
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Convex and concave micro-structured silicone controls the shape, but not the polarization state of human macrophages†
V.
Malheiro‡
a,
F.
Lehner‡
a,
V.
Dinca
bc,
P.
Hoffmann
b and
K.
Maniura-Weber
*a
aBiointerfaces, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse, St. Gallen, CH-9014, Switzerland. E-mail: katharina.maniura@empa.ch
bAdvanced Materials Processing, Empa, Swiss Federal Laboratories for Materials Science and Technology, Feuerwerkerstrasse, Thun, CH-3602, Switzerland
cNational Institute for Lasers, Plasma and Radiation Physics, Magurele-Bucharest, RO-077125, Romania
First published on 21st September 2016
Abstract
The typical foreign body response (FBR) to synthetic implants is characterized by local inflammation and tissue fibrosis. Silicone implants have been associated with the development of adverse capsular contraction (ACC); a form of excessive FBR to the material that often requires the replacement of the implant. It has been shown that surface roughening of silicone can reduce the prevalence of ACC, but the mechanisms remain poorly understood. Macrophages are key cells in FBR. They exert their control mainly by polarizing into pro-inflammatory (M1) or pro-healing (M2) cells. It is postulated that surface topography can reduce M1 polarization by limiting cell spreading and cytoskeleton organization. To test this hypothesis, we used KrF Excimer laser ablation with half-tone masks to produce convex and concave topographies with controlled surface dimensional parameters. Cells in convex and concave topographies were compared to cells in planar surfaces, with or without chemical polarization. We show that chemical polarization induced specific changes in the cell shape on planar substrates. Macrophage shape and size was different in concave and convex surfaces, but no correlation was found with the cell polarization state. The results highlight that chemical polarization of macrophages is associated with changes in the cell shape; however, topography-induced changes in macrophage shape could not be linked with a shift in macrophage polarization. Thus, the sole manipulation of cell shape does not seem to be the mechanism by which macrophage function could be controlled.
1. Introduction
A major challenge in biomaterials research is the design of implantable systems for which the cell and tissue response can be predicted and tailored, guaranteeing the long term success of an implant. An attractive way for achieving this goal is to create instructive surfaces with a combination of mechanical, chemical or topographical properties capable of directing cell behavior. However, the attainment of surfaces for which the specific response at the cellular level can be fully predicted has not been accomplished yet.Macrophages are one of the predominant cell types at the implant site after 48–72 hours of implantation,1 and have an important role in both eliciting inflammatory responses and orchestrating tissue repair. Their ability to coordinate tissue repair results from their capacity to switch between a pro-inflammatory (M1) and pro-healing (M2) state in a process known as polarization.2 Macrophage polarization in the body occurs mainly due to exposure to chemical signals, such as exposure to bacterial fragments (e.g. lipopolysaccharides (LPS)) or cytokines produced by macrophages themselves or other immune cells (e.g. IL-4 or INF-γ). Indeed, the in vitro development of M1 macrophages is achieved by exposure to IFN-γ (the main Th1 cytokine) and LPS, whereas M2 macrophages can be formed upon exposure to IL-4 (typical Th2-type cytokine). It has also been suggested that implant properties can affect macrophage polarization, and that this may be in fact a major driving force dictating the way the tissue heals at the implant interface.3,4
Out of all implant properties that can be manipulated, the surface topography is particularly interesting, for it has proven to affect tissue formation in vivo and cell function in vitro, and can be readily modified using currently available technology. The mechanistic basis for topography-induced regulation of cellular genome expression and, with this cell function, is attributed to its interference with cell adhesion,5 cytoskeleton formation6 and nuclear shape and orientation.7–9 Consequently, topographies that induce changes in the macrophages cytoskeletal organization and adhesion to a substrate can potentially alter their polarization state, steering their response at the implant–tissue interface. Modification of surface topography has already been used as a way to promote changes in macrophage behavior in in vitro studies,10–27 however the number of publications addressing this issue is rather limited compared to those found for other cell types, such as fibroblasts and osteoblasts. Whilst correlations between micro-topography, macrophage morphology and secretory activity have been established,12–14,19,20,22,24 only few studies used well-defined surface topographies for the evaluation of the specific effect of surface parameters (e.g. space between features) in macrophage polarization.12,15,25 Moreover, the type of topography conformation analyzed by others is mostly limited to random topographies or grooves, dots and posts structures. Notably, the possible effects of convex and concave topographies have not been investigated so far, despite their ability to impose distinct geometric and mechanical restrictions to the cell cytoskeleton.28 It has however been shown29–31 that other types of cells respond differently to concave and convex microstructures. The development of successful topography-based immune-modulatory surfaces dictates the need for the identification of the specific surface parameters that govern macrophage-responsiveness to topography. In the present study, we set out to investigate whether convex and concave poly-dimethylsiloxane (PDMS) structures differently affected macrophages in vitro. We have produced the different topographies in PDMS due to its relevance as an implantable material, for which the control of foreign body reactions would be beneficial in various clinical contexts. The micro-topographies were created by KrF Excimer laser ablation with half-tone masks. This technique provides a unique tool for patterning and structuring a wide variety of materials, including polymers, due to the short pulse of 20 ns which allows a non-thermal ablative material removal. As a result, structures with a depth resolution in the order of 0.1 μm and spatial resolution in the order of 1 μm or smaller can be obtained. Moreover, by carefully controlling the laser parameters and by using a grey level mask, surfaces can be multi-level processed to obtain 2 and 3 dimensional microstructures. In the present work, we used this technique to create a polycarbonate (PC) master mold from which we then produced different convex (pyramids, cones, dots) and concave (inverted pyramids, funnels, pits) structures in PDMS by replica molding. The depth/height, width, pattern and spacing between the features were kept constant to allow for a direct comparison between, and within the two groups. The effect of topography was investigated with respect to cell number, morphology and polarization using human macrophages (THP-1 monocytic leukemia cell line). Chemically-induced polarized M1 or M2 macrophages on planar substrates (PDMS and glass) were included as controls.
2. Materials and methods
2.1 Substrate fabrication and characterization
Excimer laser micromachining by mask projection with half-tone masks has been used to obtain various structures with simple and complex surface profiles. Details of the laser system are described elsewhere.32 Shortly, the experimental setup contains a KrF excimer laser (Exitech, PPM-601E Gen 6 Tool), with the following characteristics: 20 ns pulse, 248 nm wavelength, and 50 Hz repetition rate. The average fluence at the polycarbonate substrate level was 400 mJ cm−2 per pulse. The laser was collimated to illuminate a mask and then focused on the sample with the de-magnification factor of 5. By scanning the mask under the beam and the PC sample, micro-patterns of different shapes (dots, cones, pyramids, pits, funnels, inverted pyramids) with a depth/height of 10 μm and a width of 25 μm were generated on a polycarbonate (PC) substrate. The polycarbonate samples were cleaned ultrasonically for 10 min, sequentially, in isopropyl alcohol (IPA) and Millipore ultrapure water (R = 18 MΩ), followed by drying with an air gun. The final surfaces were obtained by molding the PC master with PDMS (Sylgard 184 Silicone Elastomer Kit; Dow Corning) (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10) cured for 48 h at room temperature (RT), followed by heating to 80 °C for 1 h to ensure completing curing.
:10) cured for 48 h at room temperature (RT), followed by heating to 80 °C for 1 h to ensure completing curing.
The final PDMS samples were demolded, cleaned ultrasonically twice for 10 min in IPA and finally in ultrapure water, before sterilization in 70% ethanol for 1 h. For cell culture experiments, samples were transferred to a new culture plate and washed twice with sterile warm phosphate buffered solution (PBS) prior to cell seeding. The absence of endotoxin contamination in sterilized samples was confirmed by a cell-based assay. The assay and results obtained are detailed in ESI (Fig. S1†).
A confocal laser scanning microscope (LSM780; Carl Zeiss, Germany) was used to image the PDMS topographies in order to assess the pattern transfer fidelity from the PC mold to the PDMS replica. Samples were immersed in fluorescein isothiocyanate dye to increase contrast and imaged at 488 nm.
2.2 Cell culture and seeding
All cell culture reagents were obtained from Invitrogen, and all other chemicals from Sigma, except where indicated.Human monocytic leukemia cell line THP-1, obtained from the European Collection of Cell Cultures (ECACC, https://www.phe-culturecollections.org.uk), was selected for the cell culture studies. Cells were maintained in complete medium prepared from RPMI-1640 medium supplemented with 10% (v/v) fetal calf serum (FCS), 1% (v/v) penicillin–streptomycin–neomycin (50 μg mL−1 penicillin, 50 μg mL−1 streptomycin and 100 μg mL−1 neomycin), and 0.2 mg mL−1 of L-glutamine. Cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2 and sub-cultured when reaching a concentration between 8 × 105 and 1 × 106 cells per mL. Cells from passage 10 to 17 (after thawing) were used for all experiments. Cell counting for passaging and seeding was performed using an electronic cell counting device (Casy® model TT, Roche Innovatis AG).
