
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of glycosylphosphatidylinositol (GPI)-anchor glycolipids bearing unsaturated lipids†
B.-Y.
Lee
a,
P. H.
Seeberger
ab and
D.
Varon Silva
*ab
aMax Planck Institute of Colloids and Interfaces, Biomolecular Systems, Am Mühlenberg 1, 14476 Potsdam, Germany. E-mail: daniel.varon@mpikg.mpg.de
bDepartment of Chemistry and Biochemistry, Freie Universität Berlin, Arnimallee 22, 14195 Berlin, Germany
First published on 30th November 2015
Abstract
2-Naphthyl-methyl ethers as permanent protecting groups are readily removed under acidic conditions and are key to the synthesis of complex glycosylphosphatidylinositol anchors containing unsaturated lipids. The total synthesis of the GPI pseudo-disaccharide core found on the surface of the Trypanosoma cruzi parasite serves to illustrate the power of the strategy.
GPIs are complex glycolipids that are ubiquitous in eukaryotic cells and have a common pseudo-pentasaccharide core structure 6-O-NEtP-Manα1-2-Manα1-6-Manα1-4-GlcNα1-6-Ino-1-P (Fig. 1).1 This conserved core is generally modified by additional phosphorylations, glycosylations, or acylations at the 2-O position of the myo-inositol in a cell-type dependent manner.2 GPIs are heterogeneous in the glycan as well as the lipid part. Inositol can bear sn-1-alkyl-2-acylglycerol, sn-1,2-diacylglycerol, or ceramide adorned with lipid chains of different lengths and degrees of saturation.3
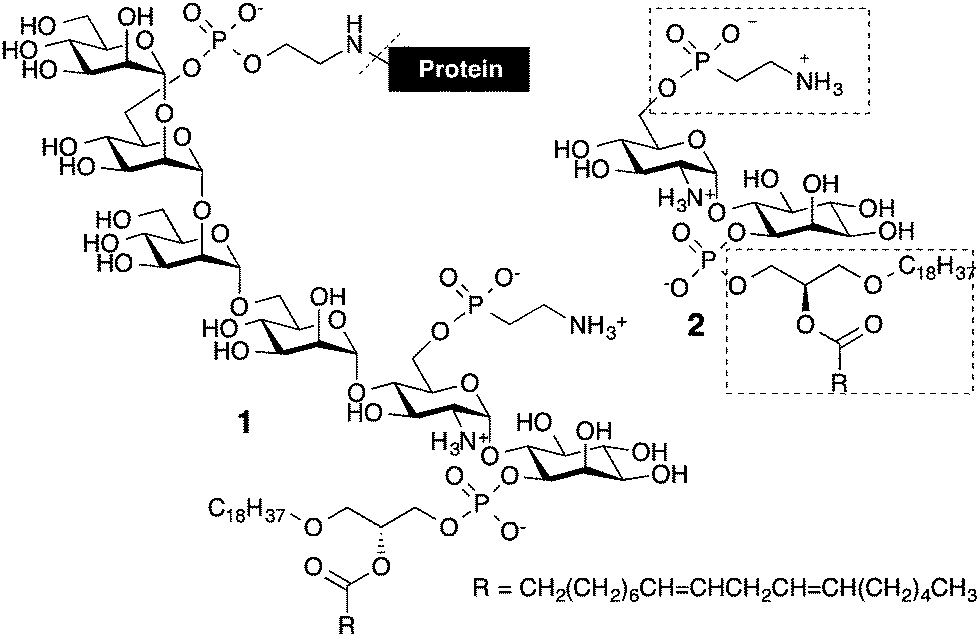 | ||
| Fig. 1 Glycosylphosphatidylinositol anchor of the parasite Trypanosoma cruzi. | ||
Chagas disease, caused by the parasite Trypanosoma cruzi (T. cruzi), is a major public health problem in Latin America, infects around 7–8 million persons worldwide, and causes more than 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 deaths each year.4 There is no vaccine for this disease and the two drugs available for treatment are used sparingly due to their cost, side effects, and low antiparasitic activity in patients with chronic infections.5
000 deaths each year.4 There is no vaccine for this disease and the two drugs available for treatment are used sparingly due to their cost, side effects, and low antiparasitic activity in patients with chronic infections.5
The cell surface of T. cruzi contains a high concentration of glycosylphosphatidylinositol (GPI) molecules, which exhibit proinflammatory activities comparable to bacterial lipopolysaccharides and are predominately attached to highly glycosylated mucins and phosphoglycans.6,7 Structural hallmarks of these GPIs are a glycan branch of galactoses, a T. cruzi specific 2-aminoethylphosphonate (2-AEP) unit at the 6-O position of the glucosamine residue and the presence of unsaturated fatty acids in the phospholipid,8,9 which have been associated with the biological activity of these molecules.6,10
