
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Rapid acquisition of high-affinity DNA aptamer motifs recognizing microbial cell surfaces using polymer-enhanced capillary transient isotachophoresis†
Shingo
Saito
*a,
Kazuki
Hirose
a,
Maho
Tsuchida
a,
Koji
Wakui
b,
Keitaro
Yoshimoto
b,
Yoshitaka
Nishiyama
a and
Masami
Shibukawa
a
aGraduate School of Science and Engineering, Saitama University, 255 Shimo-Okubo, Sakura-ku, Saitama 338-8570, Japan. E-mail: shingo@mail.saitama-u.ac.jp; Tel: +81 48 858 3559
bDepartment of Life Sciences, Graduate School of Arts and Sciences, The University of Tokyo, 3-8-1 Komaba, Meguro-ku, Tokyo, 153-8902, Japan
First published on 27th October 2015
Abstract
We present a polymer-enhanced capillary transient isotachophoresis (PectI) selection methodology for acquisition of high-affinity (kinetically inert) DNA aptamers capable of recognizing distinct microbial cell surfaces, which requires only a single electrophoretic separation between particles (free cells and cells bound with aptamers) and molecules (unbound or dissociated DNA) in free solution.
While single-stranded (ss) DNA (or RNA) aptamers1 have attracted much attention with respect to supermolecular recognition, Cell-SELEX (Systematic Evolution of Ligands by EXponential enrichment) has more recently been used to obtain DNA aptamers recognizing cell surface structures.2 DNA aptamer selection by whole Cell-SELEX consists of a binding step by mixing target cells and a library containing vast patterns of randomized-sequence DNA, each with common fixed-sequence primer regions. A separation step is needed to isolate the target–DNA complexes from free (unbound) DNA, followed by partitioning of the complexes by means of filtration or chromatographic techniques,2 and an amplification step by PCR. The DNA sequences thus obtained are re-employed as new DNA aptamer-enriched pools, followed by another series of selection steps, called a “round”. After repeated rounds, the DNA aptamers in the pools are sufficiently enriched, and ready to be sequenced and evaluated as aptamers by way of a binding assay. Cell-SELEX may require 10–20 rounds, leading to an overall procedure that is complex and time consuming (requiring one month or longer to complete all work).
To ensure the effective selection of high affinity aptamers, the key is to develop a sophisticated separation process for the cell-bound DNA sequences in order to prevent contamination by unbound DNA and to exclude loosely bound (kinetically labile) DNA. To this end, we have successfully established a semi-automated methodology consisting of a single electrophoretic separation and partitioning step using capillary electrophoresis (CE). This new methodology allows for the highly efficient separation of ultratrace, cell-bound DNA (referred to as the particle system) from a vast amount of free, unbound DNA (the molecular system) in order to extract aptamer sequences for the first time (Scheme 1). The entire procedure (including selection, partitioning, amplification, sequencing, and binding assay) can be completed within one week.
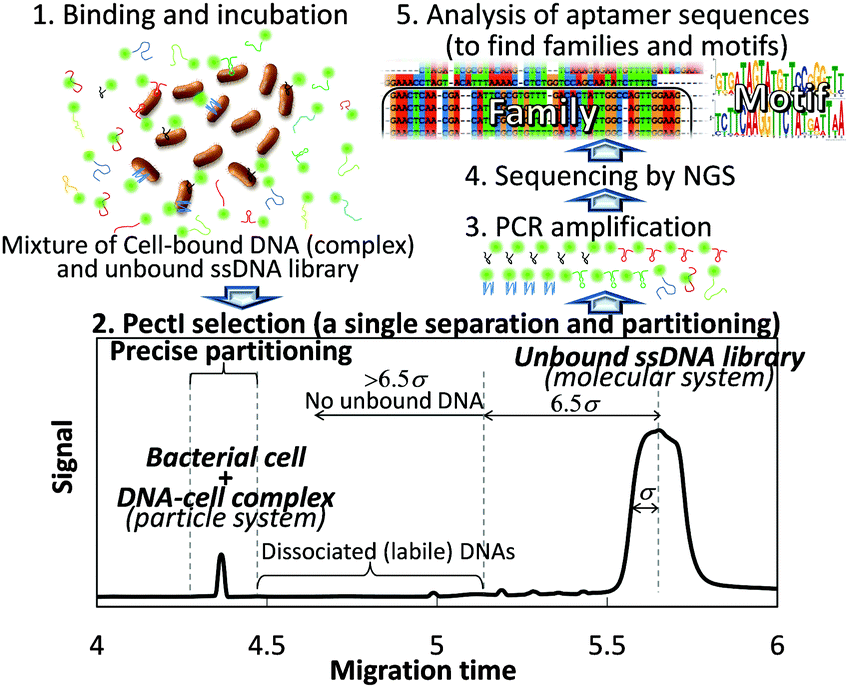 | ||
| Scheme 1 Concept of PectI selection methodology. | ||