PDMS and glass discs (15 mm diameter) were each transferred to a well of a 24-well plate and cells (7.5 × 104 cells per cm2) were plated onto the substrates in 500 μL RPMI-1640 complete medium supplemented with 100 nM phorbol-12-myristate-13-acetate (PMA) in order to induce macrophage differentiation of the monocytic THP-1 cells.33 Cells were left to differentiate for 3 days in PMA-containing medium, after which the medium was replaced by PMA-free medium. On day 4, medium was changed to either PMA-free medium, M1 medium supplemented with 100 ng mL−1 lipopolysaccharide (LPS) and 20 ng mL−1 interferon gamma (IFN-γ) or M2 medium supplemented with 20 ng mL−1 interleukin 4 (IL-4) for chemical induction of macrophage polarization on planar PDMS and glass surfaces. Cells were cultivated in contact with the surface for further 24 h and were then analyzed.
2.3 Quantification of macrophage cell number & metabolic activity
The alamarBlue® assay (Invitrogen) measures the metabolic state of a cell population by quantifying the ability to reduce resazurin, the blue (non-fluorescent) cell permeable dye, to resorufin, a red compound that is highly fluorescent.34 At day 5, each cell-seeded substrate was incubated for 4 h with fresh culture medium supplemented with 10% (v/v) alamarBlue reagent. Following incubation, 100 μL medium from each well was transferred in triplicate to a black 96-well microplate. Fluorescence (excitation 530 nm, emission 590 nm) was measured on a Mithras2 LB 943 monochromator multimode reader (Berthold Technologies GmbH & Co., Germany). Cell metabolic activity was reported as % reduction of alamarBlue and calculated as described in the ESI.†Total number of cells per area was determined by counting DAPI stained cells in confocal microscopy images (obtained as described below) with ImageJ software (National Institutes of Health, Bethesda, MD).
2.4 Macrophage morphology
:40 in 0.05% (v/v) Tween-20 in PBS (PBST)) to stain actin fibers and incubated at 37 °C for 40 min. Samples were rinsed five times with PBST, mounted with Vectashield antifade mountant containing DAPI (Vector Laboratories), to stain nuclei, and imaged using a LSM780 confocal laser scanning microscope. For cell body evaluation, samples were washed three times with PBS and incubated in the dark with 2 μg mL−1 HCS CellMask™ green stain (Invitrogen) in PBS for 30 min at room temperature. The specimens were washed three times with PBS and imaged using a LSM780 confocal laser scanning microscope.
2.5 Gene expression analysis
RNA from adherent cells was isolated by the spin column method using a commercially available RNeasy Micro Kit (Qiagen), according to the manufacturer's instructions. To prevent DNA contamination in the eluted RNA solution, genomic DNA was removed by a DNase treatment (Qiagen). The RNA concentration and quality were determined using a Nanodrop ND-1000 Spectrophotometer (ThermoScientific). Complementary DNA (cDNA) was synthesized from 100–300 ng of total RNA using iScriptTM cDNA Synthesis Kit.A temperature program of 5 min priming at 25 °C, followed by the reverse transcription at 42 °C for 30 min and the reverse transcription inactivation at 85 °C for 5 min was run. After a final cool down to 4 °C, the cDNA was diluted 1:5 in RNase-free water and stored at −80 °C for subsequent use. Primer pairs – designed over exon–exon junctions using PrimerBlast online software (http://ncbi.nlm.nih.gov/tools/primer-blast/) and manufactured by Microsynth AG (Balgach, Switzerland) – are displayed in Table S2.† Only primer pairs which resulted in a PCR efficiency between 90% and 110% using the below described program were used in this study.
Real-time PCR was performed using 5 μL of cDNA sample and 7 μL of 0.2 μM (final concentration) forward and reverse primer and iQTM SYBR® Green Supermix (Biorad Laboratories) in a CFX96™ Real-Time PCR (Biorad Laboratories). The cycling conditions were as follows: an initial 95 °C for 3 min, followed by 40 cycles of 95 °C for 10 s and 57 °C for 30 s. Then, a melting curve was constructed by heating from 65 °C to 95 °C in temperature steps of 0.5 °C. A multivariable, nonlinear regression method was used by CFX™ Manager Software (Version 2.0, Biorad Laboratories) to determine the quantification cycle values. Fold changes in gene expression were then quantified using the 2(−ΔΔCq) method35 normalized against GAPDH. Cells on M0 glass were designated as the calibrator for each experiment.
2.6 Statistical analysis
Quantitative evaluations were performed from data obtained from a minimum of three independent experiments, using two replicate samples per condition. Data are expressed as the arithmetic mean ± standard error of the mean (SEM). Statistical significance of differences in mean values was evaluated by ANOVA or Kruskal–Wallis nonparametric ANOVA (morphometric data). When values were found to be significant the t-test or Mann–Whitney U test was subsequently employed to detect differences at the multiple comparisons levels. Statistical analysis and plotting was performed with GraphPad® Prism software (GraphPad Software Inc., USA). Differences were considered statistically significant at p values of <0.05.3. Results
