
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Side chain variations radically alter the diffusion of poly(2-alkyl-2-oxazoline) functionalised nanoparticles through a mucosal barrier†
Edward D. H.
Mansfield
a,
Victor R.
de la Rosa
b,
Radoslaw M.
Kowalczyk
c,
Isabelle
Grillo
d,
Richard
Hoogenboom
b,
Katy
Sillence
e,
Patrick
Hole
e,
Adrian C.
Williams
a and
Vitaliy V.
Khutoryanskiy
*a
aSchool of Pharmacy, University of Reading, Whiteknights, Reading, Berkshire RG6 6AD, UK. E-mail: v.khutoryanskiy@reading.ac.uk
bSupramolecular Chemistry Group, Department of Organic and Macromolecular Chemistry, Ghent University, Krijgslaan 281 S4, B-9000, Ghent, Belgium. E-mail: Richard.hoogenboom@ugent.be
cChemical Analysis Facility, University of Reading, Whiteknights, Reading, Berkshire RG6 6AD, UK
dInstitut Laue-Langevin, 71 avenue des Martyrs, 38000 Grenoble, France
eMalvern Instruments Limited, London Road, Minton Park, Amesbury, Wiltshire SP4 7RT, UK
First published on 11th July 2016
Abstract
Functionalised nanomaterials are gaining popularity for use as drug delivery vehicles and, in particular, mucus penetrating nanoparticles may improve drug bioavailability via the oral route. To date, few polymers have been investigated for their muco-penetration, and the effects of systematic structural changes to polymer architectures on the penetration and diffusion of functionalised nanomaterials through mucosal tissue have not been reported. We investigated the influence of poly(2-oxazoline) alkyl side chain length on nanoparticle diffusion; poly(2-methyl-2-oxazoline), poly(2-ethyl-2-oxazoline), and poly(2-n-propyl-2-oxazoline) were grafted onto the surface of thiolated silica nanoparticles and characterised by FT-IR, Raman and NMR spectroscopy, thermogravimetric analysis, and small angle neutron scattering. Diffusion coefficients were determined in water and in a mucin dispersion (using Nanoparticle Tracking Analysis), and penetration through a mucosal barrier was assessed using an ex vivo fluorescence technique. The addition of a single methylene group in the side chain significantly altered the penetration and diffusion of the materials in both mucin dispersions and mucosal tissue. Nanoparticles functionalised with poly(2-methyl-2-oxazoline) were significantly more diffusive than particles with poly(2-ethyl-2-oxazoline) while particles with poly(2-n-propyl-2-oxazoline) showed no significant increase compared to the unfunctionalised particles. These data show that variations in the polymer structure can radically alter their diffusive properties with clear implications for the future design of mucus penetrating systems.
1. Introduction
Over recent years, nanoparticles have gained concomitant interest both in academia and industry as they provide unique physicochemical properties not shown by bulk materials.1 Due to their sub-micron size, they can exhibit quantum-like features dependent on size, morphology, and bulk composition of the material. Nanoparticles can be tuned and engineered to have specific attributes for bespoke applications and can thus have unique magnetic,2 optical,3 and physicochemical properties4 through appropriate surface functionalisation.Poly(2-oxazoline)s are versatile polymers used in pharmaceutical applications.5 There are several naming conventions given to poly(2-oxazoline)s in the literature, such as POZ, POx and PAOx. Similar to poly(ethylene glycol) (PEG), a polymer frequently used in formulations,6 they have “stealth” properties, are non-toxic and biocompatible.7 POZ offer several advantages over PEG, such as facile synthesis,8 a pendent group available for further functionalisation,8,9 a high degree of renal clearance with no bioaccumulation,10 and improved stability against oxidative degradation.11,12 Although not currently FDA approved, there is extensive research using poly(2-oxazolines) for pharmaceutical applications, and it is expected to gain regulatory clearance within the next few years.13 In fact, some POZ-based formulations are currently undergoing clinical trials.14
The gastric mucosa provides a significant barrier to oral drug delivery, including the overlying layer of glycoproteins (mucins) that form a mesh-like structure that inhibits foreign molecules, particles, and pathogens from reaching the underlying cells and systemic circulation.15 This highly viscous mucus gel comprises mucins, salts, and other biomolecules and inhibits the penetration of hydrophobic compounds.16 Given that there are numerous literature reports on mucoadhesive particles and polymers,17,18 the field of mucus-penetrating nanoparticles has emerged in recent years and gaining rapid popularity. It has been shown that, by functionalising nanoparticle surfaces with poly(ethylene glycol)19 or poly(vinyl alcohol),20 it is possible to enhance their diffusion through a mucosal barrier. Further, we have previously shown that poly(2-ethyl-2-oxazoline) enhanced nanoparticle permeation through a gastric barrier.21 Given the advantages that POZ has over other polymers such as PEG, this may allow hydrophobic drugs (which are commonly delivered by intravenous or subcutaneous administration), to be delivered orally, thus improving patient compliance. In addition to this, other systems have been studied including magnetic nanoparticles,22 poly(lactide)-PEG nanoparticles,23 and papain-modified poly(acrylic acid) nanoparticles,24 demonstrating the diversity of mucus-penetrating nanomaterials in the literature.
However, despite the popularity of mucus-penetrating nanoparticles, there are currently no systematic studies reporting the influence of polymer structure on mucus penetration of polymer decorated nanoparticles. Here, we demonstrate for the first time that minor alterations to the alkyl side chain length in POZ significantly affects the diffusion and penetration of functionalised nanoparticles through gastric mucosa. By using 50 nm silica nanoparticles as a model, known to be mucoadhesive in nature,25 any changes in diffusion can be attributed to the architecture of the polymers itself.
2. Materials and methods
2.1 Materials
3-Mercaptopropyltrimethoxysilane (MPTS), triethylamine (TEA), and porcine gastric mucin type II were purchased from Sigma-Aldrich (Gillingham, UK). Maleimide terminated Alexa 546, dimethyl sulphoxide, and fluorescein-O-methacrylate were purchased from ThermoFisher Scientific (UK), and D2O from VWR International (UK). Acetonitrile was from Acros (UK). All other chemicals used in this study were of analytical grade and purchased from Sigma-Aldrich (Gillingham, UK) unless otherwise stated.Water was ultrapure, from a Purelab UHQ water filter (Ω = 18). Propargyl benzenesulphonate was distilled prior to use. 2-Methyl-2-oxazoline, 2-ethyl-2-oxazoline and 2-n-propyl-2-oxazoline (synthesised as previously reported26) were distilled over barium oxide. Acetonitrile was dried over molecular sieves (3 Å), and tetramethylammonium hydroxide in methanol (25 wt%) was used as received.
2.2 Polymer synthesis
All reagents were stored and handled under dried nitrogen in a glove-box (Vigor gas purification technologies, Inc.). Polymer synthesis was as reported:27 a 4 M solution of the 2-alkyl-2-oxazoline monomer was prepared in acetonitrile in the presence of 1/50 equivalents of propargyl benzenesulphonate. The polymerisation mixture was heated to 100 °C in an aluminium heating block for 60 min, according to previous reports,28 cooled to 0 °C and the living polymer chains were terminated by addition of 1.1 equivalents of tetramethylammonium hydroxide under a dry nitrogen atmosphere. The polymerisation mixture was kept stirring for 18 hours at room temperature after which the solvent was evaporated under reduced pressure. The polymer was re-dissolved in dichloromethane and precipitated in cold diethyl ether, providing a white powder that was filtered and dried in a vacuum oven at 50 °C for 24 h. Polymer composition was confirmed by 1H-NMR spectroscopy, MALDI-ToF MS and size exclusion chromatography. Full characterisation details of the polymers obtained can be found in the ESI (Fig. S1†).2.3 Polymer characterisation
1H-NMR spectra were recorded in CDCl3 on a Bruker Avance 300 MHz spectrometer. Spectra were processed using Bruker software (TOPSPIN 3.0).Size exclusion chromatography (SEC) was performed on an Agilent 1260-series equipped with a 1260 ISO-pump, a 1260 Diode Array Detector, a 1260 Refractive Index Detector, using two Mixed-D 30 cm columns (Agilent) and a Mixed-D precolumn (Agilent) in series at 50 °C, with DMA containing 50 mM of LiCl as eluent, at a flow rate of 0.593 mL min−1. Molar masses and dispersity values were calculated against polymethylmethacrylate standards.