Whereas the analysis of bacterial cells by conventional CE typically results in the appearance of multiple peaks with very low repeatability, we have recently developed a CE-based separation mode for bacterial cells, called “Polymer-enhanced capillary transient Isotachophoresis (PectI)”, which yields a single peak per cell type with high repeatability.3 In PectI, microbe cells are concentrated into a narrow, single zone based on the principles of isotachophoresis (ITP) early in the migration. After the ITP stacking mode is completed, the process transitions to the capillary zone electrophoresis (CZE) mode to effect the separation of zones in PectI. The isolated single cell zone is maintained throughout the CZE mode by the effect of added polyethyleneoxide (PEO) (600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) in the migration buffer. The PEO effect was first reported by Armstrong et al.,4 but employing this method alone resulted in low repeatability in our work.3 Thus, the integrated PectI technology was applied to mixtures of randomized DNA with microbe cells in this study.
000) in the migration buffer. The PEO effect was first reported by Armstrong et al.,4 but employing this method alone resulted in low repeatability in our work.3 Thus, the integrated PectI technology was applied to mixtures of randomized DNA with microbe cells in this study.
Specifically, we employed a 5′-FAM-labeled DNA library with 3′- and 5′-primer regions of 24 nt and 19 nt, respectively, and with a 45 nt randomized sequence region (88-mer; FAM-5′-GCAATGGTACGGTACTTCC N45 CAAAAGTGCACGCTACTTTGCTAA-3′); along with three different types of microbe: Escherichia coli (BL21 competent cell), Bacillus subtilis (Bs 168) and Saccharomyces cerevisiae (Nisshin Foods, Tokyo, Japan), for generalization of our methodology to Gram-positive bacteria, Gram-negative bacteria, and fungus, respectively. Typical PectI separation results are shown in Fig. 1. We found that not only microbe cells but also the DNA library were concentrated into narrow peaks, very well resolved from one another, by using a 50 mmol L−1 Tris-300 mmol L−1 glycine (pH 8.5) separation buffer and a 50 mmol L−1 Tris-13.5 mmol L−1 HCl (pH 8.5) sample buffer, both modified by the addition of 0.0125% PEO 600000. It is expected that the cell–DNA complexes migrated together with free cells to form a single zone, since the small amount of bound DNA would not significantly affect the zeta potential of the cells (or the effective diameter of the cells) and thus, the electrophoretic mobility of the cells and cell–DNA complexes would not be significantly different. Typical theoretical plate counts for the observed microbe and DNA peaks were 260000 and 20000, respectively. Furthermore, very high resolution (Rs = 16–28) was obtained between peaks for the cell–DNA complexes and the free DNA. Theoretically, when the migration times of the complex and free DNA peaks differ by more than 6.5σ (where σ is the standard deviation of the Gaussian distribution of the free DNA peak), not even a single molecule from the free DNA zone may have co-migrated with the constituents bound in the complex zone. Given the fact that the free DNA peak appeared to be more than 30σ beyond any of the complex peaks (which consisted of cell–DNA complexes and free cells), we can conclude that any risk of contaminant DNA was suppressed in this PectI selection even though 6 × 1011 DNA molecules were injected. In addition, the cells (free or bound by DNA) migrate ahead of the free (unbound) DNA, thereby reaching the outlet of the capillary first to be collected before any free (unbound) DNA. Thus, the outlet end of the capillary would have no chance to become contaminated by carry-over or exposure to unbound DNA.