3.1 Surface topography
The different patterns created in PDMS were evaluated by microscopic analysis. In order to acquire quantitative information on the height/depth of the topographic features we immersed the surfaces in fluorescein isothiocyanate dye to increase contrast. A selected set of the height-resolved images obtained by confocal fluorescence microscopy, shown in Fig. 1, was used for the 3D reconstruction of the different PDMS topographies. Convex and concave surfaces with different topographic features could be produced. The different features have a smooth surface, are regularly distributed and equivalently spaced, and have comparable feature width (approx. 25 μm). The lateral fluorescence profile seen in the insets shows the convex and concave profiles presenting a similar height/depth of approximately 10 μm. Quantitative data of sub-micro and nanoscale roughness obtained by light interferometry and AFM, together with depth/height and width measurement obtained from the confocal images, are included in ESI (Tables S3–S5†).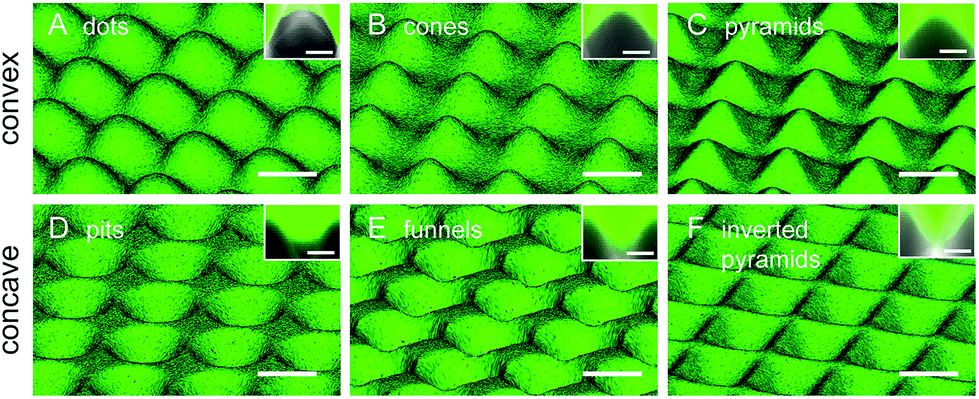 | ||
| Fig. 1 3D views of convex (A–C) and concave (D–F) PDMS topographies reconstructed from images obtained by confocal microscopy of fluorescein-immersed samples. The cross-sectional shape of each topographic feature is shown in the inset of the corresponding figure. Scale bar = 25 μm, and 10 μm in insets. | ||
3.2 Macrophage adaptation to topography
THP-1 cells were cultured on the different substrates for 5 days when the development of a mature macrophage phenotype was anticipated. Fig. 2 shows the number (a) and metabolic state (b) of macrophages cultured on different surfaces. No significant differences were observed between the numbers of macrophages attached to different topographies. The metabolic activity of the cells was decreased when cultured on PDMS, regardless of the presence or absence of topographic features.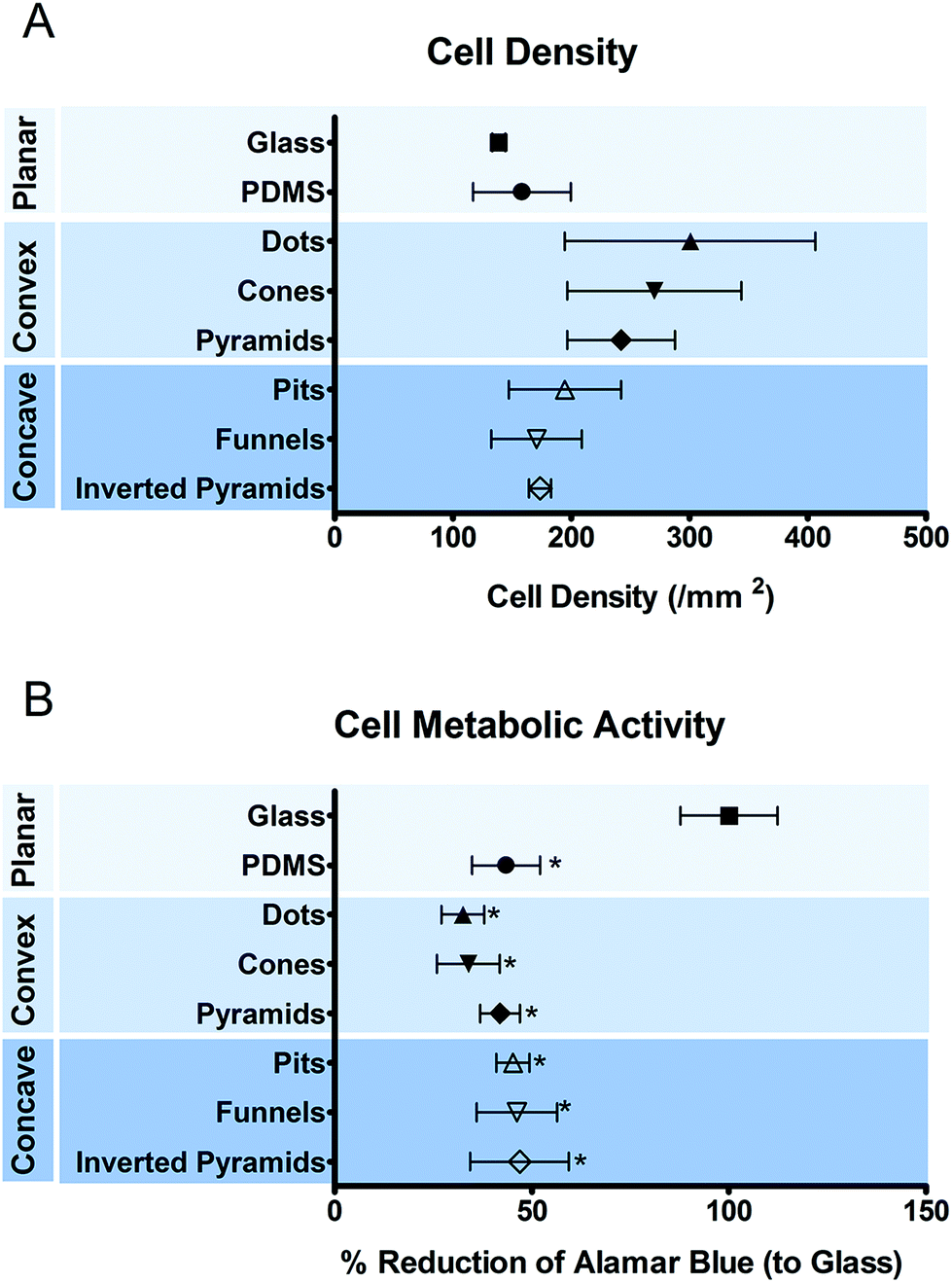 | ||
| Fig. 2 Density and metabolic state of macrophages after 5 days of culture on different surface topographies as determined by cell counting (A) and alamar blue activity (B), respectively. Data are shown as mean ± standard error of the mean, *p < 0.05 vs. glass control. | ||
Cell adhesion and morphology were also analyzed after 5 days in culture. Macrophages could adhere and spread to all different surface topographies. Cells constituted a heterogeneous population exhibiting varied morphological adaptations to the underlying topographic features. Nevertheless, some trends towards specific shape adaptations could be identified for specific surface topographies and are presented in the SEM images in Fig. 3. Fig. 3(G)–(I) shows the morphology of cells in contact with different types of convex surfaces. In general, cells on this type of topographies have a tendency to spread between the topographic features, adjusting the cell body margin to contour the sidewalls of the features. The development of star-shaped morphology was often observed, and more rarely cells bridging between topographical features. In contrast, cells in concave topographies (Fig. 3J–L) mainly attached and spread inside the concavities, typically acquiring a round morphology in pits and funnels (Fig. 3J and K) and a squarer morphology in inverted pyramids (Fig. 3L).
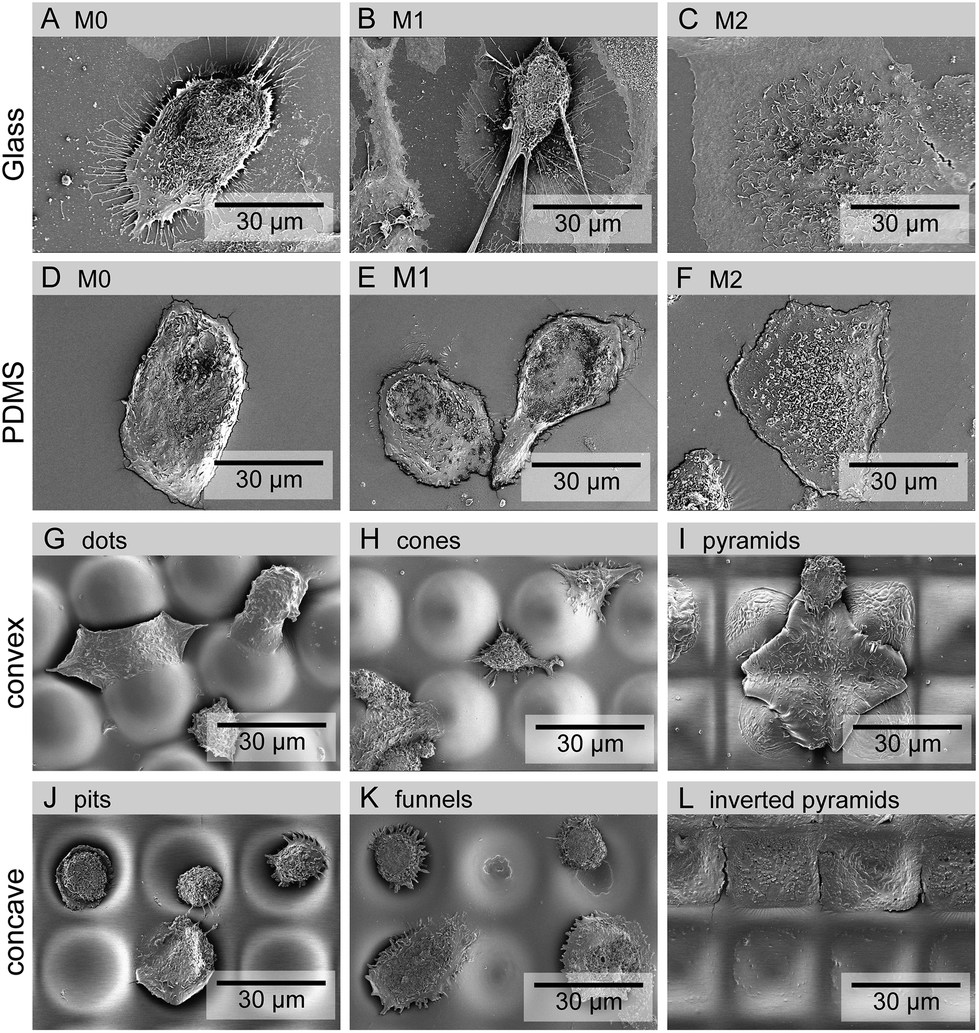 | ||
| Fig. 3 Representative SEM images of macrophages in glass (A–C) and planar PDMS (D–F), convex (G–I) or concave (J–L) PDMS topographies. Cells in planar surfaces were cultured with basal M0 (A, D), M1 (B, E) or M2 (C, F) polarization medium. Images reveal differences in cell morphology in response to topography and cultivation medium. | ||
Confocal microscopy images (Fig. 4) are in agreement with SEM observations of the cell shape adaptation to the different topographies. In addition, the images show the absence of a well-defined actin cytoskeleton and the lack of actin stress fiber formation. Side views reveal further differences between cell morphological adaptation to concave and convex topographies. Particularly, it is shown that cells acquire an angular crescent cross-sectional morphology in convex topographies (Fig. 4G–I), and a more circular or oval cross-sectional morphology in concave surfaces (Fig. 4J–L). Interestingly, the intra-cellular location of the nucleus was mostly at the places were no cell deformation was present, so that no major trends were detected in terms of nuclear adaptations to the topography.