Isolation of homogeneous GPIs is extremely difficult due to the heterogeneity and amphiphilic character of both the glycan and lipid. Biological evaluation of T. cruzi GPIs and potential applications for the diagnosis and prevention of Chagas disease require synthetic GPIs bearing unsaturated lipids. Most synthetic strategies for GPI glycolipids use benzyl ethers as permanent carbohydrate protecting groups. However, the reductive conditions required for benzyl ether removal are not compatible with the double bonds present in the lipid moiety.11 While this issue can be avoided through the use of benzoyl esters or PMB ethers,12,13 their use in this application has been limited due to the saponification of the fatty acid esters during base-mediated benzoyl ester removal and the low stability of PMB ethers under the mild acidic conditions commonly used for glycosylations.13–15
While the 2-naphthylmethyl ether (Nap) group has primarily been used as a temporary mask for hydroxyl groups in carbohydrate chemistry,16,17 its stability during glycosylation reactions and its orthogonality to silyl ethers, acyl esters, and even PMB ethers make it the ideal group for permanent protection during GPI synthesis.
Herein, we report an efficient strategy for the synthesis of GPIs bearing unsaturated lipids. The key of our strategy is the use of stable 2-naphthylmethyl ethers for permanent hydroxyl group protection, which are readily removed using acidic conditions. The strategy is illustrated for the synthesis of a portion of the GPI anchor from T. cruzi.
We considered an assembly sequence based on our recently reported strategy for accessing GPIs with saturated lipids, in which the glycan is assembled first and the phosphorylations are installed at a late-stage of the synthesis.17,18 In this strategy, Nap ethers replaced the benzyl ethers as permanent protecting groups and the Allyl and PMB ethers were included as temporary protecting groups for positions requiring phosphorylation, while an azide served as a masked amine.
To obtain the bis-phosphorylated GPI pseudo-disaccharide 2, a number of building blocks were envisioned branching from the core glycan fragment 3 containing Nap ethers as permanent protecting groups. For the subsequent phosphorylations, building blocks 4 and 5 were necessitated (Fig. 2). To assemble the glycan moiety, building blocks 6 and 7 were designed to generate the desired α-glucosylated myo-inositol. Through a series of protecting group manipulations, the pseudo-disaccharide glycan GlcN-Ino 3 was obtained with both a PMB and an allyl-protecting group. Selective step-wise removal of the PMB and allyl ethers, followed by phosphorylations and global deprotection, will complete the synthesis.