MALDI-ToF MS (Matrix-Assisted Laser Desorption and Ionisation Time of Flight Mass Spectrometry) used an Applied Biosystems Voyager-DE STR instrument equipped with a nitrogen laser operating at 337 nm, pulsed ion extraction source and reflectron detector. The laser pulse width was 3 ns with a maximum power of 20 Hz. Spectra were recorded in reflector mode with an acceleration voltage of 19 kV and delay of 400 ns. 100 single shot acquisitions were summed to give the spectra and the data were analyzed using Data Explorer software. Samples were prepared by dissolving the matrix 2-(4-hydroxyphenylazo)benzoic acid (HABA) in the solvent (acetone, 20 mg mL−1), mixing with the polymer (1 mg mL−1) and sodium iodide in acetone (15 mg mL−1) that was used as a cationising agent.
2.4 Nanoparticle synthesis
Thiolated silica nanoparticles were synthesised according to our previously published protocol.25,29 0.75 mL MPTS was reacted with 0.5 mL NaOH (0.5 mol L−1) in 20 mL DMSO. The reaction was left for 24 hours at room temperature whilst continuously bubbled through with air to aid disulphide bond formation by partial oxidation of free thiol groups. After 24 hours, the particles were purified by dialysis, using a cellulose dialysis membrane (12–14 kDa molecular weight cut-off, Medicell International, UK). The dialysis membrane was placed in a beaker containing 4 L deionised water. The water was changed, over a 48 hour period with a total of 10 changes. Samples were stored at 4 °C until further use.2.5 Determination of reactive thiol content and fluorescent labelling
Before functionalisation, the concentration of reactive thiol groups was determined by Ellman's assay. Prior to the assay, 3 × 1 mL suspensions of nanoparticles were lyophilised using a Heto PowerDry LL3000 freeze drier and the dry weight recorded to determine the initial particle concentration (mg mL−1). The samples were then pooled for solid state analysis.For Ellman's assay;30,31 3 mg of dry particles were re-suspended in 10 mL phosphate buffer (pH 8, 0.5 M), and allowed to incubate for 1 hour. 10 aliquots (0.5 mL) were individually placed in 1.5 mL Eppendorf tubes and reacted with 0.5 mL 5,5′-dithiobis-(2-nitrobenzoic acid) (0.3 mg mL−1) for 2 hours in the dark. 200 μL aliquots were then placed into a 96 well plate (three repeats for each Eppendorf tube), and the absorbance measured at 420 nm using a BioTek Epoch plate reader. L-Cysteine–HCl solutions were used as standards from 3.125 μM to 12.69 μM.
Two different fluorophores were used; maleimide terminated Alexa 546, and fluorescein-O-methacrylate. The Ellman's assay showed that the free thiol content on the surface of the unfunctionalised silica nanoparticles was 212 ± 47 μmol g−1; consequently, 10 μmol of thiol groups were reacted with fluorophore since it was previously shown that labelling 5% of the nanoparticle surface was sufficient to allow particle visibility above background fluorescence from mucins.21
For Alexa 546 labelling, 1 mg of dye was dissolved in 5 mL water. From this, 200 μL of aqueous fluorophore was added to a 2 mL aqueous suspension of nanoparticles (5 mg mL−1). The suspension was left stirring overnight, and purified by dialysis, as described in section 2.4.
For fluorescein labelling, a fluorescein-O-methacrylate solution (1.8 mM) was prepared in 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50% v/v ethanol–deionised water. From this, 2 mL was added to 5 mL of nanoparticles suspended in DMSO (10 ± 0.5 mg mL−1) along with 200 μL TEA (0.033 mM final concentration). The reaction was left to stir in a flask for 24 hours in the dark. Labelled nanoparticles were purified by dialysis, as described in section 2.4.
:50% v/v ethanol–deionised water. From this, 2 mL was added to 5 mL of nanoparticles suspended in DMSO (10 ± 0.5 mg mL−1) along with 200 μL TEA (0.033 mM final concentration). The reaction was left to stir in a flask for 24 hours in the dark. Labelled nanoparticles were purified by dialysis, as described in section 2.4.
2.6 POZylation of nanoparticles
After fluorescent labelling, particles were functionalised with POZ using a previously published protocol with minor modifications.21,32 A 5 mL aqueous suspension of nanoparticles was diluted with 5 mL of DMSO. To this, 100 mg alkyne terminated POZ and 200 μL TEA was added. The reaction was left for 96 hours at room temperature under constant stirring. Samples were then purified by dialysis, as described in section 2.4.2.7 Nanoparticle characterisation
Following functionalisation, nanoparticles were characterised for size using dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA), for surface functionality using FT-IR and FT-Raman spectroscopy, and for polymer loading using thermogravimetric analysis (TGA). Small Angle Neutron Scattering studies were used to determine the corona (and core) sizes. Transmission electron microscopy (TEM) images were also taken.DLS measurements were performed using a Zetasizer Nano-ZS (Malvern, UK). Samples were diluted 1:100 with ultrapure water to appropriate concentrations in low volume plastic cuvettes (Fisher Scientific, UK). A refractive index of 1.49 and absorbance of 0.1 was used for all recordings. Individual measurements were carried out for 10 seconds per run, with 12 runs per reading, repeated in triplicate. This was repeated for 3 separate samples at 25 °C. ζ-Potential values were also measured, using DTS-1070 folded capillary tube cuvettes (Malvern, UK). Samples were prepared to the same concentration as for sizing experiments and injected into the cuvettes ensuring no air bubbles were present. Samples were analysed using 20 sub-runs per reading, repeating 3 times for each sample. Each sample was measured three times and the results were processed using the Smoluchowski model (Fκa = 1.50).
NTA measurements used an LM10 system with temperature controlled module, green 532 nm laser, and syringe pump (Malvern, UK). NTA requires very low concentrations of nanoparticles in suspension, as each nanoparticle is individually tracked. Therefore, samples were initially diluted 1:10000 and adjusted until appropriate concentrations were found. Samples were placed in a 1 mL syringe, loaded into the system and the NTA syringe pump set to a flow rate of 30 AU. Videos were recorded for 60 seconds. 5 videos were collected per individual sample, and triplicate samples were recorded. Analysis used NTA v3.0 software. All NTA sizing experiments were performed at room temperature (25 °C) under the constant flow from the syringe pump, to obtain data representative of the dispersions.
Experiments to assess surface functionality and polymer loading were carried out with lyophilised nanoparticles. FTIR spectra were recorded using a Spectrum 100 FTIR spectrophotometer (Perkin Elmer, UK). Spectra are the average of 32 scans, between 4000 and 550 cm−1, at a resolution of 4 cm−1.
FT-Raman spectra were recorded on a Nicolet NXR 9650 Raman spectrophotometer (Thermo Scientific, UK) as an average of 1000 scans, between 4000 and 10 cm−1, at a resolution of 4 cm−1.
Thermogravimetric analysis (TGA) was performed on a Q500 instrument (TA Instruments, UK) with nitrogen as the inlet gas. Baselines were taken against an empty platinum TGA pan and Tzero aluminium DSC pans (TA Instruments, UK). Samples were placed into an aluminium DSC pan, before being loaded into the platinum TGA pan on the instrument. The initial temperature was set to 35 °C, and allowed to equilibrate for 5 minutes. Thermal decomposition was measured as the temperature ramped from 35 to 500 °C at 10 °C min−1.
TEM images were recorded using a Phillips CM20 analytical TEM, with an accelerating voltage of 200 kV and a 4 megapixel AMT camera. Samples were prepared by placing a drop of nanoparticle suspension onto a section of parafilm. A Holey Carbon film 300 mesh copper grid (HC300Cu, EMResolutions, UK) was then placed onto the drop and left for a minute. The grid was then left to air dry before being placed in the instrument.