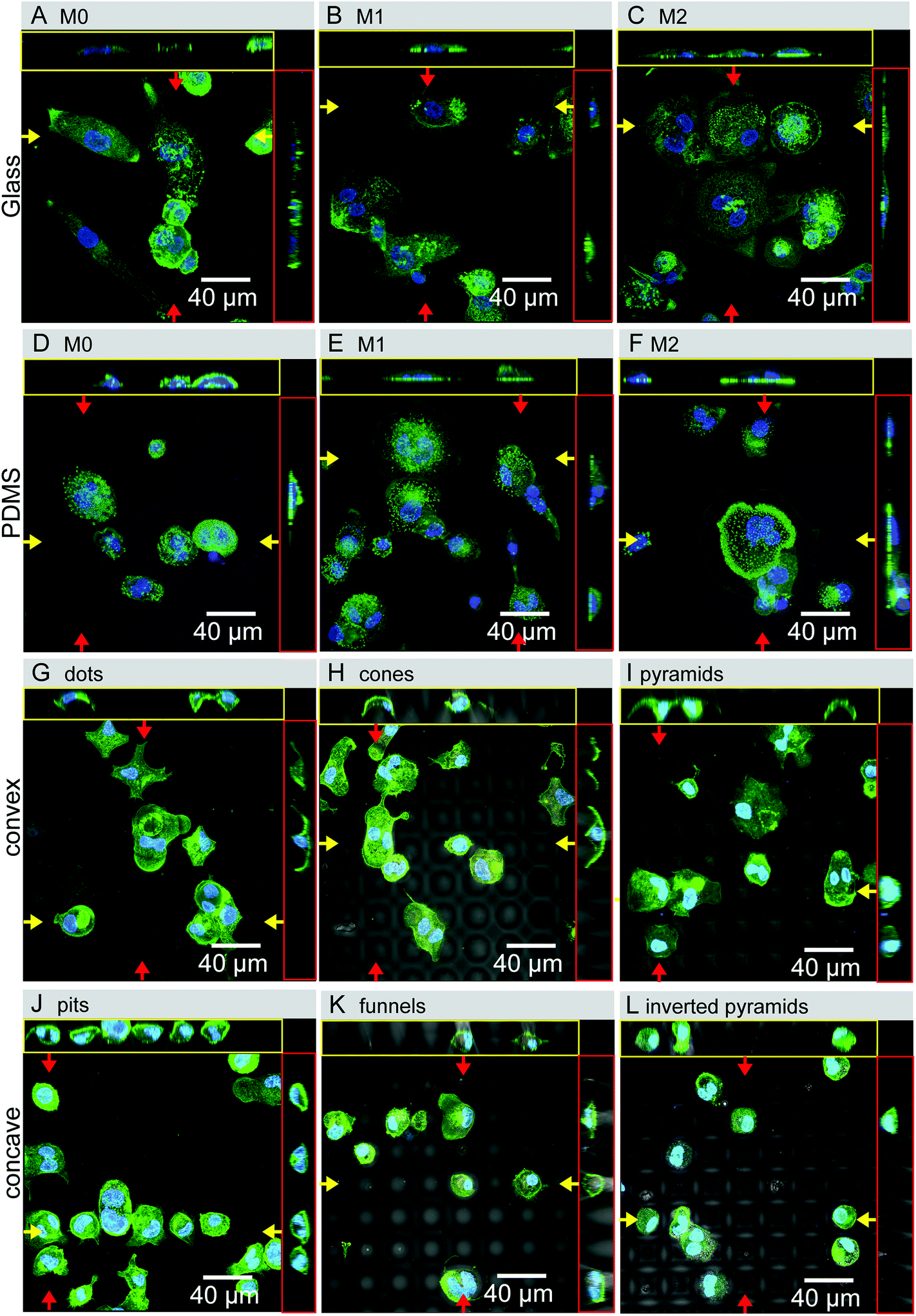 | ||
| Fig. 4 Confocal microscopy images of top and side views (yellow and red rectangles and arrows) of macrophages in glass (A–C) and planar PDMS (D–F), convex (G–I) or concave (J–L) PDMS topographies. Side views reveal differences in the cross-sectional shape of cells on convex and concave topographies. AlexaFluor488-phalloidin and DAPI stained the actin cytoskeleton green and nucleus blue, respectively. | ||
Cell morphological adaptation to a substrate, particularly in the case of a heterogeneous population, can be better distinguished by quantitative analysis of the cell shape descriptors. Cell morphology was analyzed in respect to changes in cell size (Fig. 5A), shape (Fig. 5B and C) and orientation (Fig. 5D).
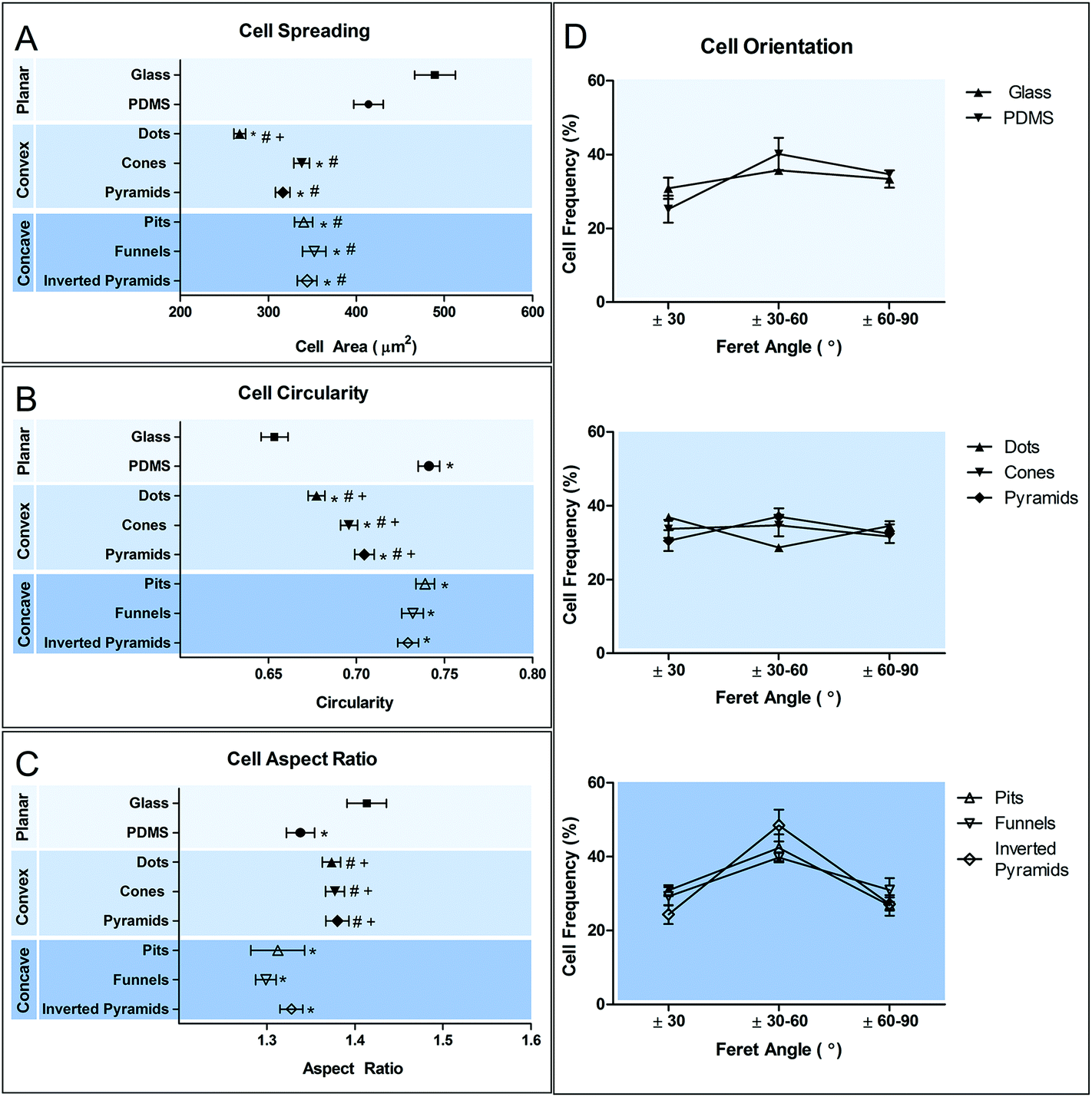 | ||
| Fig. 5 Morphometric evaluation of macrophage adaptation to different surfaces. Cell shape was quantified in terms of individual cell area (A), circularity (B), aspect ratio (C) and orientation (D). Data are shown as mean ± standard error of the mean. *p < 0.05, vs. glass control; #p < 0.05, vs. PDMS control; +P < 0.05, vs. concave counterpart. | ||
Results show that all the topographies used in this study promoted a statistically significant decreased (p < 0.05) in cell spreading in comparison to planar PDMS and glass. Convex surfaces appear to promote a further decrease in cell area compared to the concave counterpart, but only in dots such difference reached a statistical significance. Calculation of the circularity and aspect ratio (AR) allows the quantification of macrophage shape. AR is high when cell shape is elongated and decreases when cells become more polygonal. Further discrimination between polygonal and circular shapes can be achieved using the circularity parameter, where 1 represents a perfect circle. Macrophages on planar glass have the lowest degree of circularity compared to cells in PDMS. However, elongation is increased only in comparison to cells on planar glass and concave surfaces. Cell shape on concave surfaces is similar to that on planar PDMS, showing a high degree of circularity and decreased elongation. In contrast, culture of macrophages in convex PDMS surfaces decreased their circularity and increased AR. The topographies do not seem to induce any particular effect on cell orientation, except for inverted pyramids, where an increased number of cells appear to be aligned in the direction of ±30–60°. This may be a consequence not of cell alignment, but of the acquisition of a squared cell shape. A squared cell will have its longest axis (Feret's diameter) between opposing vertices and this axis will be at 45° to the x-axis of the image, within the ±30–60° range.