 | ||
| Fig. 2 Retrosynthetic analysis of the T. cruzi GPI pseudo-disaccharide containing an unsaturated lipid chain. | ||
Synthesis of the protected optically pure D-myo-inositol building block 7 started from methyl-glucoside.19,20 The primary alcohol was protected with a trityl group, followed by the overnight per-naphthyl-methylation using 2-(naphthyl)-methyl bromide and NaH (Scheme 1). After removal of the trityl ether, alcohol oxidation with SO3-pyridine complex to the aldehyde, and subsequent acetylation produced enol-ester 10. A mercury II-mediated Ferrier type-2 rearrangement afforded intermediate 11 in good yield (68%). Stereoselective reduction of ketone 11 with NaBH(OAc)3 followed by deacetylation under Zemplen conditions delivered triol 12. Regioselective allylation of 12 using bis(tributyltin) oxide and the selective protection at O-2 as a napthyl-methyl ether gave building block 7 in 19% overall yield. The Nap group did not affect the process when compared to benzyl ethers and delivered the protected myo-inositol with improved yield.20
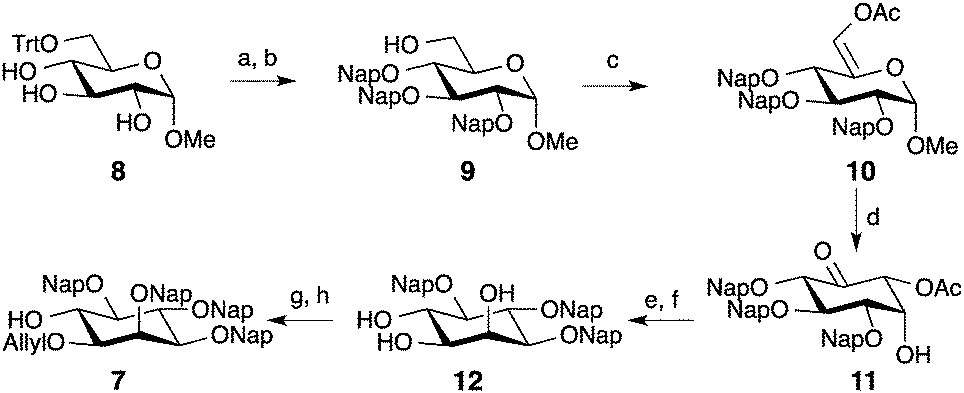 | ||
| Scheme 1 Synthesis of protected myo-inositol 7. Reaction conditions: (a) NapBr, NaH, DMF, rt, overnight, 85%; (b) p-TsOH, MeOH, rt, 16 h, 90%; (c) i. SO3-Py, DIPEA, DMSO, CH2Cl2, 0 °C, 1 h, ii. Ac2O, K2CO3, MeCN, reflux, 4 h, 80% (2 steps); (d) i. Hg(OTf)2, acetone/H2O, rt, 1 h, ii. NaOAc, NaCl, 0 °C to rt, 12 h; (e) NaBH(OAc)3, MeCN, AcOH, rt, 12 h, 68% (2 steps); (f) NaOMe, MeOH, rt, 30 min; (g) i. (Bu3Sn)2O, toluene, reflux, 5 h, ii. AllBr, TBAI, toluene 65 °C, 17 h, 68% (20% recovered starting material); (h) NapBr, NaH, DMF, −20 °C to 0 °C, 2 h, 67%. | ||
For the synthesis of GlcN-Ino pseudo-disaccharide 3, the required glucosamine donor 6 was obtained from glucosamine azide 13 by transformation of the anomeric acetyl group into a trichloroacetimidate using a two-step protocol. Three different conditions were tested to obtain the desired α-glycosidic linkage.21 Product 14 was obtained in the best α/β ratio (11:1) at room temperature, using Et2O/CH2Cl2 as solvent and TMSOTf as an activator (Scheme 2).