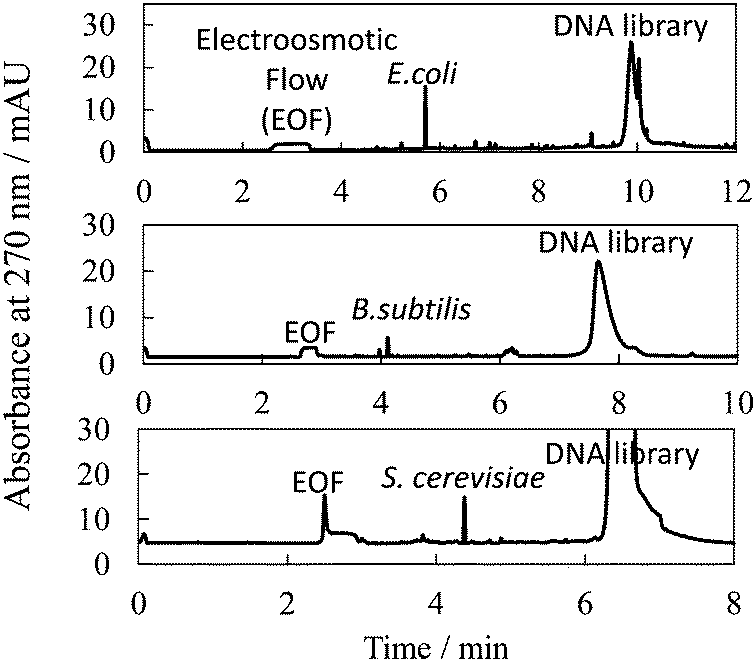 | ||
| Fig. 1 Typical electropherogram for separation between microbe cells bound with DNA and unbound DNA (library) by PectI. Sample, 8 × 107, 8 × 107, and 1 × 105 cfu mL−1 microbe; 2.0 × 10−6, 2.0 × 10−6 and 5.0 × 10−6 mol L−1 DNA library, for E.coli, B subtilis and S. cerevisiae, respectively; 50–13.5 mmol L−1 Tris-HCl, 0.0125% PEO at pH 8.6. Injection volume, 500 nL. Applied voltage, 20 kV. Other conditions are described in the ESI.† | ||
It is very important to be able to partition only the narrow cell–DNA complex zone to avoid contamination of the collected sample by any loosely bound DNA sequences, which may have dissociated from the complex peak and which would migrate immediately after the cell peak. However, it is well known that precise fractionation in CE is very difficult due to characteristically high efficiencies, small volumes, and possible drifts in migration times.5 This is especially true for an on-capillary concentration technique like tITP, since the migration velocity of each zone necessarily changes until the CZE separation mode takes over (after completion of the tITP stacking mode), at which point the velocity of each zone is then constant. Thus, the coupling of separation modes (ITP with CZE) essential to PectI posed an operational dilemma for the desired partitioning. While there is no report of precise partitioning following a tITP method, we managed to solve this problem by installing a second UV/Vis detector positioned 10 cm ahead of the first detector preset in the CE instrument. By means of this simple dual-detector system, it was possible to precisely measure the velocity and the position of the cell–DNA complex zone after the CZE mode began. The detection times for the cell–DNA zone were measured at two positions along the capillary during the CZE portion of the separation (see the ESI†). Thus, the precise time when the zone of interest was to be eluted could be determined. Using this technique, partitioning with a fluctuation of about 0.4% of migration time was achieved (unpublished data).
Using this partitioning system with a typical injection of 500 nL of 1 μmol L−1 DNA library (6 × 1011 DNA molecules), no contamination from the DNA library was observed in the fraction representing the cell–DNA complex. This lack of contamination was confirmed by the partitioning of a negative-control sample, followed by two rounds of PCR, which resulted in no detection of a contaminant DNA molecule (below the detection limit of approximately below 105 in this case) as shown in Fig. S1 (ESI†). The cell–DNA complex peaks shown in Fig. 1 were precisely partitioned using intervals of 12 and 18 s before and after the peak maximum, respectively. Bound DNA molecules were clearly included in the partitioned fraction for E.coli, B. subtilis and S. cerevisiae, as confirmed by PCR amplification and PAGE. Each amplified DNA pool (originating from the partitioned cell–DNA complex zone) was subjected to a binding assay with the relevant microbe using PectI-laser-induced fluorescence detection (LIF) or filtering techniques (data not provided). It was found that 97 ± 2% and 64 ± 2% of the partitioned pool bound to E. coli and B. subtilis, respectively, while the initial randomized DNA library showed no binding in these assays. That is, DNA aptamer sequences for E. coli and B. subtilis were successfully enriched with a single selection according to our method.