3.3 Macrophage adaptation to chemical stimuli
The effect of M1 and M2 chemical polarizers on macrophage response to planar surfaces was analyzed in control experiments using two different substrate materials (glass and PDMS). On day 5, no differences were observed in the number of cells attached to planar PDMS and glass, irrespectively of the presence or absence of chemical polarizers (Fig. 6). The metabolic activity of the cells cultured on PDMS was decreased when compared to cells on glass.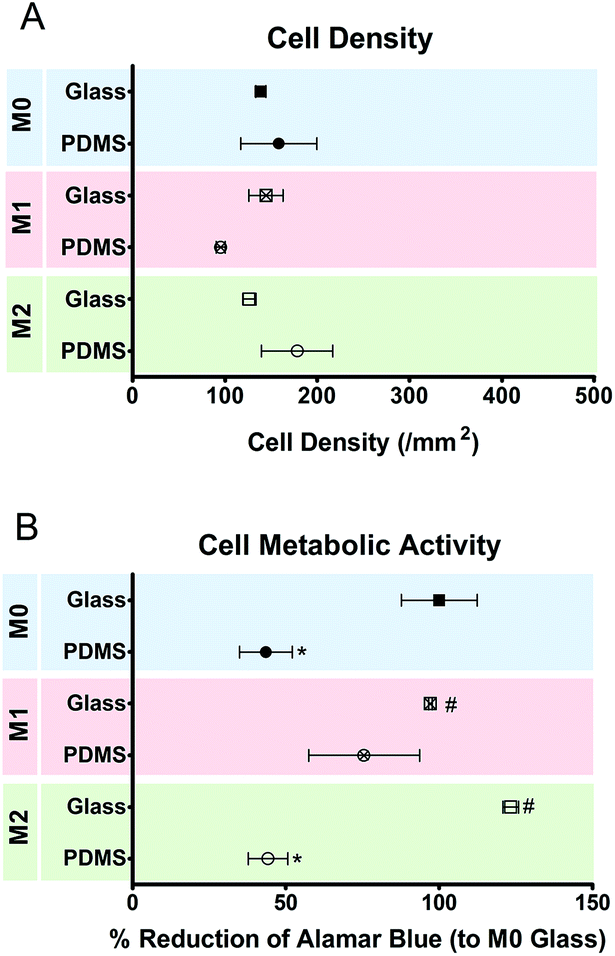 | ||
| Fig. 6 Density and metabolic state of macrophages after 5 days of culture on planar glass and PDMS surfaces with or without chemical polarization, as determined by cell counting (A) and alamar blue activity (B), respectively. Data are shown as mean ± standard error of the mean; *p < 0.05 vs. M0 glass, \p < 0.05 vs. M0 PDMS. | ||
The effect of chemical polarization on macrophage morphology is shown in Fig. 7. Macrophage spreading and AR is increased when cultured on glass compared to PDMS surfaces (p < 0.05), whereas the circularity is decreased. Chemical polarization also influences cell shape. M2 polarization increases cell spreading and decreases circularity (p < 0.05) on both substrates, while M1 polarization has no effect on cell spreading but promotes likewise a decrease (p < 0.05) in circularity. The effect of polarization in cell AR is not as clear, and it appears to be substrate dependent as it was only influenced by M2 polarization on glass. No effect on cell orientation was observed in response to chemical stimulation.
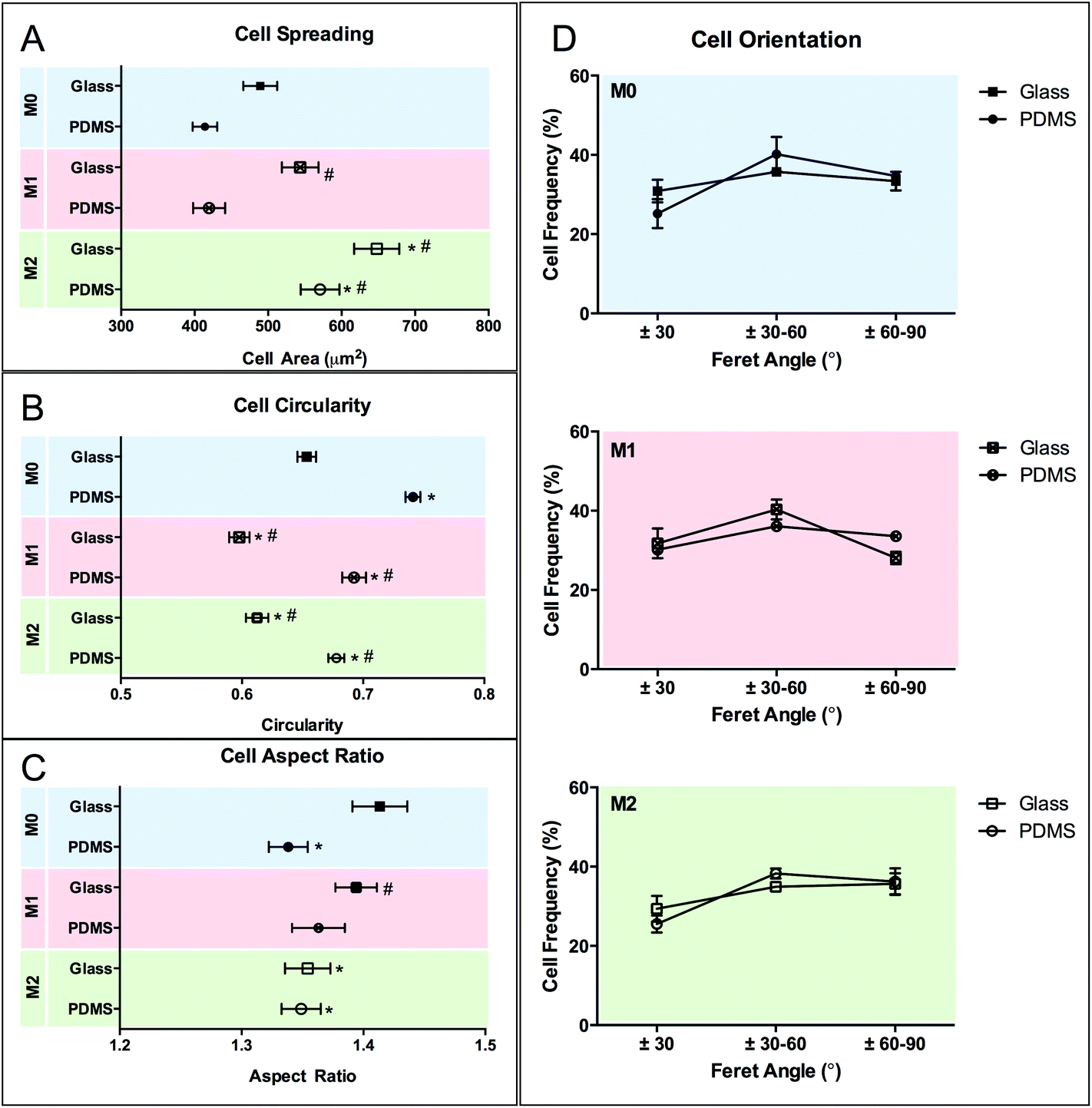 | ||
| Fig. 7 Morphometric evaluation of macrophage adaptation in response to chemical polarization on two different planar surface materials (glass and PDMS). Cell shape was quantified in terms of individual cell area (A), circularity (B), aspect ratio (C) and orientation (D). Data are shown as mean ± standard error of the mean. *p < 0.05, vs. M0 glass control; #p < 0.05, vs. M0 PDMS control. | ||
3.4 Macrophage polarization in response to topographic and chemical stimulation
Macrophage polarization in response to topographic and chemical stimulation was evaluated by using surface markers (CD197, CD206), effector cytokines (TNF-alpha, IL-10) and chemokines (CCL22, CXCL10) expressed by particular M1 or M2 macrophage populations (Fig. 8). When subjected to M1 chemical polarization, we observed an up-regulation in all three inflammatory markers and of CCL22 anti-inflammatory markers, together with a down-regulation of CD206 both on planar PDMS and glass. M2 chemical polarization resulted in an up-regulation of CD206 on both planar surfaces, whereas IL-10 expression was slightly down-regulated, and CCL22 expression was up-regulated in cells on glass, but not on PDMS. Macrophages cultured on PDMS in M0 medium presented a distinct state to those cultured on glass, characterized by a down-regulation of CXCL10 and CD197 (n/d) and a slight up-regulation of anti-inflammatory markers CD206 and IL-10. Surface topography does not seem to exert a relevant effect on the expression of the selected genes, compared to planar PDMS.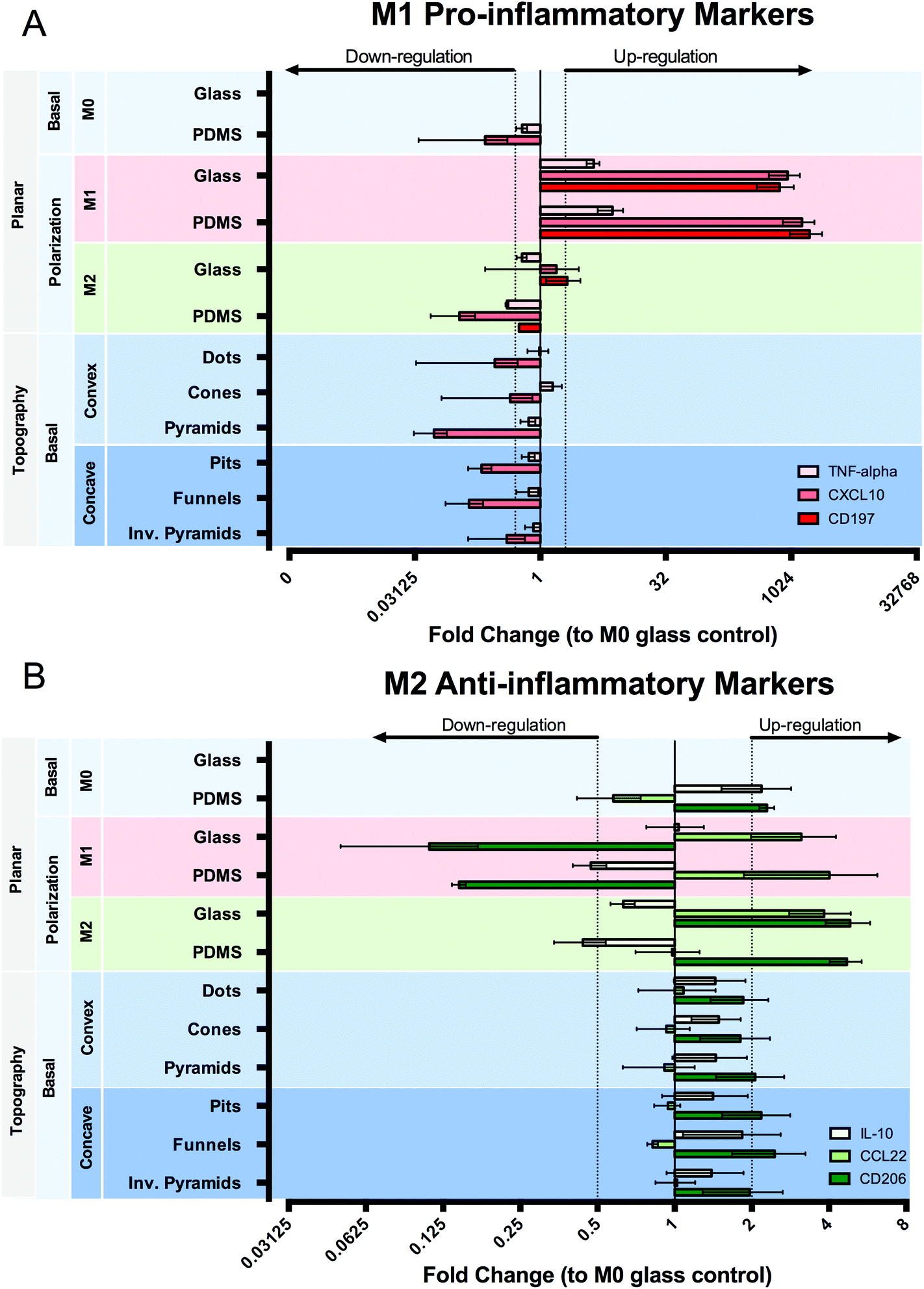 | ||
| Fig. 8 Quantitative reverse transcription-PCR analysis of mRNA expression of (A) pro- and (B) anti-inflammatory genes in macrophages cultured on different surfaces, with or without chemical polarization. Data normalized to GAPDH mRNA. Values represent fold change relative to M0 glass control. Data are shown as mean ± standard error of the mean in a logarithmic scale. Genes considered up-regulated for fold changes ≥2 and down-regulated for fold changes ≤0.5. The dotted lines represent the regulation thresholds set at 2-fold change. When no bar is shown it means the gene was not detected (n/d). | ||
4. Discussion