 | ||
| Scheme 2 Synthesis of Glc-Ino pseudo-disaccharide 3. Reaction conditions: (a) i. NH3(g), CH3CN, 0 °C to rt, 1 h, ii. CCl3CN, DBU, CH2Cl2, 0 °C, 15 min (α/β mixture donor, for b and c) or CCl3CN, K2CO3, CH2Cl2, rt, 5 h (β donor, for d); (b)TMSOTf, Et2O/CH2Cl2(6:1), 0 °C, 1 h, 87%; (c) TMSOTf, Et2O/CH2Cl2(6:1), rt, 1 h, 85%; (d) TMSOTf, CH2Cl2, rt, 30 min, 91%; (e) i. NaOMe, MeOH, rt, 1 h, ii. Anisaldehyde dimethylacetal, CSA, DMF, rt, overnight, 86% (2 steps); (f) NapBr, NaH, TBAI, DMF, rt, overnight, 88%; (g) NaCNBH3, TFA, THF/CH2Cl2, rt, 9 h, 80%; (h) NapBr, NaH, DMF, rt, overnight, 82%. PMP: p-methoxyphenyl. | ||
After separation of the α/β isomers of 14 by silica gel chromatography, the acetyl groups were removed using Zemplen conditions and the 4-O and 6-O hydroxyls were protected as a p-methoxybenzylidene acetal by reacting the generated triol with anisaldehyde dimethylacetal under acidic conditions. Nap protection of the free 3-O position of disaccharide 15, followed by selective opening of the 4,6-acetal group using NaCNBH3 under acidic conditions, and protection of the 4-O position delivered the fully protected 3 in good yield (Scheme 2).
With the glycan part in hand, the synthesis of the phosphorylated building blocks 4 and 5 was advanced. Etherification of alcohol 17 using 1-bromooctadecane and NaH, followed by the hydrolysis of the isopropylidene acetal and subsequent protection of the primary alcohol using a TBS group furnished 19 (Scheme 3a).
 | ||
| Scheme 3 Synthesis of phosphorylation synthons. Reagents and conditions: (a) 1-Bromooctadecane, NaH, Toluene, 97%; (b) i. HCl, MeOH, 15%, ii. TBSCl, Imidazole, DMF, 75%; (c) i. linoleic acid, DCC, DMAP, CH2Cl2, r.t., ii. BF3–Et2O, CH2Cl2, 0 °C, 60% (over two steps); (d) bis(diisopropylamino)(2-cyanoethoxy)phosphine, 1H-tetrazole, CH2Cl2, 90%; (e) LiBr, 2-pentanone, 110 °C, 2 h, quant.; (f) Oxalylchloride, DMF cat., CH2Cl2, rt, 1 h; (g) TMSBr, CH2Cl2, 0 °C to rt, overnight. | ||
Acylation of alcohol 19 with linoleic acid using DCC/DMAP and deprotection of the silyl group provided the desired alkylacylglycerol 20 (Scheme 3). To obtain the required phosphoamidite building block, glycerol derivative 20 was transformed into 4 using the commercially available bis(diisopropylamino)(2-cyanoethoxy)phosphine and 1H-tetrazole.22
Ethyl-(2-azidoethyl)phosphonochloridate (5) and bis(chloro)(2-azidoethyl)phosphonate (5a) were obtained from commercially available diethyl (2-bromoethyl)phosphonate. Starting with the conversion of bromide into the corresponding azide 21,12 the obtained ethyl phosphonate 21 was converted into chloro-phosphonate 5 using a two-step protocol. First, ethylphosphonate 21 was hydrolyzed with LiBr to provide phosphonic acid mono ethyl ester 22,23 which was converted into 5 by treatment with oxalylchloride.24 Phosphonodichloridate 5a was synthesized from ethyl phosphonate 21via silylated intermediate 23, which underwent reaction with oxalyl chloride.25
Two phosphorylation sequences of 3 were evaluated. In the first case, the PMB group of pseudo-disaccharide 3 was selectively removed under acidic conditions and without affecting the Nap groups to obtain alcohol 24.16 This compound was further phosphitylated with 5 in the presence of 1H-tetrazole and DIPEA. Following reduction of the azides using DTT/DIPEA and amine protection with Boc anhydride the phosphonate compound 26 was delivered, which was deallylated using PdCl2 and NaOAc in AcOH. To complete the synthesis of the fully modified pseudo-disaccharide, the free hydroxyl group of 27 was phosphitylated with 4 and oxidized to the corresponding phosphorylated product 28 by using tert-butylhydroperoxide (Scheme 4).