2.8 NMR spectroscopy
Solid state NMR spectroscopic analysis was carried out using freeze-dried thiolated silica and poly(2-ethyl-2-oxazoline) decorated silica nanoparticles. The 29Si solid state NMR spectra were recorded on Bruker 500 MHz Avance III spectrometer at a Larmor frequency of 99.35 MHz (11.75 T) for 29Si. Both single pulse (SP) and cross-polarisation magic angle spinning (CPMAS) spectra were recorded. The standard bore 4 mm MAS probe was spun at 10 kHz. The 29Si 90° pulse length was 6.25 μs (at the power level of 67.86 W) and the relaxation delay time was 150 s for all SP experiments. For all CPMAS experiments, the 1H 90° pulse length was 3.7 μs (at the power level of 38 W), the CP contact time was 3 ms and the relaxation delay time was 5 s. Between 500 and 4096 signal transients were accumulated and averaged into a single spectrum. All spectra were referenced to external kaolinite signals as a secondary reference (well resolved peaks at −90.0 ppm and −91.4 ppm with respect to TMS).2.9 SANS
Small angle neutron scattering (SANS) experiments were performed on the D11 instrument at the Institute Laue-Langevin (Grenoble, France). The detector distance and flux were set in order to assess the core–shell structure of the functionalised and unfunctionalised nanoparticles across a large Q range, defined by eqn (1), where θ is the incidence angle, and λ is the neutron wavelength;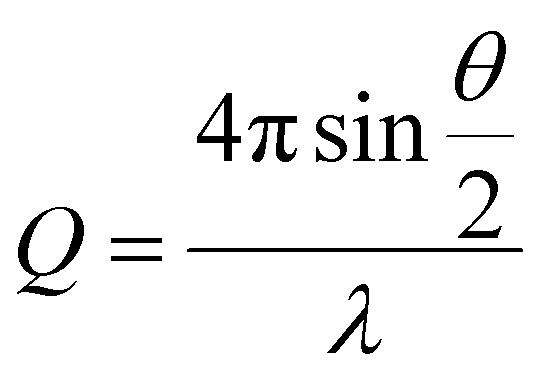 | (1) |
Prior to SANS experiments, both functionalised and unfunctionalised nanoparticles were dialysed into D2O using a 7 kDa molecular-weight cut-off membrane (Medicell International, UK). This was carried out to remove the incoherent scattering obtained from particles suspended in H2O. A 2 mL suspension of particles (5 mg mL−1) was dispensed into the membrane and sealed, before being placed into a vial containing ∼30 mL D2O. D2O was replaced every 6 hours with a total of 3 changes. Following dialysis, samples were stored in glass vials, sealed with parafilm at 4 °C. For SANS experiments, samples were diluted 1:10 into D2O, placed into quartz 700 μL Hellma cuvettes and sealed with a PTFE stopper. Samples were then placed in the beam-line. An incident wavelength of 8 Å was used and 3 detector distances of 1.2 m, 8 m and 28 m were used to cover a large Q-range from 2 × 10−3 to 0.3 Å−1 (DOI: 10.5291/ILL-DATA.9-12-422).
Data was fitted using the SasView programme (http://www.sasview.org). Both DLS and TEM data were used to define the core particle diameter and polydispersity index (PDI). A scattering length density (SLD) of 3.2 × 10−6 Å−1 was used for the core silica, based on previous SANS experiments using these particles,33 along with an SLD of 6.23 × 10−6 Å−1 for the nanoparticle corona. Initially scattering profiles were fitted to a spherical model (built into the software with no further modification), followed by a core–shell form to determine the thickness of the corona.
2.10 Nanoparticle diffusion
Diffusion experiments used the NTA LM10 system described above for size measurements. Prior to analysis, 3 × 25 mL gastric mucin type II dispersions (1% w/v) in ultrapure water were prepared, and left to stir overnight to ensure complete hydration. Alexa 546 labelled nanoparticles were diluted by a factor of 1:100 to form a stock solution. 10 μL of these suspensions was then added into 1 mL of the mucin dispersion for analysis, resulting in a total dilution of 1:10000.
Samples were injected into the NTA system and the flow-rate set to 50 AU in order to minimise fluorescent bleaching of the nanoparticles during analysis. All videos were recorded through a 565 nm cut-on filter. 6 × 60 second videos were recorded at 25 and 37 °C for each sample. Each independent dispersion of mucin was employed three times with each nanoparticle type, resulting in a total of 9 × 6, 60 second videos for each temperature. A viscosity of 25 cP at 25 °C and 28 cP at 37 °C was used for analysis.21
2.11 Mucosal penetration and histology
The mucus penetration study was carried out according to a previously published protocol, with minor modifications.21Fresh porcine stomach was obtained from a local abattoir (P.C. Turner Abattoirs, Farnborough, UK), and dissected to remove connective tissue and muscle, leaving only the mucosa, submucosa, and stomach lining intact. 4 × 1 cm2 sections were cut and placed on a glass plate, with the mucosal layer facing upward. 200 μL of fluorescein labelled nanoparticles were pipetted onto the sections; deionised water was also administered as a (blank) control. Samples were left to incubate for 0, 15, 30, 45, and 60 minutes at 37 °C. Following each time point, tissue sections were placed mucus side up into a small (3.5 × 5.5 cm) weighing boat (Fisher Scientific, UK), half filled with OCT, a cryoprotective embedding medium. They were then placed on dry ice, before being completely embedded in OCT to preserve the particle-loaded mucus membrane. Once all sections were embedded, samples were left on dry ice for 3–4 hours, before storage at −80 °C until processing.
For sectioning, samples were removed from the −80 °C freezer and placed on dry ice. Each sample was mounted onto a 22 mm standard solid object holder using OCT, and placed on dry ice for 30 min until completely frozen. 20 μm sections were cryosectioned transversely using a standard 189 × 27 × 10 mm blade at 22°, placed onto superfrost charged slides (Life Technologies, UK) and left to air dry for 30 minutes before being stored in a slide box. All sections were cut from interior to exterior (i.e. upwards through the mucosal layer) in order to avoid carriage of particles into the biological tissue during cutting. All sections were cut using a Bright 5040 cryostat in a Bright Model PTF freezing chamber at −20 °C (Bright Instrument Co. Ltd, UK).
Sections were placed under a Leica MZ10F stereomicroscope (Leica Microsystems, UK). All images were taken through an ET-GFP filter (Leica Microsystems, UK) using an exposure time of 0.8 ms. 10 images were taken for each particle type, at each time point from a separate section of tissue.
ImageJ software (National Institute of Health, USA, v1.43) was used to quantify penetration of the nanoparticles. For each image, the background was subtracted, a line drawn across the mucus barrier, and the “plot profile” measured. A diagrammatical representation of this process can be found in ESI (Fig. S2†). This was repeated 5 times at random locations along the mucosal barrier for each image, providing a total of 50 (10 × 5) profiles for each sample. Each individual profile was then assessed for fluorescent particle penetration using the width of the predominant peak. After each profile had been analysed, mean thickness was then calculated for each sample. Auto fluorescence was corrected for using values from the blank (water control) treated tissue at the appropriate time point, subtracted from the nanoparticle treated values.
In addition to fluorescence microscopy, histology of the stomach mucosa was carried out. Sections were initially immersed in a 1% v/v hematoxylin solution for 5 minutes. The samples were then washed with deionised water to remove excess solution, followed by a wash with 0.1 M HCl in ethanol for 10 seconds and a final wash with deionised water. The section was then counterstained with 1% v/v eosin for 2 minutes before a final washing step with deionised water and left to dry. Samples were examined under a light microscope (Leica DM2500 M, Leica, UK), and images obtained using an Infinity 1-1C camera (Lumenera, UK).
2.12 Statistics
Statistical analysis used GraphPad Prism, v 5.0. Means ± standard deviations were determined and assessed for significance using 2-way ANOVA with a Bonferoni post hoc test. Values of p < 0.05 were considered to be significant.3. Results and discussion
3.1 Nanoparticle characterisation
Here, we report that systematic variation of the alkyl side chain length of POZ grafted on nanoparticle surfaces radically alter their penetration into and diffusion through a gastric mucosal barrier. These structural modifications have minimal impact on the particles’ sizes, surface charge, or free thiol contents.The thiolated silica nanoparticles were monodisperse (PDI < 0.1), and contained a high concentration of reactive thiol groups for further functionalisation and fluorescent labelling. The particles were functionalised with 3 different variants of POZ; poly(2-methyl-2-oxazoline) (PMOZ), poly(2-ethyl-2-oxazoline) (PEOZ), and poly(2-n-propyl-2-oxazoline) (PNPOZ). The reaction mechanism for grafting is shown in Fig. 1; the TEA hydrogen bonds to the hydrogen atom in the thiol, generating a stronger nucleophile that attacks the reactive alkyne species on the terminal group of the polymer, forming a covalent bond between the two. It is possible (dependent on the proximity of the thiol groups to each other) that the resulting alkene formed in the reaction can further react with a thiol group, eliminating all double bonds in the system. However, this secondary reaction is highly dependent on the proximity of the thiol groups, and therefore unlikely, while it will not influence the grafting efficiency nor grafting density.
