightforward room temperature ultrasound-assisted synthesis could dramatically enhance the battery cycling performance. Our future work will concentrate on the preparation of heterometallic alkoxides containing both Li and the desired transition metal in a single precursor in an effort to obtain highly crystalline nanostructures with excellent battery performance in a single step.
The authors gratefully acknowledge the allocation of beamtime at the I11 beamline at the Diamond Light Source. This work was supported by funding from the EPSRC (EP/K026290/1 and EP/N001982/1). We thank the School of Chemistry at the University of Glasgow for PhD funding and support.
Notes and references
- J.-M. Tarascon and M. Armand, Nature, 2001, 414 Search PubMed; B. Kang and G. Ceder, Nature, 2009, 458 Search PubMed.
- H. Liu, F. C. Strobridge, O. J. Borkiewicz, K. M. Wiaderek, K. W. Chapman, P. J. Chupas and C. P. Grey, Science, 2014, 344, 6191 CrossRef PubMed.
- A. Manthiram, A. Vadivel Murugan, A. Sarkar and T. Muraliganth, Energy Environ. Sci., 2008, 1, 621 CAS.
- V. Aravindan, J. Gnanaraj, Y.-S. Lee and S. Madhavi, J. Mater. Chem. A, 2013, 1, 3518 CAS.
- H. Kim, D.-H. Seo, H. Kim, I. Park, J. Hong, K.-Y. Park and K. Kang, Chem. Mater., 2012, 24, 720 CrossRef CAS.
- H. Wang, L.-F. Cui, Y. Yang, H. Sanchez Casalongue, J. Tucker Robinson, Y. Liang, Y. Cui and H. Dai, J. Am. Chem. Soc., 2010, 132, 13978 CrossRef CAS PubMed.
- S. Luo, H. Wu, Y. Wu, K. Jiang, J. Wang and S. Fan, J. Power Sources, 2014, 249, 463 CrossRef CAS.
- C. Chen, H. Jian, X. Fu, Z. Ren, M. Yan, G. Qian and Z. Wang, RSC Adv., 2014, 4, 5367 RSC.
- J. Yue, X. Gu, L. Chen, N. Wang, X. Jiang, H. Xu, J. Yang and Y. Qian, J. Mater. Chem. A, 2014, 2, 17421 CAS.
- Z. Bai, X. Zhang, Y. Zhang, C. Guo and B. Tang, J. Mater. Chem. A, 2014, 2, 16755 CAS.
- Y. Luo, S. Fan, N. Jao, S. Zhong and W. Liu, Dalton Trans., 2014, 43, 15317 RSC.
- I. Bilecka, A. Hintennach, I. Djerdj, P. Novák and M. Niederberger, J. Mater. Chem., 2009, 19, 5125 RSC; I. Bilecka, A. Hintennach, M. D. Rossell, D. Xie, P. Novák and M. Niederberger, J. Mater. Chem., 2011, 21, 5881 RSC; T. E. Ashton, J. Vidal Laveda, D. A. MacLaren, P. J. Baker, A. Porch, M. O. Jones and S. A. Corr, J. Mater. Chem. A, 2014, 2, 6238 Search PubMed.
- F. Yu, L. Zhang, M. Zhu, Y. An, L. Xia, X. Wang and B. Dai, Nano Energy, 2014, 3, 64 CrossRef CAS.
- M. Mantymaki, M. Ritala and M. Leskela, Coord. Chem. Rev., 2012, 256, 854 CrossRef CAS.
- G. B. Biddlecombe, Y. Guńko, J. M. Kelly, S. C. Pillai, J. M. D. Coey, M. Venkatesan and A. P. Douvalis, J. Mater. Chem., 2001, 11, 2937 RSC; S. A. Corr, Y. K. Gun’ko, A. P. Douvalis, M. Venkatesan and R. D. Gunning, J. Mater. Chem., 2004, 14, 944 RSC; S. A. Corr, Y. Guńko, A. P. Douvalis, M. Venkatesan, R. D. Gunning and P. D. Nellist, J. Phys. Chem. C, 2008, 112, 1008 Search PubMed.
- G. Qin, S. Xue, Q. Maa and C. Wang, CrystEngComm, 2014, 16, 260 RSC.
- L.-H. Hu, F.-Y. Wu, C.-T. Lin, A. N. Khlobystov and L.-J. Li, Nat. Commun., 2013, 4, 1687 CrossRef PubMed.
- R. von Hagen, H. Lorrmann, K.-C. Möller and S. Mathur, Adv. Energy Mater., 2012, 2, 553 CrossRef CAS.
- H. Huang, L. Zhang, Y. Xia, Y. Gan, X. Tao, C. Liang and W. Zhang, New J. Chem., 2014, 38, 4743 RSC.
- J. Gao, M. A. Lowe and H. D. Abruña, Chem. Mater., 2011, 23, 3223 CrossRef CAS.
- Y. Liu, W. Wang, Y. Wang, Y. Ying, L. Sun and X. Peng, RSC Adv., 2014, 4, 16374 RSC.
- I. Nam, N. D. Kim, G.-P. Kim, J. Park and J. Yi, J. Power Sources, 2013, 244, 56 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc07732j |
| This journal is © The Royal Society of Chemistry 2016 |