 | ||
| Scheme 4 Reagents and conditions: (a) 15% TFA, CH2Cl2, 0 °C, 15 min, 91%; (b) 5, DIPEA, 1H-tetrazole, toluene, 0 °C to rt, overnight, 72%; (c) i. dithiothreitol, DIPEA, MeCN/H2O, rt, 2 h, ii. Boc2O, DIPEA, MeOH, rt, 2 h, 86% (2 steps); (d) PdCl2, NaOAc, AcOH, rt, 6 h, 71%; (e) i. 4, 1H-tetrazole, CH2Cl2/MeCN, rt, 5 h, ii. t-BuOOH, Me2S, −40 °C, 2 h, 64% (2 steps). | ||
Removal of the protecting groups from 2-aminoethylphosphonate proved to be more challenging than expected. High stability of the phosphorous ethyl ester in 28 was observed after reduction of the azide and Boc protection of the formed amines. A variety of conditions were evaluated for the removal of the ester (see ESI†); however they resulted in non-reaction or in the decomposition of the starting material when long reaction times and strong reaction conditions were used. As such, an alternative assembly/deprotection sequence was needed. First, the allyl group of pseudo-disaccharide 3 was removed in a two-step protocol involving an iridium catalyzed isomerization and hydrolysis with mercury chloride. The resulting free hydroxyl group was phosphitylated with phosphoamidite 4 and oxidized with t-BuOOH to deliver phospholipidated pseudo-dissacharide 30.
To install the 2-azidoethylphosphonate unit, the PMB on the glucosamine of 30 was selectively removed under acidic conditions using 15% TFA in DCM (Scheme 5). Then the hydroxyl group was reacted with 5a using 1H-tetrazole and DIPEA activation at 0 °C, followed by the hydrolysis of the remaining chloride by addition of water. The remaining protecting groups on the fully decorated pseudo-disaccharide were removed via a three-step protocol. Efficient removal of the cyanoethoxy group of the phospholipid with DBU was followed by azide reduction with zinc in AcOH. Different conditions were evaluated for the final removal of the Nap groups. Methods involving oxidative conditions (DDQ) delivered a mixture of compounds due to incomplete deprotection. However, by using TFA/toluene 8:1 at 0 °C, target 2 was obtained in high yield (72% for the three deprotection steps) after purification using size exclusion chromatography.26
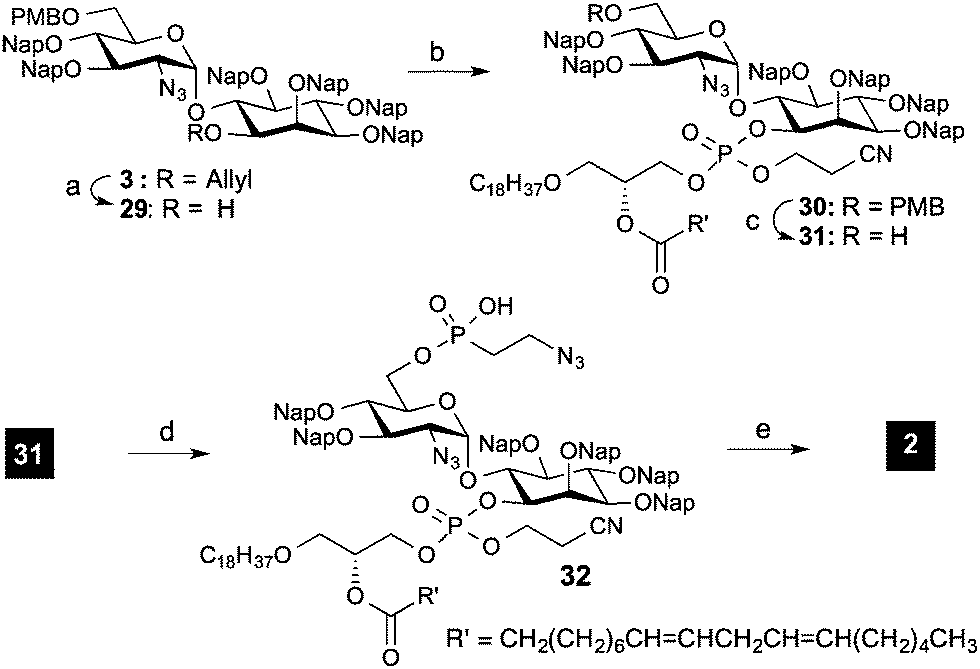 | ||