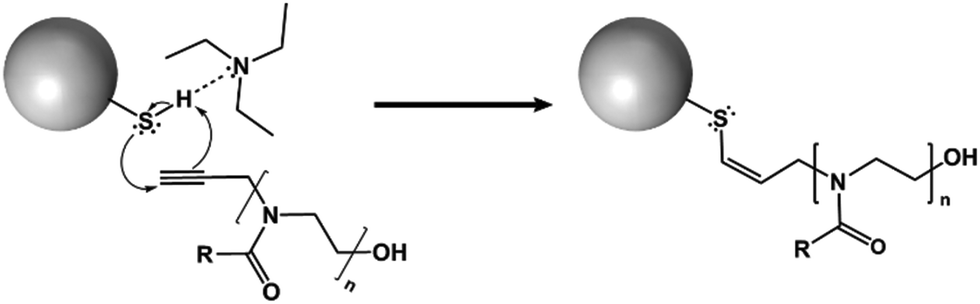 | ||
| Fig. 1 Proposed reaction mechanism for the functionalisation of silica nanoparticles by POZ. | ||
Immediately following synthesis and purification, the particles were sized using DLS and NTA, their surface functionality assessed using FT-IR and FT-Raman spectroscopy, and polymer loading determined using TGA. The DLS size distributions (ESI, Fig. S3†) show that, as expected, upon functionalisation particle size increases, suggesting successful conjugation. Further DLS information (PDI and ζ-potential) and NTA (size and diffusion coefficient) can be found in Table 1. In addition to size analysis, the particles were tested for their functionality by FT-IR and FT-Raman. Fig. 2a shows the FT-IR spectra for the functionalised and unfunctionalised nanoparticles, and the Raman spectra can be found in Fig. 2b.
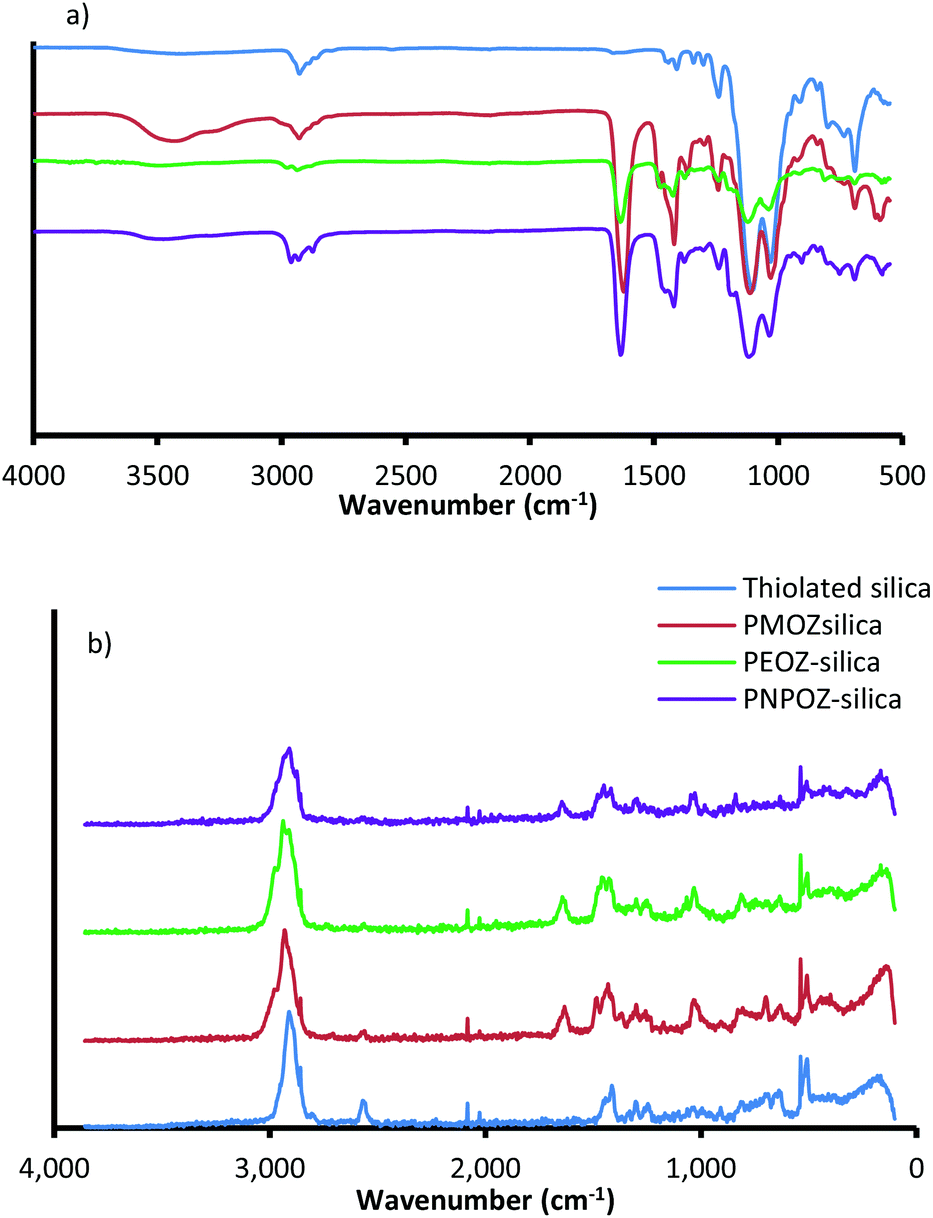 | ||
| Fig. 2 (a) FT-IR and (b) FT-Raman spectra for thiolated silica (blue), and functionalised silica nanoparticles. | ||
| Nanoparticle | z-Averagea (nm) | PDIa | ζ-Potentiala (mV) | Mode diameterb (nm) | Diffusion coefficient in waterb (×104 nm2 s) | Concentration of polymerc (%) | Free thiol (μmol g−1) |
|---|---|---|---|---|---|---|---|
| a Shows values determined by DLS. b Shows values determined by NTA. c Shows values determined by TGA. | |||||||
| Thiolated silica | 52 ± 1 | 0.072 | −46 ± 2 | 54 ± 1 | 788 ± 20 | 0 | 212 ± 47 |
| PMOZ-silica | 61 ± 4 | 0.188 | −23 ± 1 | 61 ± 4 | 811 ± 33 | 29 | 24 ± 6 |
| PEOZ-silica | 59 ± 1 | 0.106 | −20 ± 2 | 59 ± 1 | 798 ± 2 | 37 | 27 ± 3 |
| PNPOZ-silica | 61 ± 1 | 0.194 | −21 ± 2 | 63 ± 3 | 787 ± 28 | 33 | 14 ± 1 |
All spectra show strong vibrational modes at ∼1032 and ∼1102 cm−1, consistent with Si–O–Si stretching from disiloxane in the nanoparticle core. Due to the complex nature of the siloxane moiety in the nanoparticles (either Si–O–Si–O–Si, or C–O–Si–O), additional stretching modes appear as relatively weak features between 1300 and 1000 cm−1.34 Other minor modes from the silica core are seen at 2923 cm−1 (C–H stretch), 1237 cm−1 (C–O stretch), and 687 cm−1 (Si–S stretch). The nanoparticle structure proposed by Irmukhametova et al.25 is in agreement with these spectral features.
Upon functionalisation, the spectra for POZ-silica show modes attributed to the polymer; 2978 cm−1 (CH2 stretch), 1629 cm−1 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch), 1463 cm−1 (C–H deformation), 1416 cm−1 (CH3 symmetrical deformation/CH2 bending), and 1236 cm−1 (C–N stretch). The spectra for the different polymer variants (PMOZ, PEOZ, and PNPOZ) essentially show the same molecular modes, most noticibly the strong CO stretching mode around ∼1630 cm−1. These features confirm the presence of POZ in the samples, and successful conjugation of the polymer to the particle surface.
O stretch), 1463 cm−1 (C–H deformation), 1416 cm−1 (CH3 symmetrical deformation/CH2 bending), and 1236 cm−1 (C–N stretch). The spectra for the different polymer variants (PMOZ, PEOZ, and PNPOZ) essentially show the same molecular modes, most noticibly the strong CO stretching mode around ∼1630 cm−1. These features confirm the presence of POZ in the samples, and successful conjugation of the polymer to the particle surface.
Additionally, the molecular modes were investigated by FT-Raman spectroscopy (Fig. 2b). For thiolated silica (blue), there are characteristic Raman peaks at 2898 cm−1 (C–H), 2555 cm−1 (S–H), 1406 cm−1 (CH2), 1294 cm−1 (C–C), 1242 cm−1 (C–C), 627 cm−1 (C–S), and ∼500 cm−1 (Si–O–Si and S–S). After functionalisation with PMOZ, PEOZ, and PNPOZ there are two notable changes to the spectra; firstly a decrease in the intensity of the peak at 2555 cm−1 (S–H stretch), suggesting a decrease in the number of reactive thiol groups (confirmed with Ellman's assay), and secondly the emergence of two new features at 1631 cm−1 (CO stretch), and 1024 cm−1 (C–C stretch). The emergence of these two peaks further confirms successful conjugation of POZ to the surface of the nanoparticles.