The control of cell function through substrate/implant topography is an attractive method for the improvement of the overall biological response to implants. Albeit extensive research on the topic, there is still a lack of definition on which are the geometrical parameters that can truly influence cell response to a material. Furthermore, the influence of topography on immune cells has only been seldom addressed, which is surprising as these cells play a determinant role in how, and whether at all, the implant is accepted by the body. In this study, we hypothesized that micro-sized convex and concave surface topographies would induce distinct morphological deformations to macrophages, and that those would dictate a change in the way the cell responds to the surface. We have extended the subject of our study by including different types of convex and concave topographies, so that the impact of factors such as the base geometry and the curvature of the lateral walls could be simultaneously investigated. We have produced different surfaces topographies by KrF Excimer laser micromachining with half-tone masks. While we used the technique for the production of the master topographies, it is also suitable for direct creation of the topographies in a wide variety of materials (i.e. from ceramics, glasses to polymers) making it an appealing method for future systematic investigations on the role of topography versus chemistry in macrophage function. In the current study, the selection of this production method permitted the creation of the desired topographies with a strict control over the shape, distribution and dimension of the topographic features.The quantitative and qualitative analysis of the cell shape adaptation to the different topographies showed an evident decrease in cell spreading, independently of the shape of topography investigated in the present work. The effect of topography on macrophage size and shape seems to be largely dependent on the scale of topography, geometric parameters, material and cell type. Poly(methyl methacrylate)36 grooves (0.5 μm depth, 10 μm width) promoted an increase in murine macrophages mean area compared to flat surface from 380 to 650 μm2. On the other hand, similar structures (0.8–1.33 μm depth, 10 μm width) in Ti had no significant impact in macrophage spreading.37 These examples provide evidence that not only topography can have a significant impact in the size of macrophages, but also that this capability depends on factors other than the geometric parameters of the topographic features. The decrease in spreading that has been observed in response to the topographies in the present work and those reported elsewhere38,39 may indicate that there is possibly a specific combination of material and topography that converts the discontinuities into energy barriers which hinder cell spreading.28 Other systems11,17,22 may facilitate cell spreading by a combination of properties that lead to increase in the density of contact points, protein adsorption and/or due to contact guidance mechanisms. The role of the topographic features as energy barriers is supported by the observation that cells preferentially adhered and spread either within or between the features depending on whether the topography was concave or convex, respectively. Bartneck et al.25 showed similar behavior of human peripheral blood-derived macrophages in response to perfluoropolyether posts (20 μm height, 20 μm diameter). Convex versus concave surface topography also induced, as predicted, the development of characteristic cell shapes, but no significant differences were observed in response to the individual topographic features within each group. Changes in morphology were not reflected by significant changes in the number of cells attached to the substrate and neither in their metabolic state, when compared to planar PDMS.