| Scheme 5 Reagents and conditions: (a) i. Ir(COD)2Cl2, THF, rt, 5 h, ii. HgCl2, HgO, acetone, rt, 1 h, 91%; (b) i. 4, 1H-tetrazole, CH2Cl2, rt, ii. t-BuOOH, Me2S, −40 °C, 2 h, 87% (2 steps); (c) TFA, CH2Cl2, 0 °C, 80%; (d) i. 5a, 1H-tetrazole, DIPEA, toluene, 0 °C, 2 h, ii. H2O, rt, 1 h, 87%; (e) i. DBU, CH2Cl2, rt, ii. Zn, AcOH, CH2Cl2, rt, iii. TFA, toluene, 0 °C, 72% (calculated from 32). | ||
In summary, a new strategy to obtain glycosylphosphatidyl-inositol anchors containing unsaturated lipid chains has been disclosed. The strategy is based on the use of 2-napthyl-methyl ethers for permanent protection and allyl and PMB ethers as orthogonal groups for masking positions requiring late stage modifications. The synthesis of the required building blocks demonstrated that the size of the Nap group did not affect the reactivity of donors and acceptors and can easily replace benzyl ether in established protocols. We described the synthesis of the complex Glc-Ino pseudo-disaccharide core of the T. cruzi parasite GPI, which bears a lipid moiety, and is modified by a linoleic acid ester and a 2-aminophosphoethylamine. We demonstrated the high stability of the Nap group in glycosylation and phosphorylation reactions and its removal under acidic conditions without affecting the phosphorylations, the alkyl ether on alkylacylglycerol, or the fatty acid containing the double bond. This strategy will enable the synthesis of diverse glycosylphosphatidylinositols and other glycolipids requiring unsaturated lipids and give access to pure GPIs for biological investigation.
We thank the Max Planck Society and the RIKEN-Max Planck Joint Center for Systems Chemical Biology for financial support. We thank Ankita Malik for her support in the deprotection and characterization of the final compound.
Notes and references
- M. G. Paulick and C. R. Bertozzi, Biochemistry, 2008, 47, 6991–7000 CrossRef CAS PubMed.
- M. A. J. Ferguson and A. F. Williams, Annu. Rev. Biochem., 1988, 57, 285–320 CrossRef CAS PubMed.
- M. J. McConville and M. A. Ferguson, Biochem. J., 1993, 294, 305–324 CrossRef CAS PubMed.
- A. Rassi Jr, A. Rassi and J. A. Marin-Neto, Lancet, 2010, 375, 1388–1402 CrossRef.
- E. Chatelain, J. Biomol. Screening, 2015, 20, 22–35 CrossRef PubMed.
- I. C. Almeida, M. M. Camargo, D. O. Procopio, L. S. Silva, A. Mehlert, L. R. Travassos, R. T. Gazzinelli and M. A. J. Ferguson, EMBO J., 2000, 19, 1476–1485 CrossRef CAS PubMed.