TGA was used to assess polymer loading on the silica particles. Weight loss during thermal decomposition (between 350 and 400 °C) showed that the polymer accounted for 29–37% of the nanoparticle weight (Table 1). TGA curves can be found in Fig. S4. To show that comparable amounts of polymer were grafted to the nanoparticles in all cases, the grafting density was determined. Grafting densities of 0.13, 0.11, and 0.12 μg nm−2 were calculated for PMOZ, PEOZ, and PNPOZ, respectively. It can therefore be concluded that comparable amounts of polymer were grafted to the nanoparticles in each case, ensuring any changes in diffusion coefficient/mucus penetration were due to changes in side-chain structure of the polymer and not grafting density.
3.2 NMR spectroscopy
NMR spectroscopic studies were undertaken to further confirm successful surface modification and, importantly, to also confirm that decorating the nanoparticle surfaces had no adverse effects on the silica core dimensions. Fig. 3 shows the CPMAS NMR spectrum of the silica precursor, highlighting two peaks at −57.9 ppm and −67.8 ppm. These resonances are attributed to the 29Si nuclei at three and two silicon–oxygen–silicon bridge coordination (so called T3 and T2 species), respectively.35 The SP and CPMAS spectra do not show any significant differences and yield similar intensity ratios IT3/IT2 of ca. 8.6.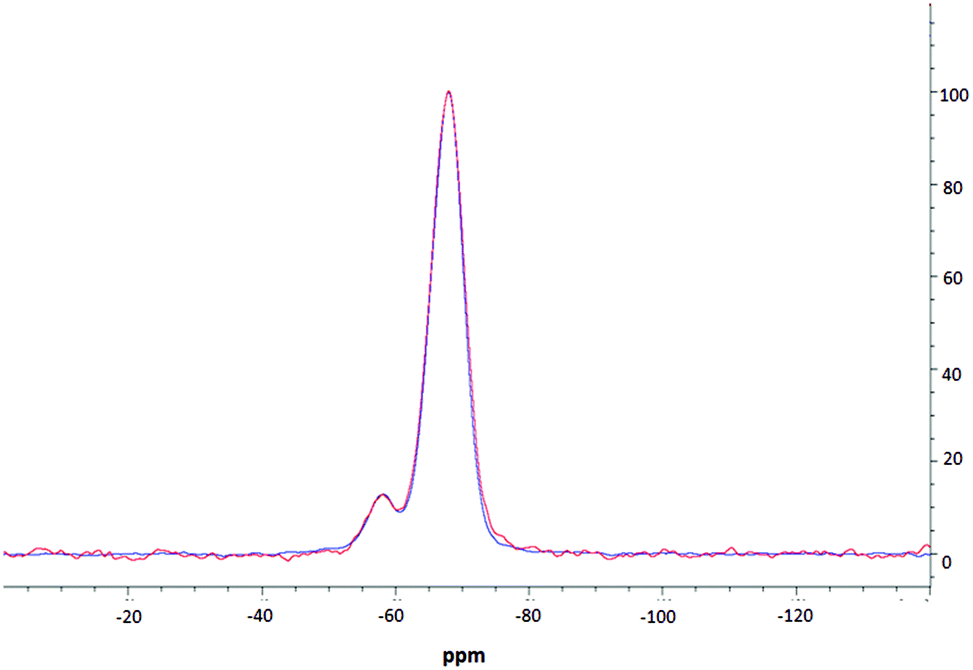 | ||
| Fig. 3 MAS NMR spectra of thiolated silica; one pulse spectrum is shown in red, CPMAS spectrum is shown in blue. | ||
Fig. 4 compares CPMAS spectra of the thiolated nanoparticles and PEOZ functionalised silica. The resonance at −67.8 ppm remains unchanged whereas the peak at −57.9 ppm shifts upfield to −58.9 ppm after functionalising the surface of the silica nanoparticles with PEOZ. The intensity ratio IT3/IT2 decreases from ca. 8.6 for the SP experiment, to ca. 7.6 for the CPMAS experiment. The increase in the T2 intensity suggests proximity of additional protons which will cause enhancement of the signal in the CPMAS experiment.36 Both the upfield shift and increase in the intensity of the T2 signal are consistent with the presence of the PEOZ molecules (additional source of protons) on the nanoparticle surface.
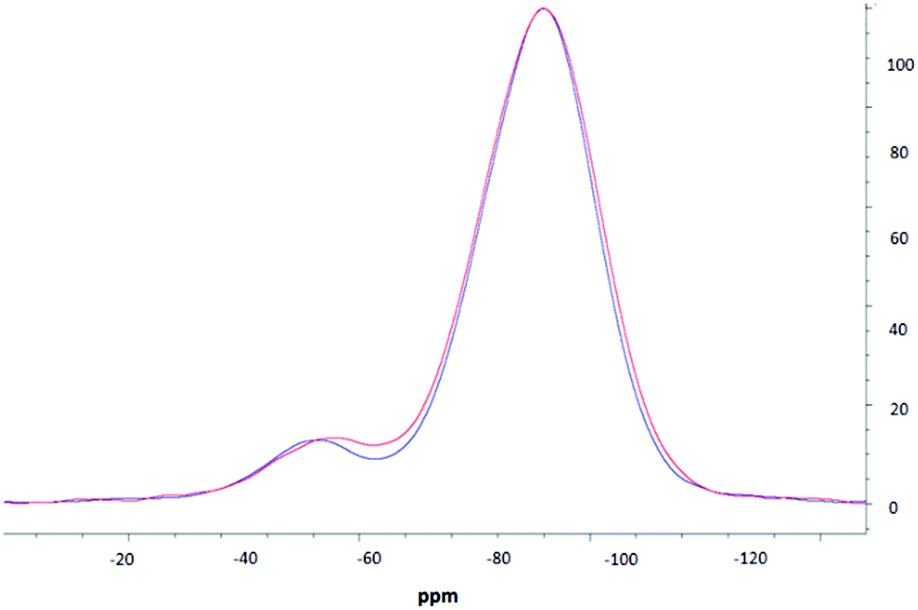 | ||
| Fig. 4 The CPMAS NMR spectra of thiolated silica (blue) and PEOZylated silica (red). | ||
The changes to the T2 resonance suggest that Si–O–Si bridges are predominantly present on the surface of the nanoparticle and are thus more sensitive to its modification. Assuming that the nanoparticles are spherical (as shown by TEM) and that T2 species are present only on the surface, the radius of the nanoparticle can be estimated. The intensity ratio IT3/IT2 (the core and surface silica) can provide a crude estimate of the size because the ratio of the volume to the surface area of sphere changes as the function of its radius. The unfunctionalised thiolated silica nanoparticle had a radius estimated to be ca. 26 nm (both from SP and CPMAS experiments), and the PEOZ functionalised nanoparticles had an estimated core radius also of ca. 26 nm. Both sizes appear the same because there are no changes to the number of silicon moieties on the surface, and the single pulse experiment does not rely on signal enhancement (proton-carbon cross-polarisation transfer). With the CPMAS experiment, the estimated size is smaller ca. 23 nm, as expected for the enhanced T2 signal intensity. These sizes are in good agreement with the DLS data; however, the DLS shows that the Dh of the particles is 52 nm, including the hydrated polymer chains. As the size determined by NMR spectroscopy is in the solid-state (and therefore not in the presence of water) and only takes into account the silica particle and not the polymer chains, it would be expected that the size would be smaller. However, this is not the case. It is likely that the freeze-drying process causes this discrepancy, as water diffusing into the core of the nanoparticle would freeze forming ice crystals, which would warp the internal structure of the particle, thus creating pockets of empty space which would result in a larger particle size. A similar result was reported previously,37,38 where the size of suspended silica nanoparticles was larger than the original particle size after lyophilisation, without the presence of any cryoprotectants.
3.3 SANS analysis
Small angle neutron scattering can probe both the external and internal structure of nanoparticles. Previously we have employed SANS to look at the core–shell structure of PEGylated silica nanoparticles.33 Here, SANS was used to determine the structure of functionalised silica nanoparticles and to confirm that decorating the particle surface with POZ has no detrimental effects to the core particle itself.The scattering profiles for these particles are shown in Fig. 5; curve fitting for each is given in ESI (Fig. S4†). The scattering profile for thiolated silica was initially fitted to a spherical form factor, using DLS and TEM data as a guide. The particle was found to have a radius of 137 Å, resulting in a diameter of 28 nm, which is significantly (P < 0.05) smaller than the size determined by DLS (52 nm) but is in good agreement with the NMR data. Previous studies by Mun et al.33 (who studied the core–shell structure of PEGylated MPTS silica nanoparticles) showed sizes of 30 nm for these particles using SANS. The discrepancy between SANS data with that from light scattering is due to the presence of a solvation shell around the particle, which is detected by DLS, but not by SANS. Due to the abundance of surface –SH and –Si–O− groups, there are strong interactions between water molecules and the nanoparticle surface, leading to a relatively large hydration shell.