Although 88-mer DNA was likewise amplified and detected (by PCR and PAGE) in the partitioned cell–DNA complex fraction for S. cerevisiae, the DNA pool thus obtained did not demonstrate appreciable binding with the microbial cell in a subsequent binding assay (data not provided). This suggests that a large number of non-specific binding DNA sequences were also partitioned by the single PectI selection along with the population of ultratrace, specifically binding DNA sequences in the library. Here, we employed very simple counter selection by centrifugation using E. coli cells prior to PectI selection: a 1.0 × 10−7 mol L−1 initial DNA library solution was mixed with 8 × 109 cfu mL−1E. coli, which nonspecifically bound to members of the DNA library with a low dissociation constant, followed by centrifugation. The resulting supernatant was directly subjected to PectI selection with S. cerevisiae. In this manner, it was found that 26 ± 1% of the selected DNA clearly bound with S. cerevisiae.
Selected DNA pools for E. coli and S. cerevisiae, as obtained by the PectI selection method, were sequenced using the Ion PGM™ next generation sequencer by Life Technologies. The sequence reads obtained for E. coli and S. cerevisiae were 12175 and 113964, respectively, and these were analyzed by ClustalX software6 to find highly analogous sequences (families). Consequently, certain family sequences were found to be unusually enriched for both microbes: 7.7% and 2.7% of all sequences were composed of 37 and 395 families for E. coli and S. cerevisiae, respectively (as shown in Tables S1 and S2, ESI†). This result was surprising since Bowser et al. reported that the heterogeneity of the selected DNA pool in CE-SELEX for proteins was retained and that the highest abundance contiguous sequence was found after four rounds.7 Using the MEME software,8 it was found that there are specific motifs in the extracted analogue sequences (0.11–7%) (see Fig. S2 and S3 and Tables S3 and S4, ESI†). The unusual enrichment suggests that a single PectI selection is sufficient due to highly efficient separation and partitioning.
Some sequences chosen from the families were then subjected to a binding assay by PectI-LIF and fluorescence spectroscopy (see the ESI† for experimental conditions), as shown in Fig. 2A and Fig. S4 (ESI†), respectively. The microbe peak was significantly enhanced with the addition of ssDNA in PectI-LIF. The determined dissociation constants, Kd, as summarized in Table 1 and Table S5 (ESI†), are in the range of several nmol L−1 to 100 nmol L−1 (see also Fig. 2B, Fig. S4 and S6, ESI†). These values are sufficiently low to be suitable for aptamers. Crossover binding experiments were also conducted for S. cerevisiae with aptamers of E. coli, and vice versa. As a result, no significant enhancement in PectI-LIF (Fig. 2A, a–c), fluorescence spectroscopy (Fig. S7, ESI†) and fluorescence microscopy (Fig. S5d, ESI†) was observed; i.e., the DNA aptamers selected for a certain species showed no reactivity towards another species. In addition, the binding experiment was conducted for another strain of E. coli (BW25113) with aptamers of E. coli (BL21). As a result, while Ec 2 and Ec 3 demonstrated an ability to bind to the BW25113 E. coli strain, Ec 1 showed no binding to the other strain (see Fig. S7, ESI†). This fact suggests that there is a possibility of acquiring aptamers that are selective towards different strains using PectI selection, although it was reported that no selective aptamer was selected for different strains of E. coli using conventional whole cell-SELEX even after 10 rounds.9 Thus, a more detailed investigation designed to improve our understanding of the characteristics of the PectI selection will be necessary.