- R. M. de Lederkremer, C. Lima, M. I. Ramirez, M. A. Ferguson, S. W. Homans and J. Thomas-Oates, J. Biol. Chem., 1991, 266, 23670–23675 CAS.
- A. A. Serrano, S. Schenkman, N. Yoshida, A. Mehlert, J. M. Richardson and M. A. Ferguson, J. Biol. Chem., 1995, 270, 27244–27253 CrossRef CAS PubMed.
- J. O. Previato, C. Jones, M. T. Xavier, R. Wait, L. R. Travassos, A. J. Parodi and L. Mendonca-Previato, J. Biol. Chem., 1995, 270, 7241–7250 CrossRef CAS PubMed.
- M. M. Camargo, I. C. Almeida, M. E. Pereira, M. A. Ferguson, L. R. Travassos and R. T. Gazzinelli, J. Immunol., 1997, 158, 5890–5901 CAS.
- Y. H. Tsai, X. Y. Liu and P. H. Seeberger, Angew. Chem., Int. Ed., 2012, 51, 11438–11456 CrossRef CAS PubMed.
- D. V. Yashunsky, V. S. Borodkin, M. A. J. Ferguson and A. V. Nikolaev, Angew. Chem., Int. Ed., 2006, 45, 468–474 CrossRef CAS PubMed.
- B. M. Swarts and Z. W. Guo, J. Am. Chem. Soc., 2010, 132, 6648–6650 CrossRef CAS PubMed.
- B. M. Swarts and Z. W. Guo, Chem. Sci., 2011, 2, 2342–2352 RSC.
- L. Yan and D. Kahne, Synlett, 1995, 523–524 CrossRef CAS.
- J. Xia, S. A. Abbas, R. D. Locke, C. F. Piskorz, J. L. Alderfer and K. L. Matta, Tetrahedron Lett., 2000, 41, 169–173 CrossRef CAS.
- M. J. Gaunt, J. Yu and J. B. Spencer, J. Org. Chem., 1998, 63, 4172–4173 CrossRef CAS.
- A. Lipták, A. Borbás, L. Jánossy and L. Szilágyi, Tetrahedron Lett., 2000, 41, 4949–4953 CrossRef.
- Z. J. Jia, L. Olsson and B. Fraser-Reid, J. Chem. Soc., Perkin Trans. 1, 1998, 631–632 RSC.
- X. Y. Liu, B. L. Stocker and P. H. Seeberger, J. Am. Chem. Soc., 2006, 128, 3638–3648 CrossRef CAS PubMed.
- Y.-H. Tsai, S. Gotze, I. Vilotijevic, M. Grube, D. Varon Silva and P. H. Seeberger, Chem. Sci., 2013, 4, 468–481 RSC.
- H. Q. Nguyen, R. A. Davis and J. Gervay-Hague, Angew. Chem., Int. Ed., 2014, 53, 13400–13403 CrossRef CAS PubMed.
- D. Sikora, T. Nonas and T. Gajda, Tetrahedron, 2001, 57, 1619–1625 CrossRef CAS.
- H. P. Dijkstra, H. Sprong, B. N. H. Aerts, C. A. Kruithof, M. R. Egmond and R. J. M. K. Gebbink, Org. Biomol. Chem., 2008, 6, 523–531 CAS.
- L. Rigger, R. L. Schmidt, K. M. Holman, M. Simonovic and R. Micura, Chem. – Eur. J., 2013, 19, 15872–15878 CrossRef CAS PubMed.
- During the preparation of this report a new method for the acidic removal of the Nap group was published. Although we did not try this method in our compounds, it is an additional method for global deprotection. see: A. G. Volbeda, H. A. V. Kistemaker, H. S. Overkleeft, G. A. van der Marel, D. V. Filippov and J. D. C. Codée, J. Org. Chem., 2015, 80, 8796–8806 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc07694c |
| This journal is © The Royal Society of Chemistry 2016 |