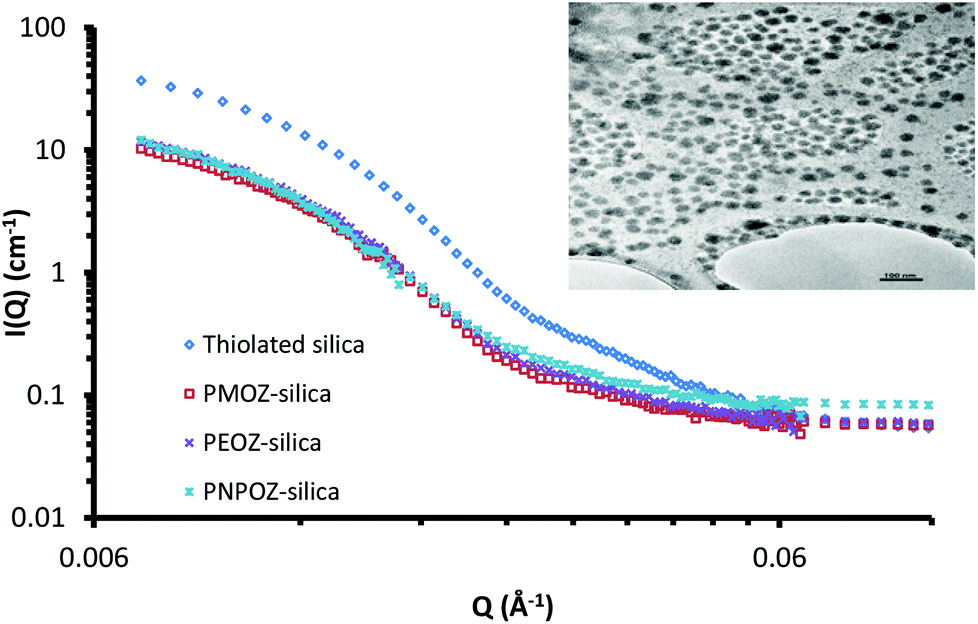 | ||
| Fig. 5 SANS profiles for thiolated silica (blue), PMOZ-silica (red), PEOZsilica (green), and PNPOZ-silica (purple) nanoparticles. The insert shows the TEM image for unfunctionalised thiolated silica, used for the fitting. The scale-bar represents 100 nm. | ||
Upon functionalisation, only minor changes in the scattering pattern arise (at high Q values ∼0.03 Å−1). A study of the swelling behaviour of PMOZ and PEOZ showed that POZ macromolecules are highly hydrated and bind tightly to water, and that PMOZ was significantly more hydrated than PEOZ.39 This phenomenon explains why the shell is barely visible using SANS; it is highly hydrated and offers no contrast to the D2O background, and so cannot be seen. It was found during curve fitting that changing the size of the shell (in a core–shell model) did not affect the curve. Based on this, the particles were fitted to a spherical form-factor, and displayed a size of 28 nm for all particles. With no significant change in size or scattering observed, it can be concluded that the addition of the polymer shell does not change the nanoparticles core architecture during the functionalisation reaction.
Although the SANS data could not be fitted to a core–shell model, the size of the hydrated polymer shell can be estimated. From the SANS data, the core diameter is 28 nm. Light scattering sizes include the hydration shell and provide a diameter of 52 nm for the thiolated particles; from this, a hydration layer of ∼13 nm for the non-polymer coated particles can be postulated. The particle diameters from DLS data in Table 1 for the functionalised nanoparticles then indicate shell thicknesses of 4, 3, and 5 nm for PMOZ, PEOZ, and PNPOZ, respectively. Similar values have been reported in the literature for PMOZ functionalised silica.40 It should be noted that these sizes are estimates based on the difference in hydration shell thickness. Clearly it is feasible that some polymer is present within the hydration shell, which would suggest a larger shell thickness.
3.4 Diffusion through a mucus dispersion
Previously we used nanoparticle tracking analysis to measure the diffusion of fluorescently labelled nanoparticles through solutions of polymers,33 and mucin dispersions.21 Here, POZ decorated nanoparticle diffusion coefficients were measured in 1% w/v gastric mucin dispersions at 25 and 37 °C; Fig. 6 shows the mean ± SD values for functionalised and unfunctionalised nanoparticles.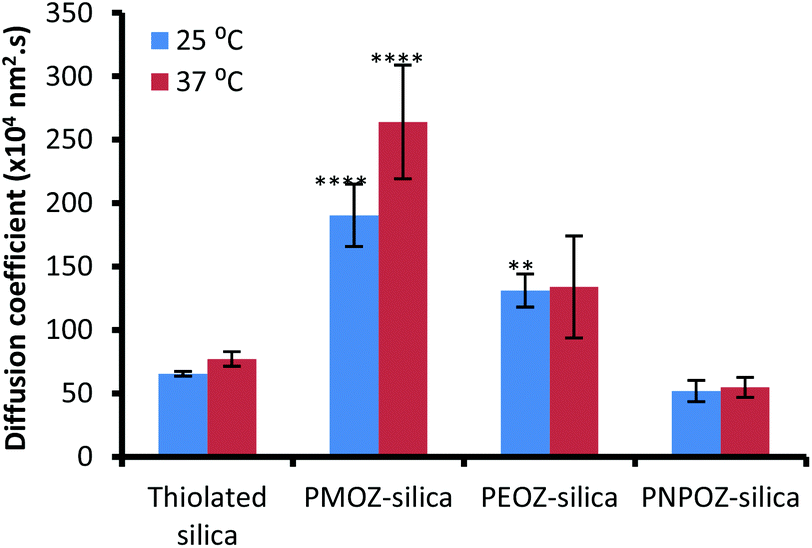 | ||
| Fig. 6 Diffusion coefficients for thiolated, and POZylated silica nanoparticles through a 1% gastric mucin dispersion at 25 and 37 °C. Error bars represent the mean ± standard deviation of 3 repeats. | ||
The diffusion coefficients vary radically with only minor alterations to the POZ alkyl side groups. The unfunctionalised thiolated particles are mucoadhesive in nature, and correlate with the diffusion coefficients determined in our previous study. Upon functionalisation, the PMOZ-coated particles are significantly more diffusive than the unfunctionalised particles and also have a significantly higher diffusion coefficient than PEOZ-coated particles (p < 0.05), at both temperatures. PEOZ-particles are also significantly more diffusive than the unfunctionalised particles (p < 0.05), although to a lesser degree. PNPOZ-silica nanoparticles were no more diffusive than the unfunctionalised particles (p > 0.05). The trend to decreasing diffusion coefficient with the addition of methylene bridges in the polymer can be ascribed to less efficient hydration and was corroborated by studies evaluating penetration of the nanoparticles into gastric mucosa.