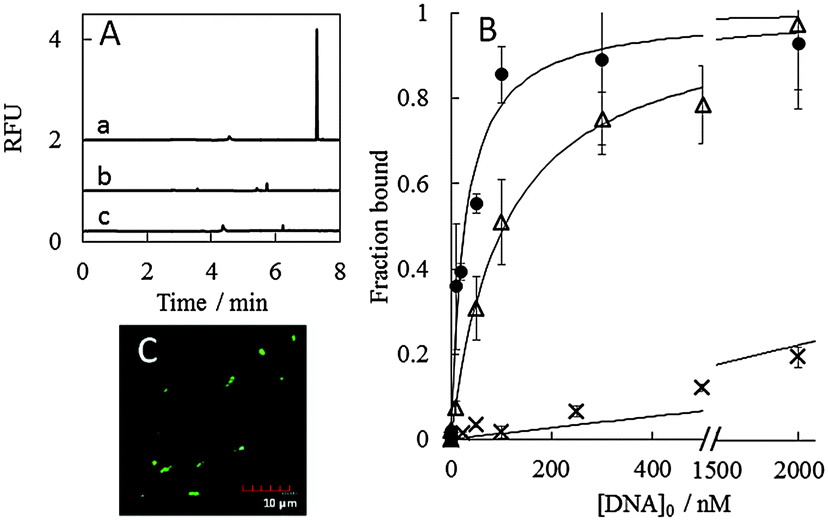 | ||
| Fig. 2 PectI-LIF assay (A), binding assay (B) and fluorescence microscopy (C) for obtained DNA sequences mixed with microbes. (A) Sample, 10 nmol L−1Ec 3 incubated with 1.6 × 108 cfu mL−1E. coli (a); 1.6 × 108 cfu mL−1E. coli (b); 10 nmol L−1Ec 3 incubated with 1.1 ×105 cfu mL−1S. cerevisiae (c). (B) Sequences of Ec 2 (●), Ec 1 (△) and randomized DNA library (×) were employed. (C) Sample, E. coli with 500 nmol L−1Ec 3. | ||
| Species | Sequence (5′ to 3′) | K d/nmol L−1 | |
|---|---|---|---|
| Ec 1 | E. coli | ACTCATCACCACTAGTGATAGTATGTTCCGGGTTTCTCTGCACTA | 106 ± 9 |
| Ec 2 | E. coli | CGCTAGTGCACGTCTTCAAGGTTCTATGATTAATTTATACATTGG | 27 ± 4 |
| Ec 3 | E. coli | GAACTCAACGACATTCGCGTGTTTGACACTATTGGCAGTTGGAAG | 9 ± 6 |
| Sc 2 | S. cerevisiae | CTGCTTCCAACGATTCAAAACGTTTTAGGTGACATGAATATATGA | 170 ± 60 |
| Sc 5 | S. cerevisiae | TTATACAGTAACATCTTGTACTTTCACTTGGATCATGATTTAATA | 30 ± 10 |
Recently, Gold et al. developed the Slow Off-rate Modified Aptamer (SOMAmer) strategy (called the kinetic challenge) and showed the importance of slow dissociation kinetics for clinical applications.10 Our PectI selection strategy is also oriented toward the acquisition of aptamers with kinetically strong interactions with cells. This is because, in principle, it was the DNA aptamers remaining bound to bacterial cells during migration that were selected. Typical theoretical plate numbers of 230000–450000 for peaks of cell–DNA complexes in the partitioning step mean that washing of the complex a number of times, and partitioning of only non-dissociated complexes occurred on the PectI time scale.
The kinetic inertness of selected aptamer complexes with microbe cells was confirmed by PectI-LIF detection (see Fig. 2A, Fig. S6 and S7, ESI†) and fluorescence microscopy with more than three washings (Fig. 2C and Fig. S5, ESI†). The rate constant for dissociation was estimated to be less than (2 ± 4) × 10−4 s−1 judging from the PectI-LIF detection time scale (see the ESI†).
There are several advantages of our PectI selection strategy over conventional Cell-SELEX2 and CE-SELEX.11 First, it should be emphasized that this is the first-ever report of DNA aptamer selection by cell-based CE, with high reproducibility and high resolution between cells and DNA sequences, with evidence of DNA motif enrichment. Second, this is also the first-ever report of aptamer selection for cell-based samples by way of a single simple CE fractionation in free solution. Although 10–20 rounds are usually conducted in Cell-SELEX, Bowser et al. reported that an increase in byproducts occurred after 7 or more rounds while a decrease in affinity occurred after 5–6 rounds in CE-SELEX for protein and small molecule targets.7 In addition, it was recently reported that multiple rounds of selection significantly biased the types of sequences in some studies.12 Thus, a process requiring fewer rounds for selection is desirable. Although there are some reports of single selection methods of DNA aptamers for proteins13 and viruses,14 those included somewhat complex procedures compared to our method, such as using chromatographic techniques with immobilization of targets or the construction and operation of specialized devices, or cutting of the separation column, etc. In contrast to conventional Cell-SELEX, with a very complex set of operations, the PectI selection is completed within one day, and the fluorescently-labeled and sequence-determined aptamers can be obtained within one week using a simple and semi-automated CE procedure. Third, since the cell–DNA complexes could be separated from protoplasmic elements released from disrupted or dead cells,15 this PectI selection provides aptamers selective only towards intact cell surfaces, while traditional Cell-SELEX is unable to avoid contamination from complexes formed between protoplasmic substances and DNA.
The authors acknowledge helpful comments from Prof. C.L. Colyer (Wake Forest University) during the manuscript preparation stage. This work was supported by JSPS KAKENHI Grant No. 25410139.