On the other hand, the substrate material set as control, played a significant role both in the shape and metabolic activity of cells. Cells cultured on glass showed a higher degree of spreading, decreased circularity and increased metabolic activity compared to PDMS. The increased spreading on glass – interestingly correlating with an increased TNF-alpha production – compared to other materials (chitosan, polyurethane and hyaluronic acid) has been shown previously.40 The authors in this study explained the differences based on the chemical nature of the surfaces; i.e. that was the difference between the negatively charged nature of the glass, relatively inert polyurethane, positively and negatively charged polymer brushed that induced differences in the adhesiveness of the cells. We used PDMS in this study, which also differs greatly from glass in its electrochemical properties, and this may be the reason for the differences in cell morphology observed in the present study.
It can further not be excluded that the differentiation of monocytes to macrophages on TCP in our study resulted in macrophages with a different phenotypical plasticity when compared to cells differentiated on PDMS. Such different approach might theoretically result in macrophages with different sensitivity to chemical stimulation or topography. However, macrophages cultivated on glass as well as smooth or structured PDMS substrates in our study were of the same origin and differentiation state and show the discrepancy between the morphological adaptations as well as response to chemical induction of polarization.
Chemical polarization induced changes in cell morphology independently of the substrate material. The relationship between chemical polarization and changes in the morphology of macrophages has been shown previously.11,12,41 McWhorter et al.12 reported that M2 polarization induced murine macrophage elongation, but no changes in cell size. Nishio et al.42 reported a similar observation with human cells for IL-4 polarized cells, but showed in addition that tumor-associated macrophages expressed M2 markers, increased area but not elongation. Others11,43 have also shown that primary human monocyte-derived macrophages expressing M2 markers display a more irregular, but not elongated morphology. In our system, we observed an increase in cell spreading (particularly for M2) and a slight decrease in AR, which signifies an increase in cell irregularity but not elongation.
It is possible that the discrepancy between the morphological adaptations in our study and those reported by McWhorter et al.12 result from substrate chemistry or inter-species differences; i.e. between human and murine macrophages. The intracellular mechanisms that govern changes in macrophage shape in response to chemical polarization are not well understood, and it is therefore possible that other reasons contribute to the disparity of behaviors between the different studies. One of the most obvious reasons why macrophages would adapt different shapes is to facilitate their ability to migrate, phagocyte and degrade matrix degradation. All of these functions depend on the intracellular organization of actin. Thus, the importance of actin modulators such as Rac is under investigation and has already proved to be important modulators of macrophage function.44 However, other mechanisms, such as the inhibition of histone deacetylases explored by Cabanel et al.45 can also lead to a change in macrophage shape and polarization, suggesting the existence of nuclear epigenetic mechanisms in the regulation of cell shape.
In the last stage of our study, we tried to establish whether differences in cell morphology affected the gene expression of selected genes, known to be constitutively expressed in M1 or M2 macrophages (reviewed in ref. 46 and 47). We showed a clear polarization towards an M1 phenotype in response to M1 chemical polarization, as detected by an up-regulation of all inflammatory markers selected. Interestingly, of the anti-inflammatory markers selected for this study, only CD206 showed a clear distinguishable response to M2 polarization. IL-10 gene expression was instead slightly down-regulated, and CCL22 expression was up-regulated on glass, not on PDMS, and also appeared up-regulated upon M1 polarization. CCL22 is a chemokine that attracts Th2 cells and is expressed in M2 macrophages. Nonetheless, as shown by El Chartouni and Rehli,48 the exposure of macrophages to the strong innate activation signals (LPS) can alter the typical cytokine expression in both phenotypes. They showed that up-regulation of CCL22 can occur even in M1 macrophages when polarization is primed by both INF-γ and LPS, but not with INF-γ alone. In the same study, the authors also failed to detect an increased production of IL-10 in response to exposure to IL-4 alone. The clear observation of an up-regulation of CD206, an important M2 associated receptor, indicates that nonetheless an alternative state of polarization was induced in spite of the lack of change in the expression of IL-10 and CCL22. Differences between the macrophage phenotype in basal medium were detected between glass and PDMS. Under this culture condition, cells on PDMS showed a decrease in the inflammatory state compared to glass, since CXCL10 was down-regulated and CD206 and IL-10 slightly up-regulated. The indication that cells on glass are more inflammatory than on PDMS, correlates with the observed increase in metabolic activity and cell size. As discussed earlier, the increased spreading on glass has been previously associated with an increase in the production of the inflammatory TNF-alpha cytokine.40 We have used resazurin reduction to resorufin as a way to estimate the metabolic activity of macrophages, which means we effectively measured cellular redox metabolism. Increased inflammation has also been associated with an increase in the reductive environment (discussed in ref. 49).
The differences in topography-induced morphology did not promote any significant changes in the gene expression of the selected pro- and anti-inflammatory genes. It is postulated that cell sensitivity to surface topography is promoted by changes in cell adhesion, cytoskeleton formation or nuclear shape and orientation. In the present study we did not observe topography-induced nucleus deformation. In addition, and unlike other studies,12,25 our topographies did not promote dramatic changes in cell elongation. Thus, only changes in cell cytoskeleton and/or degree of adhesion might lead to the results presented here. However, macrophages have a distinct cytoskeleton arrangement compared to that of mesenchymal cells, such as fibroblasts. The development of actin stress fibers and focal adhesions is less frequent in macrophages than in fibroblasts50 whereas podosomes, distinct circular adhesion structures, are only formed in the earlier.51 Podosomes found in the basal face of the cell exert perpendicular forces to the substrate, sense the substrate stiffness and increase their force in response, which is a hallmark of mechanosensing activity.51 The differences in stiffness between glass and PDMS may result in different activation of mechanosensing pathways, constituting another possible mechanism leading to the diverse macrophage responses to glass and PDMS. However, this different mechanotransduction process may also mean that macrophages differ in sensitivity to substrate topographic characteristics compared to mesenchymal cells, and that in an already low state of adhesion to the soft PDMS substrate, fail to respond to the distinct cell body deformations promoted by concave and convex topographies. In support of this notion is the observation that M2 macrophages reach an average spreading area of approx. 600 μm2 (Fig. 7A) whereas macrophages in all PDMS topographies have <400 μm2 spreading area (Fig. 5A) revealing a considerable decreased interaction with the surface in the topographies.
5. Conclusion
In this study, we successfully produced a variety of convex and concave PDMS surface topographies. By culturing macrophages on either substrate we were able to induce distinct cell morphological adaptation to PDMS. Cells were not greatly affected by the different 3D shape of the individual topographic features within each group. The changes in cell size and morphology promoted by concave and convex microtopographies could not be correlated with alterations in the macrophage polarization state. On the other hand, the substrate material has an impact on macrophage response to planar surfaces. Namely, cells on PDMS have decreased metabolic activity, elongation and spreading and increased circularity than those on glass, which was correlated to a decrease in metabolism and expression of M1 polarization markers in basal medium. We have also confirmed that chemical polarization induces specific changes in the cell morphology on planar substrates, independently on the substrate material.Acknowledgements
The authors would like to acknowledge the EU Marie Curie Co-funding programme (PCOFUND-GA-2010-267161) for providing financial support to this project. The authors would like to thank the assistance of Karl Boehlen and Erdem C. Siringil for the laser micromachining experiments, and Ursina Tobler with RT-PCR.References
- J. M. Anderson, Cardiovasc. Pathol., 1993, 2, 33–41 CrossRef.
- T. Kruis, A. Batra and B. Siegmund, Front. Immunol., 2013, 4, 510 Search PubMed.
- B. N. Brown, R. Londono, S. Tottey, L. Zhang, K. A. Kukla, M. T. Wolf, K. A. Daly, J. E. Reing and S. F. Badylak, Acta Biomater., 2012, 8, 978–987 CrossRef CAS PubMed.
- E. M. Sussman, M. C. Halpin, J. Muster, R. T. Moon and B. D. Ratner, Ann. Biomed. Eng., 2013, 1–9 Search PubMed.
- B. K. K. Teo, S. T. Wong, C. K. Lim, T. Y. S. Kung, C. H. Yap, Y. Ramagopal, L. H. Romer and E. K. F. Yim, ACS Nano, 2013, 7, 4785–4798 CrossRef CAS PubMed.
- R. J. McMurray, A. K. T. Wann, C. L. Thompson, J. T. Connelly and M. M. Knight, Sci. Rep., 2013, 3 Search PubMed.
- C. T. McKee, V. K. Raghunathan, P. F. Nealey, P. Russell and C. J. Murphy, Biophys. J., 2011, 101, 2139–2146 CrossRef CAS PubMed.
- S. Pagliara, K. Franze, C. R. McClain, G. W. Wylde, C. L. Fisher, R. J. M. Franklin, A. J. Kabla, U. F. Keyser and K. J. Chalut, Nat. Mater., 2014, 13, 638–644 CrossRef CAS PubMed.