3.5 Penetration into gastric mucosa and tissue histology
In order to assess penetration of the functionalised nanoparticles into gastric mucosa, fluorescence microscopy was employed. The summarised data in Fig. 7 illustrates that PMOZ-silica particles are significantly more diffusive than both PEOZ, and PNPOZ functionalised silica (p > 0.05) at all time points, a relationship that was found to be linear with time (with R2 values of 0.95, 0.98, and 0.92, respectively, Fig. S5†). The variance in rate of penetration accords with the diffusion coefficients for the particles in the mucin dispersion (Fig. 6). As the alkyl chain length increases (from methyl to propyl) there is a significant (P < 0.05) change in both penetration and diffusion coefficient of PMOZ and PEOZ-silica nanoparticles, again a trend which was found to be linear (R2 = 0.9975, Fig. S6†). For PNPOZ-silica nanoparticles, there is no significant difference in diffusion coefficient to the unmodified silica particles, however there is a significant difference observed in their mucosal penetration at longer time periods. This is attributed to interactions between the mucus and the particles. Although the diffusion coefficients of the nanoparticles in the mucin dispersions are similar (77 ± 6 or 51 ± 8 × 104 nm2 s for thiolated silica and PNPOZ-silica, respectively), the thiolated silica is highly mucoadhesive and will therefore bind to the mucosal surface and hence its penetration is retarded as illustrated in Fig. 7. PNPOZ however is less mucoadhesive; the functionalisation greatly reduces the nanoparticles surface thiol groups that are responsible for mucoadhesion and so the particles permeate through the mucus, although slower than the other POZylated particles.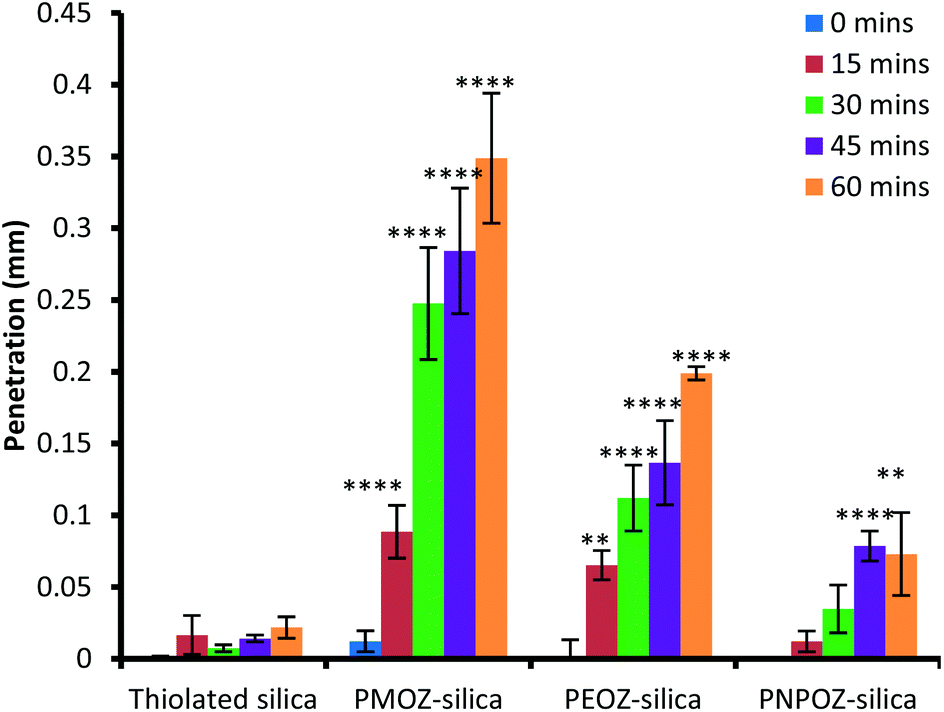 | ||
| Fig. 7 Penetration of thiolated and functionalised silica nanoparticles over 1 hour. Values represent the mean penetration across 10 separate tissue sections ± standard deviation. | ||
Based on the physicochemical characterisation data and previous reports, the difference in diffusion coefficient and penetration is likely due to the hydration of the polymer. PMOZ is significantly more hydrated than PEOZ, with PNPOZ being the least hydrated of the series. Thus, the hydrophobicity of the polymer grafted onto the surface of a nanoparticle is a major factor in the penetration of surface modified nanoparticles through mucus barriers, and so minor structural alterations can have unexpectedly large impacts. There are other factors which could affect this process (such as size), however these were corrected for as much as possible. It is well known that hydrophobic molecules are poorly penetrating through mucosal barriers,16 and our data are consistent with this effect. In fact, PMOZ is more hydrophilic than PEG,41 whereas PNPOZ exhibits a lower critical solution temperature (LCST) behaviour at 25 °C.26 Based on the data presented here, and the findings of our previous work,21 PMOZ appears to facilitate transmucosal diffusion to a greater extent than PEG.
To confirm the effect of polymer hydrophobicity on mucus penetration, the hydrophobic–hydrophilic balance (HHB) was calculated for each polymer, using the equation described in ref. 42. Values of 5.0, 6.0, and 7.1 were calculated for PMOZ, PEOZ and PNPOZ, respectively; showing the hydrophobicity changes with increased alkyl chain length.
Enhanced diffusion by polymer-functionalised nanoparticles through mucus has been attributed to “stealth properties”. However, our study shows that if a polymer is designed to be highly hydrophilic as well as containing “stealth” properties, the diffusion of the particles will be significantly enhanced. The thiolated silica nanoparticles themselves do not show any mucopenetration, and are well known to be mucoadhesive in nature,43 as was also shown in a previous study.21
In addition to fluorescence microscopy, some sections of stomach mucosa (chosen at random) were subjected to histological analysis. Fig. 8 shows representative images of the penetration of PEOZ functionalised nanoparticles into fresh porcine mucosa over time. Sections of a porcine stomach were stained with hematoxylin and eosin, and images were taken under a fluorescence microscope. Additional fluorescent images can be found in the ESI Fig. SI.7† for each particle type used in the penetration study.
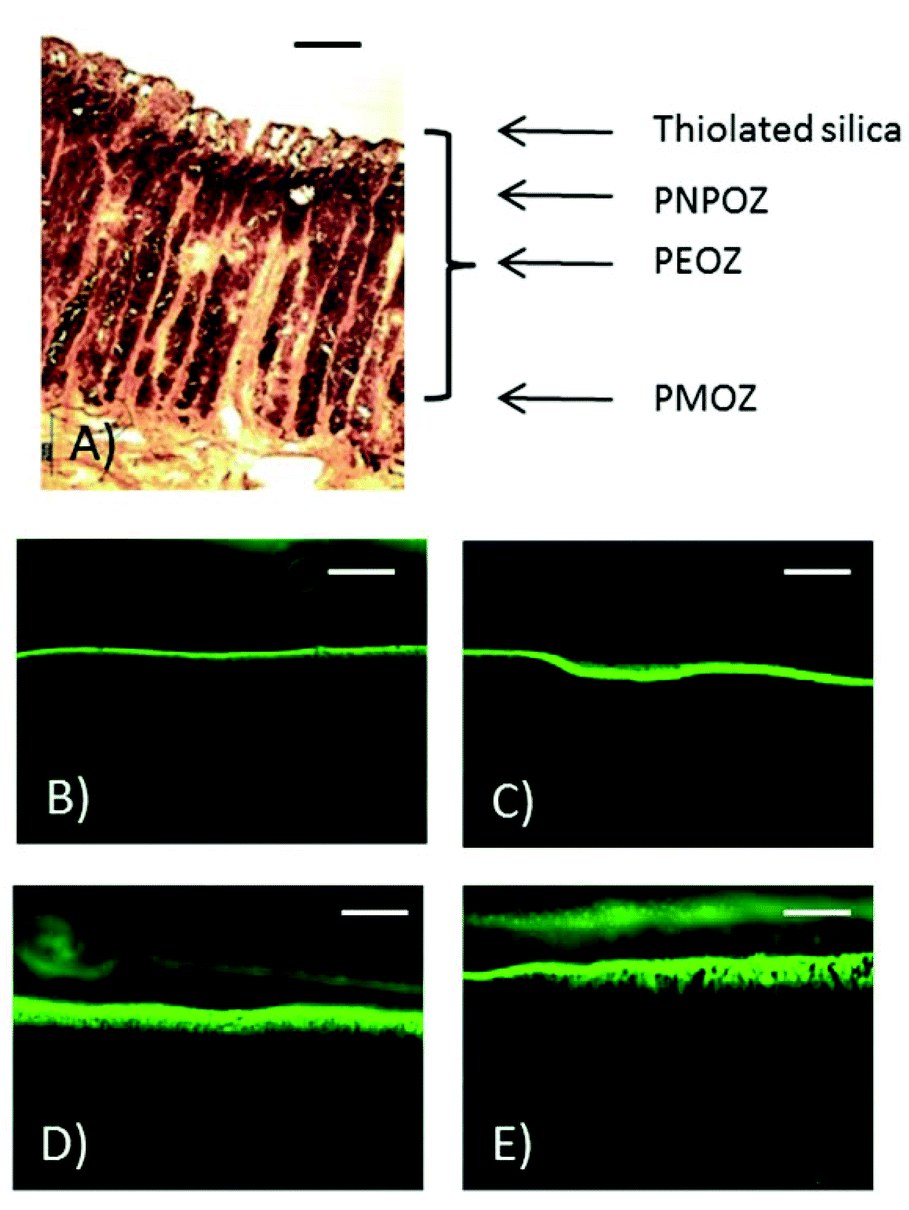 | ||
| Fig. 8 Microscopy images of porcine stomach mucosa. (A) Section stained with H&E showing the penetration of POZ-silica nanoparticles after 1 hour. The arrows indicate potential depth of penetration for different nanoparticles. Scale bar represents 300 μm. (B–E) Fluorescence microscopy images showing the penetration of PEOZ particles through the mucosa at (B) 15 min, (C) 30 min, (D) at 45 min, and (E) at 60 min. Scale bar represents 1 mm. | ||
Fig. 8 shows the histology of a porcine stomach lining, with the gastric pits and underlying muscle tissue evident. From the fluorescent images at 60 min (Fig. 8E) nanoparticle penetration into the gastric pits can be seen.
These data correlate with the diffusion coefficients determined using NTA, and show that the addition of a single CH2 group onto the alkyl side chain of polymer-functionalised nanoparticles can have dramatic effects on the diffusion coefficient of the particles.
4. Conclusions
This study is the first to report on the systematic modification to alkyl side-chain length in nanoparticle diffusion. It provides a fundamental framework in the future design of mucus penetrating systems, by showing that increasing the hydrophilicity of the POZ macromolecule causes the particles diffuse significantly faster through a mucus dispersion, and as a result penetrate further through a mucosal barrier. The addition of single methylene groups to the polymer side-chain was found to induce a linear decrease in permeation and diffusion coefficient. The hydrophobicity of polymers decorating nanoparticles is a major influence in their diffusion through mucous and their mucosal penetration.It should be noted that in this study, silica nanoparticles were used as a model system due to their facile synthesis, monodisperse nature, and high abundance of reactive thiol groups for further modification. However, POZ could be readily grafted to other particle types, such as lipid nanocarriers for enhanced drug delivery, or gold nanoparticles/quantum dots for mucus-penetrating bioimaging.
Acknowledgements
The authors are grateful to Malvern Instruments and the University of Reading for funding this project. Chemical Analysis Facility (University of Reading) is acknowledged for providing access to FT-IR and FT-Raman spectroscopy, TGA, TEM and NMR. P. C. Turner Abattoirs (Farnborough, UK) is also thanked for supplying the stomach tissues used in this work, along with the Institute Laue-Langevin (proposal number 9-12-422) for allowing us access to D11 for neutron studies. V. R. R. would like to thank IWT (project 140800) for financial support. Dr Michael Cook is also acknowledged for his help during the beam-time experiments.References
- E. Roduner, Chem. Soc. Rev., 2006, 35, 583–592 RSC.
- A. Akbarzadeh, M. Samiei and S. Davaran, Nanoscale Res. Lett., 2012, 7, 144 CrossRef PubMed.
- F. Iskandar, Adv. Powder Technol., 2009, 20, 283–292 CrossRef CAS.
- A. Albanese, P. S. Tang and W. C. W. Chan, Annu. Rev. Biomed. Eng., 2012, 14, 1–16 CrossRef CAS PubMed.
- V. R. de la Rosa, J. Mater. Sci. Mater. Med., 2014, 25, 1211–1225 CrossRef CAS PubMed.
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed.
- R. Hoogenboom, Angew. Chem., Int. Ed., 2009, 48, 7978–7994 CrossRef CAS PubMed.
- K. Lava, B. Verbraeken and R. Hoogenboom, Eur. Polym. J., 2015, 65, 98–111 CrossRef CAS.
- H. Wei and C. Yu, Biomater. Sci., 2015, 3, 1050–1060 RSC.
- F. C. Gaertner, R. Luxenhofer, B. Blechert, R. Jordan and M. Essler, J. Controlled Release, 2007, 119, 291–300 CrossRef CAS PubMed.
- Y. Chen, B. Pidhatika, T. von Erlach, R. Konradi, M. Textor, H. Hall and T. Lühmann, Biointerphases, 2014, 9, 031003 CrossRef PubMed.
- J. Ulbricht, R. Jordan and R. Luxenhofer, Biomaterials, 2014, 35, 4848–4861 CrossRef CAS PubMed.
- M. N. Macgregor-Ramiasa, A. A. Cavallaro and K. Vasilev, J. Mater. Chem. B, 2015, 3, 6327–6337 RSC.
- Clinical trial number: NCT02579473. Accessed from https://clinicaltrials.gov/show/NCT02579473, May 2016.
- G. C. Hansson, Curr. Opin. Microbiol., 2012, 15, 57–62 CrossRef CAS PubMed.
- R. A. Cone, Adv. Drug Delivery Rev., 2009, 61, 75–85 CrossRef CAS PubMed.
- M. Davidovich-Pinhas and H. Bianco-Peled, Expert Opin. Drug Delivery, 2010, 7, 259–271 CrossRef CAS PubMed.
- S. Sakuma, R. Sudo, N. Suzuki, H. Kikuchi, M. Akashi and M. Hayashi, Int. J. Pharm., 1999, 177, 161–172 CrossRef CAS PubMed.
- S. K. Lai, D. E. O'Hanlon, S. Harrold, S. T. Man, Y.-Y. Wang, R. Cone and J. Hanes, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1482–1487 CrossRef CAS PubMed.
- M. Yang, S. K. Lai, T. Yu, Y.-Y. Wang, C. Happe, W. Zhong, M. Zhang, A. Anonuevo, C. Fridley, A. Hung, J. Fu and J. Hanes, J. Controlled Release, 2014, 192, 202–208 CrossRef CAS PubMed.
- E. D. H. Mansfield, K. Sillence, P. Hole, A. C. Williams and V. V. Khutoryanskiy, Nanoscale, 2015, 7, 13671–13679 RSC.
- X. Gong, Q. Xu, X. Song, T. Yu, B. Schuster, J.-C. Yang, N. W. Lamb, J. Chang and J. Hanes, Nanomed. Nanotechnol., Biol. Med., 2016, 12, 521 CrossRef.
- A. Popov, L. Schopf, J. Bourassa and H. Chen, Int. J. Pharm., 2016, 502, 188–197 CrossRef CAS PubMed.
- C. Müller, K. Leithner, S. Hauptstein, F. Hintzen, W. Salvenmoser and A. Bernkop-Schnürch, J. Nanopart. Res., 2013, 15, 1353 CrossRef.
- G. S. Irmukhametova, G. A. Mun and V. V. Khutoryanskiy, Langmuir, 2011, 27, 9551–9556 CrossRef CAS PubMed.
- M. M. Bloksma, C. Weber, I. Y. Perevyazko, A. Kuse, A. Baumgärtel, A. Vollrath, R. Hoogenboom and U. S. Schubert, Macromolecules, 2011, 44, 4057–4064 CrossRef CAS.
- M. W. M. Fijten, C. Haensch, B. M. van Lankvelt, R. Hoogenboom and U. S. Schubert, Macromol. Chem. Phys., 2008, 209, 1887–1895 CrossRef CAS.
- F. Wiesbrock, R. Hoogenboom, M. A. M. Leenen, M. A. R. Meier and U. S. Schubert, Macromolecules, 2005, 38, 5025–5034 CrossRef CAS.
- G. Irmukhametova, B. Fraser, J. Keddie, G. Mun and V. V. Khutoryanskiy, Langmuir, 2012, 28, 299–306 CrossRef CAS PubMed.
- G. L. Ellman, Arch. Biochem. Biophys., 1959, 82, 70–77 CrossRef CAS PubMed.
- I. Bravo-Osuna, D. Teutonico, S. Arpicco, C. Vauthier and G. Ponchel, Int. J. Pharm., 2007, 340, 173–181 CrossRef CAS PubMed.
- M. N. Ganivada, P. Kumar and R. Shunmugam, RSC Adv., 2015, 5, 50001–50004 RSC.
- E. A. Mun, C. Hannell, S. E. Rogers, P. Hole, A. C. Williams and V. V. Khutoryanskiy, Langmuir, 2014, 30, 308–317 CrossRef CAS PubMed.
- A. L. Smith, Spectrochim. Acta, 1960, 16, 87–105 CrossRef CAS.
- T. Sen and I. J. Bruce, Sci. Rep., 2012, 2, 564 Search PubMed.
- R. Faulkner, J. DiVerdi, Y. Yang, T. Kobayashi and G. Maciel, Materials, 2012, 6, 18–46 CrossRef.
- M. Sameti, G. Bohr, M. N. V. Ravi Kumar, C. Kneuer, U. Bakowsky, M. Nacken, H. Schmidt and C.-M. Lehr, Int. J. Pharm., 2003, 266, 51–60 CrossRef CAS PubMed.
- W. Abdelwahed, G. Degobert, S. Stainmesse and H. Fessi, Adv. Drug Delivery Rev., 2006, 58, 1688–1713 CrossRef CAS PubMed.
- F. Rehfeldt, M. Tanaka, L. Pagnoni and R. Jordan, Langmuir, 2002, 18, 4908–4914 CrossRef CAS.
- G. Bissadi and R. Weberskirch, Polym. Chem., 2016, 7, 1271–1280 RSC.
- T. X. Viegas, M. D. Bentley, J. M. Harris, Z. Fang, K. Yoon, B. Dizman, R. Weimer, A. Mero, G. Pasut and F. M. Veronese, Bioconjugate Chem., 2011, 22, 976–986 CrossRef CAS PubMed.
- V. V. Khutoryanskiy, G. A. Mun, Z. S. Nurkeeva and A. V. Dubolazov, Polym. Int., 2004, 53, 1382–1387 CrossRef CAS.
- E. A. Mun, P. W. J. Morrison, A. C. Williams and V. V. Khutoryanskiy, Mol. Pharm., 2014, 11, 3556–3564 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6bm00375c |
| This journal is © The Royal Society of Chemistry 2016 |