Notes and references
- C. Tuerk and L. Gold, Science, 1990, 249, 505 Search PubMed; L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermass and J. J. Toole, Nature, 1992, 355, 564 CrossRef CAS PubMed.
- W. Tan, M. J. Donovan and J. Jiang, Chem. Rev., 2013, 113, 2842 CrossRef CAS PubMed; C. L. A. Hamula, H. Zhang, F. Li, Z. Wang, X. C. Le and X.-F. Li, Trends Anal. Chem., 2011, 30, 1587 CrossRef; K. Sefah, D. Shangguan, X. Xiong, M. B. O’Donoghue and W. Tan, Nat. Protoc., 2010, 5, 1169 CrossRef PubMed; S. Ohuchi, Bioresearch, 2012, 1, 265 Search PubMed.
- S. Saito, T. Massie, T. Maeda, H. Nakazumi and C. L. Colyer, Anal. Chem., 2012, 84, 2454 Search PubMed; S. Saito, T. Maeda, H. Nakazumi and C. L. Colyer, Anal. Sci., 2013, 29, 157 CrossRef CAS PubMed.
- D. W. Armstrong, G. Schulte, J. M Schneiderheinze and D. J. Westenberg, Anal. Chem., 1999, 71, 5465 CrossRef CAS PubMed; D. W. Armstrong and J. M. Schneiderheinze, Anal. Chem., 2000, 72, 4474 CrossRef PubMed.
- K. R. Riley, S. Saito and C. L. Colyer, J. Chromatogr. A, 2013, 1368, 183 CrossRef CAS PubMed; Z. Luo, H. Zhou, H. Jiang, H. Ou, X. Li and L. Zhang, Analyst, 2015, 140, 2664 RSC; B. J. Huge, R. J. Flaherty, O. O. Dada and N. J. Dovichi, Talanta, 2014, 130, 288 CrossRef PubMed.
- R. Chenna, H. Sugawara, T. Koike, R. Lopez, T. J. Gibson, D. G. Higgins and J. D. Thompson, Nucleic Acids Res., 2003, 31, 3497 CrossRef CAS PubMed.
- M. Jing and M. T. Bowser, Anal. Chem., 2013, 85, 10761 CrossRef CAS PubMed.
- T. L. Bailey, N. Williams, C. Misleh and W. W. Li, Nucleic Acids Res., 2006, 34, W369 CrossRef CAS PubMed.
- Y. S. Kim, M. Y. Song, J. Jurng and B. C. Kim, Anal. Biochem., 2013, 436, 22 CrossRef CAS PubMed.
- L. Gold, D. Ayers, J. Bertino, A. Bock and C. Bock, et al. , PLoS One, 2010, 5, e15004 CAS; S. Kraemer, J. D. Vaught, C. Bock, L. Gold and E. Katilius, et al. , PLoS One, 2011, 10, e26332 Search PubMed.
- S. D. Mendonsa and M. T. Bowser, J. Am. Chem. Soc., 2005, 127, 9382 CrossRef CAS PubMed; M. V. Berezovski, M. U. Musheev, A. P. Drabovich, J. V. Jitkova and S. N. Krylov, Nat. Protoc., 2006, 1, 1359 CrossRef PubMed.
- M. Blind and M. Blank, Mol. Ther.–Nucleic Acids, 2015, 4, e223 CrossRef CAS; T. Schütze, B. Wilhelm, N. Greiner, H. Braun, F. Peter and M. Mörl, et al. , PLoS One, 2011, 6, e29604 Search PubMed; A. K. Orpana, T. H. Ho and J. Stenman, Anal. Chem., 2012, 84, 2081 CrossRef PubMed.
- X. Lou, J. Qian, Y. Xiao, L. Viel and A. E. Gerdon, et al. , Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 2989 CrossRef CAS PubMed; M. Fan, S. R. Mcburnett, C. J. Andrews, A. M. Allman, J. G. Bruno and J. L. Kiel, J. Biomol. Tech., 2008, 19, 311 Search PubMed.
- A. Nitsche, A. Kurth, A. Dunkhorst, O. Pänke and H. Sielaff, et al. , BMC Biotechnol., 2007, 7, 48 CrossRef PubMed.
- C. Meng, X. Zhao, F. Qu, F. Mei and L. Gu, J. Chromatogr. A, 2014, 1358, 269 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section including precise partitioning, detailed results of sequencing, binding assay, and kinetics. See DOI: 10.1039/c5cc07268a |
| This journal is © The Royal Society of Chemistry 2016 |