- L. E. McNamara, R. Burchmore, M. O. Riehle, P. Herzyk, M. J. P. Biggs, C. D. W. Wilkinson, A. S. G. Curtis and M. J. Dalby, Biomaterials, 2012, 33, 2835–2847 CrossRef CAS PubMed.
- D. Khang, P. Liu-Snyder, R. Pareta, J. Lu and T. J. Webster, Acta Biomater., 2009, 5, 1425–1432 CrossRef CAS PubMed.
- Q.-L. Ma, L.-Z. Zhao, R.-R. Liu, B.-Q. Jin, W. Song, Y. Wang, Y.-S. Zhang, L.-H. Chen and Y.-M. Zhang, Biomaterials, 2014, 35, 9853–9867 CrossRef CAS PubMed.
- F. Y. McWhorter, T. Wang, P. Nguyen, T. Chung and W. F. Liu, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17253–17258 CrossRef CAS PubMed.
- J. Meyle, K. Gültig and W. Nisch, J. Biomed. Mater. Res., 1995, 29, 81–88 CrossRef CAS PubMed.
- C. C. G. Moura, D. Zanetta-Barbosa, P. Dechichi, V. F. Carvalho and P. B. F. Soares, Braz. Dent. J., 2014, 25, 96–103 CrossRef PubMed.
- N. E. Paul, C. Skazik, M. Harwardt, M. Bartneck, B. Denecke, D. Klee, J. Salber and G. Zwadlo-Klarwasser, Biomaterials, 2008, 29, 4056–4064 CrossRef CAS PubMed.
- J. Rice, J. A. Hunt, J. A. Gallagher, P. Hanarp, D. S. Sutherland and J. Gold, Biomaterials, 2003, 24, 4799–4818 CrossRef CAS PubMed.
- S. Chen, J. A. Jones, Y. Xu, H.-Y. Low, J. M. Anderson and K. W. Leong, Biomaterials, 2010, 31, 3479–3491 CrossRef CAS PubMed.
- S. Svensson, M. Forsberg, M. Hulander, F. Vazirisani, A. Palmquist, J. Lausmaa, P. Thomsen and M. Trobos, Int. J. Nanomed., 2014, 9, 775 CrossRef PubMed.
- J. Takebe, C. M. Champagne, S. Offenbacher, K. Ishibashi and L. F. Cooper, J. Biomed. Mater. Res., Part A, 2003, 64, 207–216 CrossRef CAS PubMed.
- K. S. Tan, L. Qian, R. Rosado, P. M. Flood and L. F. Cooper, Biomaterials, 2006, 27, 5170–5177 CrossRef CAS PubMed.
- B. Wójciak-Stothard, A. Curtis, W. Monaghan, K. MacDonald and C. Wilkinson, Exp. Cell Res., 1996, 223, 426–435 CrossRef PubMed.
- B. Wójciak-Stothard, Z. Madeja, W. Korohoda, A. Curtis and C. Wilkinson, Cell Biol. Int., 1995, 19, 485–490 CrossRef PubMed.
- T. D. Zaveri, N. V. Dolgova, B. H. Chu, J. Lee, J. Wong, T. P. Lele, F. Ren and B. G. Keselowsky, Biomaterials, 2010, 31, 2999–3007 CrossRef CAS PubMed.
- K. A. Barth, J. D. Waterfield and D. M. Brunette, J. Biomed. Mater. Res., Part A, 2013, 101, 2679–2688 CrossRef PubMed.
- M. Bartneck, V. A. Schulte, N. E. Paul, M. Diez, M. C. Lensen and G. Zwadlo-Klarwasser, Acta Biomater., 2010, 6, 3864–3872 CrossRef CAS PubMed.
- M. Mohiuddin, H.-A. Pan, Y.-C. Hung and G. S. Huang, Nanoscale Res. Lett., 2012, 7, 1–9 CrossRef PubMed.
- S. Lee, J. Choi, S. Shin, Y.-M. Im, J. Song, S. S. Kang, T.-H. Nam, T. J. Webster, S.-H. Kim, D. Khang, S. Lee, J. Choi, S. Shin, Y.-M. Im, J. Song, S. S. Kang, T.-H. Nam, S.-H. Kim and D. Khang, Acta Biomater., 2011, 7, 2337–2344 CrossRef CAS PubMed.
- K. Anselme, L. Ploux and A. Ponche, in Surface and Interfacial Aspects of Cell Adhesion, CRC Press, 2011, pp. 43–64 Search PubMed.
- J. Y. Park, D. H. Lee, E. J. Lee and S.-H. Lee, Lab Chip, 2009, 9, 2043–2049 RSC.
- M.-H. Kim, Y. Sawada, M. Taya and M. Kino-oka, J. Biol. Eng., 2014, 8, 1–9 CrossRef PubMed.
- A. M. Green, J. A. Jansen, J. Van der Waerden and A. F. Von Recum, J. Biomed. Mater. Res., 1994, 28, 647–653 CrossRef CAS PubMed.
- A. M. Schmidt, S. R. Fagerer, K. Jefimovs, F. Buettner, C. Marro, E. C. Siringil, K. L. Boehlen, M. Pabst and A. J. Ibanez, Analyst, 2014, 139, 5709–5717 RSC.
- S. Tsuchiya, Y. Kobayashi, Y. Goto, H. Okumura, S. Nakae, T. Konno and K. Tada, Cancer Res., 1982, 42, 1530–1536 CAS.
- M. V. Lancaster and R. D. Fields, US5501959, 1996 Search PubMed.
- K. J. Livak and T. D. Schmittgen, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- B. Wójciak-Stothard, Z. Madeja, W. Korohoda, A. Curtis and C. Wilkinson, Cell Biol. Int., 1995, 19, 485–490 CrossRef PubMed.
- T. U. Luu, S. C. Gott, B. W. K. Woo, M. P. Rao and W. F. Liu, ACS Appl. Mater. Interfaces, 2015, 7, 28665–28672 Search PubMed.
- A. M. B. Collie, P. C. S. Bota, R. E. Johns, R. V. Maier and P. S. Stayton, J. Biomed. Mater. Res., Part A, 2011, 96, 162–169 CrossRef PubMed.
- S. Ghrebi, D. W. Hamilton, J. D. Waterfield and D. M. Brunette, J. Biomed. Mater. Res., Part A, 2013, 101, 2118–2128 CrossRef PubMed.
- H.-S. Lee, S. J. Stachelek, N. Tomczyk, M. J. Finley, R. J. Composto and D. M. Eckmann, J. Biomed. Mater. Res., Part A, 2013, 101, 203–212 CrossRef PubMed.
- V. Ballotta, A. Driessen-Mol, C. V. C. Bouten and F. P. T. Baaijens, Biomaterials, 2014, 35, 4919–4928 CrossRef CAS PubMed.
- M. Nishio, N. Urakawa, M. Shigeoka, N. Takase, Y. Ichihara, N. Arai, Y. Koma and H. Yokozaki, Pathol. Int., 2016, 66, 83–93 CrossRef CAS PubMed.
- V. Ballotta, A. Driessen-Mol, C. V. C. Bouten and F. P. T. Baaijens, Biomaterials, 2014, 35, 4919–4928 CrossRef CAS PubMed.
- S. Joshi, A. R. Singh, M. Zulcic, L. Bao, K. Messer, T. Ideker, J. Dutkowski and D. L. Durden, PLoS One, 2014, 9, e95893 Search PubMed.
- M. Cabanel, C. Brand, M. C. Oliveira-Nunes, M. P. Cabral-Piccin, M. F. Lopes, J. M. Brito, F. L. de Oliveira, M. C. El-Cheikh and K. Carneiro, PLoS One, 2015, 10, e0132984 Search PubMed.
- A. Mantovani, A. Sica, S. Sozzani, P. Allavena, A. Vecchi and M. Locati, Trends Immunol., 2004, 25, 677–686 CrossRef CAS PubMed.
- F. O. Martinez, L. Helming and S. Gordon, Annu. Rev. Immunol., 2009, 27, 451–483 CrossRef CAS PubMed.
- C. El Chartouni and M. Rehli, Immunobiology, 2010, 215, 780–787 CrossRef CAS PubMed.
- S. K. Biswas and A. Mantovani, Cell Metab., 2012, 15, 432–437 CrossRef CAS PubMed.
- W. E. Allen, G. E. Jones, J. W. Pollard and A. J. Ridley, J. Cell Sci., 1997, 110, 707–720 CAS.
- A. Labernadie, A. Bouissou, P. Delobelle, S. Balor, R. Voituriez, A. Proag, I. Fourquaux, C. Thibault, C. Vieu, R. Poincloux, G. M. Charrière and I. Maridonneau-Parini, Nat. Commun., 2014, 5 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6bm00425c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |