
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An in vitro evaluation of fibrinogen and gelatin containing cryogels as dermal regeneration scaffolds
I. U
Allan
a,
B. A.
Tolhurst
a,
R. V.
Shevchenko
b,
M. B.
Dainiak
c,
M.
Illsley
*a,
A.
Ivanov
d,
H.
Jungvid
d,
I. Y.
Galaev
e,
S. L.
James
a,
S. V.
Mikhalovsky
af and
S. E.
James
a
aSchool of Pharmacy and Biomolecular Sciences, University of Brighton, Brighton, UK. E-mail: mi18@bton.ac.uk
bPharmidex Pharmaceutical Services Ltd., London, W1S 1YH, UK
cDept. of Protein Purification, Novo Nordisk A/S, Novo Nordisk Park, 2760 Maaloev, Denmark
dProtista Biotechnology AB, Lund, Sweden
eLund University, Lund, Sweden
fNazarbayev University, School of Engineering, Astana, 010000, Kazakhstan
First published on 3rd May 2016
Abstract
Macroporous cryogels containing mixtures of two key components of the dermal extracellular matrix, fibrinogen and collagen-derived gelatin, were evaluated for use as dermal tissue regeneration scaffolds. The infiltration of human dermal fibroblasts into these matrices was quantitatively assessed in vitro using a combination of cell culture and confocal laser scanning microscopy. The extent of cellular infiltration, as measured by the number of cells per distance travelled versus time, was found to be positively correlated with the fibrinogen concentration of the cryogel scaffolds; a known potentiator of cell migration and angiogenesis within regenerating tissue. An analysis of the proteins expressed by infiltrating fibroblasts revealed that the cells that had migrated into the interior portion of the scaffolds expressed predominantly F-actin along their cytoplasmic stress fibres, whereas those present on the periphery of the scaffolds expressed predominantly α-smooth muscle actin, indicative of a nonmotile, myofibroblast phenotype associated with wound contraction. In conclusion, the cryogels produced in this study were found to be biocompatible and, by alteration of the fibrinogen content, could be rendered more amenable to cellular infiltration.
Introduction
Dermal tissue regeneration matrices are now considered as a treatment option to assist in the healing of full thickness burns, they also offer potential for the replacement of dermal tissue following surgical excision of chronic ulcers or skin tumours.1,2 Such scaffolds can be in the form of acellular matrices that provide a suitable biocompatible framework for the attachment and infiltration of the various peripheral cells in vivo. Alternatively, scaffolds can be pre-seeded with dermal cells (e.g. fibroblasts, endothelial cells) in vitro that can remodel the material to provide a biocompatible matrix to facilitate further cellular infiltration in vivo. These have been described in considerable detail in the literature.3–5 The ultimate goal in the implantation of a dermal scaffold material should be cell infiltration, degradation and remodelling of the material to restore the pre-trauma condition.Skin is a multifunctional organ, with roles in fluid retention, antimicrobial barrier function, temperature control, sensory perception and excretion.6 Of primary importance following trauma and the creation of an open wound is the restoration of a barrier function to prevent fluid loss and infection i.e. wound closure. Wound closure by epithelial cells is not possible unless these cells are provided with the necessary nutrients and cytokines from the various cell types present in the underlying dermis.7 In order to enable keratinocyte-mediated wound closure via grafting, dermal scaffolds must be adequately infiltrated with underlying host cells whilst retaining a sufficiently interconnected pore structure to facilitate free movement of growth factors and vascularisation.
Macroporous cryogels synthesised from biocompatible materials offer potential as dermal replacement scaffolds. The production of these materials exploits the reactions between gel precursors and crosslinking agents under freezing conditions. When the mixture is exposed to a suitably low temperature, ice crystals form from pure water, simultaneously increasing the relative concentration of the crosslinking agent in unfrozen voids. Thus, concentrated unfrozen polymer is crosslinked to form the cryogel pore walls. Following thawing and expulsion of unbound melt water, the resultant cryogel has a relatively increased wall strength, with a highly porous, interconnected structure.8
Cryogels can be produced with interconnected pores of controllable dimensions, for example in the range 100–200 μm, optimal for cellular infiltration. These materials have the potential to be rapidly infiltrated and remodelled by dermal cells from the wound bed, or subsequent to seeding in vitro.
The aim of this study was to examine the interactions of primary human dermal fibroblasts with various formulations of cryogels containing mixtures of fibrinogen (FB) and gelatin. FB has a crucial role in blood clotting immediately after injury and in subsequent wound repair where it a key component of the regenerating dermal matrix.9 FB and its cleavage products are chemoattractants for a variety of cells types, including fibroblasts, which have been shown to specifically adhere to, and spread upon, FB containing surfaces.10–12 FB cleavage products are reported to have a mitogenic effect on a variety of cell types including endothelial cells and fibroblasts.11,13
Gelatin, of piscine origin in this instance, is a non-immunogenic, partially denatured form of collagen, the most abundant protein of the extracellular matrix (ECM).14 Collagen provides important mechanical properties for the ECM as well as ligand receptors recognised by cells as sites for adhesion, via membrane bound integrins.15–17 Therefore, FB-gelatin scaffolds potentially offer surfaces suitable for attracting cells from the surrounding wound bed, with the provision of appropriate cell adhesion sites from which attached cells may further spread and proliferate. These proteins are also fully degradable in vivo, and therefore should be re-modelled and replaced with nascent tissue some weeks after implantation.
In this study, we quantitatively assessed the infiltration of primary human dermal fibroblasts from a confluent, contact-inhibited cell layer (equivalent to the quiescent state at the wound bed) into the cryogel scaffolds over a defined time period in order to model both cellular infiltration of the materials in vitro, and from the wound bed in vivo, prior to potential clinical use. The functionality of the cells within these matrices was investigated by examining the orientation and location of cells within the scaffolds and by assessing the production of ECM proteins, using a combination of immunofluorescent staining and confocal laser scanning microscopy (CLSM). We hypothesised that increasing the concentration of the fibrinogen component of the cryogel would increase cellular proliferation and infiltration rates.
Materials and methods
Fibrinogen-gelatin cryogel synthesis
Fibrinogen (FB) from human plasma (Sigma) at a final concentration of 0.06% (low (LFB)), 0.84% (middle (MFB)) or 1.67% w/v (high (HFB)) was added with stirring to a solution of cold water fish gelatin (Sigma) giving final concentrations of the latter of 4.3, 3.5, 2.5% (w/v), respectively. The samples were then held on ice for 15 min. Glutaraldehyde (GTA) was added, with stirring, to give a final concentration of 0.3% (v/v). Aliquots of 0.25 ml of the solution were then added to glass tubes (20 × 7 mm internal diameter) which were sealed at the bottom with a silicone plug. These were placed at −12 °C in a cooling chamber (Arctest), and the samples held at this temperature overnight before thawing at room temperature. The resultant cylindrical cryogels were washed with 10 ml deionised water. In order to neutralise any free GTA, 2 ml of freshly prepared NaBH4 solution (0.1 M in sodium carbonate buffer, pH 9.2) was passed through each cryogel followed by extensive washing with sterile deionised water.Cryogel preparation for cell culture
Prior to cell culture, the cryogels were vertically bisected with a scalpel blade forming two half cylinders and placed in separate wells of a series of 12-well tissue culture (TC) plates containing Dulbecco's Modified Eagle Medium plus Glutamax (DMEM, Invitrogen) supplemented with antibiotics (penicillin, streptomycin, gentamicin and amphotericin B, all at ×5 concentration, Invitrogen) and quarantined for 48 h at 37 °C to eradicate any possible microbial contamination. Samples were then rinsed (×3) in 10 ml sterile Hank's Buffered Salt Solution (Invitrogen) with rolling for 24 h, and suspended in growth medium without antibiotics. Sterility was confirmed by cultures remaining free from contamination after 48 h in antibiotic free culture at 37 °C and 5% CO2.Cell culture and cryogel seeding
Primary human dermal fibroblasts (SKF371) (from University of Brighton cell bank funded by DTI grant number MMP4.5) were grown in DMEM supplemented with 10% heat inactivated bovine foetal calf serum (FCS) (Invitrogen) in tissue culture (TC) treated flasks (Greiner Bio-one) at 37 °C in 100% relative humidity (RH), 5% CO2. No antimicrobials were used during cell propagation. At 80% confluence, cells were harvested with trypsin and seeded in 2 ml volumes at a concentration of 1 × 104 cells per ml into 12-well TC plates (Greiner Bio-one) and incubated as above for 5 days until confluent, and therefore contact inhibited.18To assess cellular proliferation and infiltration into the cryogel samples, cells were allowed to migrate against gravity from the TC surface into the cryogels by placing the bisected half-cylinder gels, flat surface down, onto the confluent cell layer (Fig. 1) with incubation at 37 °C, 100% RH, 5% CO2 for 4 hours. The cell infiltrated cryogels were then removed and suspended in fresh wells containing 2 ml growth medium and incubated for a further 0, 7, 14 or 28 days with growth medium replacement every second day. At the appropriate time point, the medium was removed and the samples washed with phosphate buffered saline (PBS) (×2, Invitrogen), fixed with 10% neutral buffered formalin (NBF) (Fisher) for 15 minutes, washed and stored in PBS at 4 °C, prior to immunofluorescent staining and viewing with (CLSM).
 | ||
| Fig. 1 A schematic of a synthesised cryogel and its preparation for cell culture. All cryogels were bisected to present a nascent internal surface for the infiltration of confluent dermal fibroblasts from a TC plate, over a 4 h period. The materials were then removed to fresh medium for continued propagation. | ||
Quantification of cell infiltration into the cryogels
To assess the infiltration of cells into the cryogels, each sample was sectioned and scanned in three areas (Fig. 2 – areas labelled 1, 2 and 3); two scans near each outer edge and one from the centre. The samples were stained with PI to visualise the cell nuclei and re-oriented 90° for viewing with CLSM (Fig. 2). To visualise the cryogels structures, the samples were excited by a laser at 405 nm and the resultant autofluorescent light visualised by capture in the range 450–490 nm. An example of a section visualised by CLSM is shown in Fig. 3, with cell nuclei shown as white dots with the grey cryogel pore-walls visible amongst black voids (pores). By scanning along the cross-sectional plane to a distance of 1.0 mm and to a depth of 50 μm, a z-stack image was then produced extending from the seeded surface to 1.0 mm into the interior of each sample. This information enabled the calculation of cell migration in 100 μm increments (0–1.0 mm) by superimposition of an appropriately labelled grid (using Microsoft Excel) corresponding to the dimensions of the CLSM image (Fig. 3) as described elsewhere.18 | ||
| Fig. 2 Viewing of cells and the cryogel scaffold in cross-section using CLSM for the quantification of cell infiltration versus depth. Following the desired propagation time in culture, each half cylinder section of gel was fixed and sliced into equal portions, with the cut surfaces (1–3) placed face-down for scanning with CLSM. | ||
 | ||
| Fig. 3 Diagram showing the enumeration of cell nuclei within a cryogel in cross section using a measuring template. The image of the sample (MFB formulation represented here) is orientated such that the seeded surface is at the bottom. Cell nuclei (PI stained) are visible as white dots against the autofluorescent scaffold background. The number of cells was counted in each row of a superimposed template (each row representing 100 μm) allowing the number of cells versus distance from the seeded surface to be calculated. The image represents scanned dimensions of 1.55 × 1.0 × 0.1 mm. | ||
Immunofluorescent staining of the cell seeded cryogels
The expression of intra and extracellular proteins produced by fibroblasts infiltrating the cryogels was studied using a combination of immunofluorescent staining and CLSM. The cryogel scaffold samples were fixed with 10% NBF rinsed with PBS (×2), permeabilised with Triton-X 100 and blocked with 10% FCS in PBS. The samples were then immunostained with primary mouse monoclonal antibodies: anti-human collagen I clone COL-1 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2000 in PBS), anti-fibronectin clone FN-3E2 (1:400), or α-smooth muscle actin (α-sma) clone 1A4 (1:400) (all Sigma Aldrich) for 1 hour at RT. The cryogel samples were then washed in PBS (×2) to remove unbound antibodies and then incubated in the blocking solution, followed by the application of a secondary antibody, goat anti-mouse immunoglobulin tagged with Alexafluor-633 (1:500) (Invitrogen). For F-actin staining, phalloidin tagged with Alexafluor-488 (Invitrogen) was used (replacing or following the antibody stains above, as appropriate) and incubated for 20 min. All cryogels were then washed in PBS (×2) and PI added to stain the cell nuclei (assigned to a blue or red colour as indicated, with CLSM software). CLSM settings were as described previously, with the protein of interest shown as a green or blue colour, as indicated.18 The autofluorescent properties of the cryogels were exploited to enable viewing of the scaffold structure; fluorescent output converted to white (Fig. 4) or magenta (Fig. 7).
:2000 in PBS), anti-fibronectin clone FN-3E2 (1:400), or α-smooth muscle actin (α-sma) clone 1A4 (1:400) (all Sigma Aldrich) for 1 hour at RT. The cryogel samples were then washed in PBS (×2) to remove unbound antibodies and then incubated in the blocking solution, followed by the application of a secondary antibody, goat anti-mouse immunoglobulin tagged with Alexafluor-633 (1:500) (Invitrogen). For F-actin staining, phalloidin tagged with Alexafluor-488 (Invitrogen) was used (replacing or following the antibody stains above, as appropriate) and incubated for 20 min. All cryogels were then washed in PBS (×2) and PI added to stain the cell nuclei (assigned to a blue or red colour as indicated, with CLSM software). CLSM settings were as described previously, with the protein of interest shown as a green or blue colour, as indicated.18 The autofluorescent properties of the cryogels were exploited to enable viewing of the scaffold structure; fluorescent output converted to white (Fig. 4) or magenta (Fig. 7).
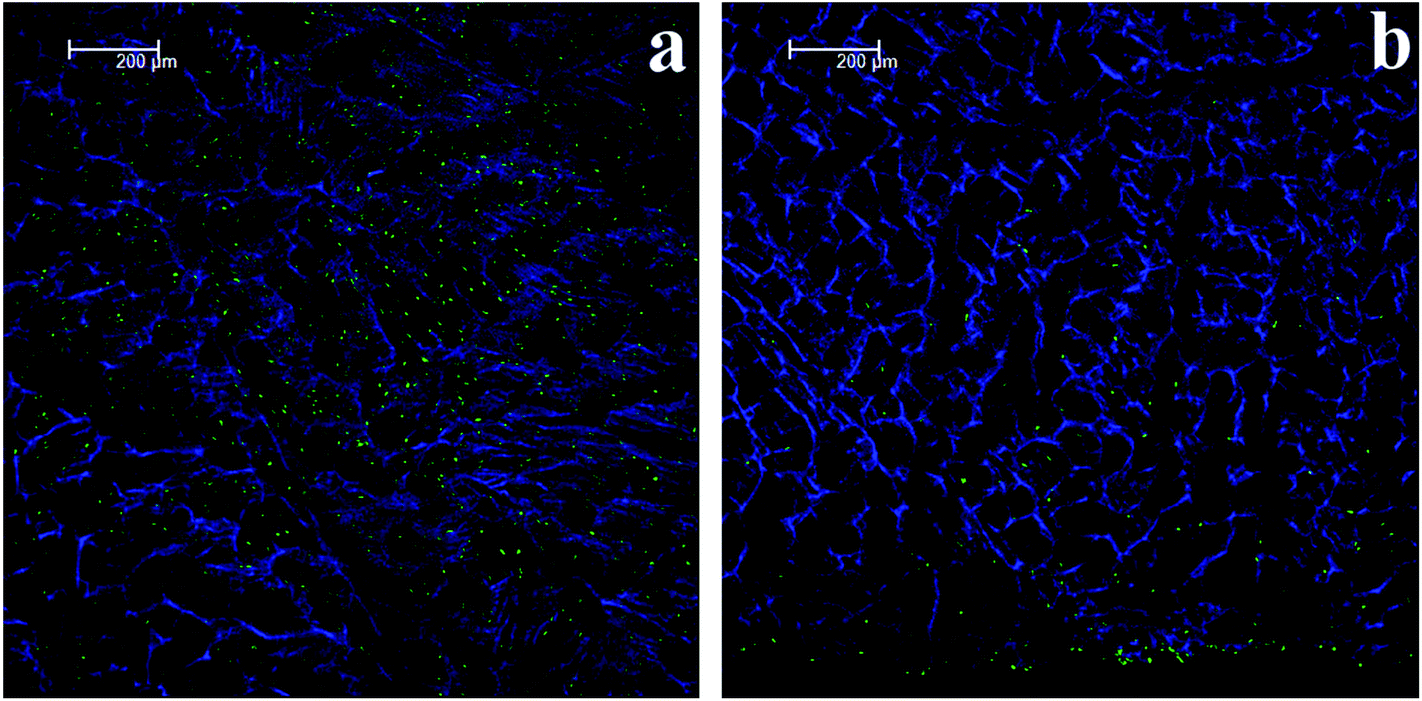 | ||
| Fig. 4 An example of the relative cell numbers on the cryogel exterior compared with interior. The figure shows images of a LFB cryogel cultured for 14 days. (a) Shows the exterior surface where many cell nuclei (green) are present, whereas (b) shows cells in the interior that relatively decrease in number as the distance from the seeded (lower) surface increases. The same effect was noted in all of the cryogel formulations. | ||
Statistical analyses
Multiple regression analysis, within a generalised linear model (GLM) was used to determine whether there was a difference between cryogel formulations (fibrinogen concentrations) in terms of the number of cells cultured. The effect of time (day of the experiment) was controlled for via inclusion in the model. The response variable ‘cell count’ was regressed against the explanatory variables ‘cryogel formulation’ (a categorical variable with three levels – low, medium and high) and ‘day’ of experiment’, referring to the day of each cell count (0, 7, 14, 28) and a two-way interaction term. Because the response variable was non-normal and over dispersed (dispersion parameter > 2, variance ≫ mean), the data were modelled with a negative binomial error distribution and a log-link function. Data for each day were then analysed separately using the same process. All analyses were computed using the statistics package R version 3.2.0 (R Foundation for Statistical Computing, Vienna, Austria).Results and discussion
All synthesised FB-gelatin cryogels were able to support cell attachment and growth; those containing elevated fibrinogen concentrations favouring an increased infiltration of cells from the scaffold surface to the interior. When we visualised our cryogels with CLSM (Fig. 4), we observed a fundamental difference in cell number between the surface of the scaffolds and the interiors. This was also noted when we used this method to evaluate cellular infiltration of a commercially available dermal replacement scaffold material, Integra.18 These differences in cell distribution, with the majority of cells on the external surfaces of the scaffolds, represent a challenge in biomaterial design. In a clinical setting, it is important that dermal scaffolds provide a suitable environment for the proliferation and migration of the overlying epidermal cells that are ultimately responsible for wound closure.19,20 Therefore, any dermal scaffolds must be adequately infiltrated and remodelled by the appropriate dermal cells, such as fibroblasts, in order to produce the appropriate physiological matrix and cell to cell communications necessary for the subsequent epithelial regrowth and wound closure.21–24We quantified the in vitro compatibility of the scaffolds by taking virtual sections with a confocal microscope. This analysis allowed us to enumerate the cells on the surface, as well as those cells that are penetrating the interior of the scaffold. It was hypothesised that fibroblasts would infiltrate more rapidly through cryogels containing increasing fibrinogen concentrations.25
Quantification of total cells numbers within cryogels (0–1.0 mm cross sections)
In the LFB cryogel formulation, proliferation occurred rapidly over the first 7 days (Fig. 5), however, between 14 and 28 days the total cell number had reached equivalence. The final cell numbers at day 28 in the two cryogel formulations, with the highest FB concentrations (middle FB and high FB), were more than double that of the low FB concentration. | ||
| Fig. 5 Total cell counts within the cryogel formulations (from a depth of 0–1 mm from the seeded surface). For each of the cryogel formulations, the results of post hoc (z) tests for individual days are shown. Error bars indicate Standard Error of the Mean (SEM). For each day, bars with the same letter indicate no significant differences, while different letters denote statistically significant results at the 0.05 level. 1 Maximum Likelihood Parameter Estimate (MLPE) = 1.42, z = 2.42, p < 0.05; 2 MLPE = 0.44, z = 2.23, p < 0.05; 3 MLPE = 0.49, z = 2.09, p < 0.05; 4 MLPE = 0.88, z = 3.04, p < 0.01; 5 MLPE = 0.79, z = 2.92, p < 0.01. Thus, after 28 days in culture, the cryogel formulations MFB and HFB accommodated a significantly greater number of cells compared with the formulation with low FB content (LFB). | ||
The HFB gel was infiltrated more rapidly than MFB, but final cell numbers were similar.
Cell counts overall differed significantly between cryogel formulations (GLM Anova table, change in deviance = 12.971, df = 2, p < 0.01) where high FB cryogels cultured more cells than all other formulations (Table 1). A significant interaction between day of experiment and cryogel formulation was also detected (GLM Anova table, change in deviance = 20.80, df = 6, p < 0.01), indicating that the effect of the cryogel formulation on cell count varied across the different days of incubation. Separate analyses for each day support this finding: on day 0 HFB contained significantly more cells than LFB; on days 7 and 14 HFB contained significantly more cells than MFB and on day 28 the cell numbers in both the MFB and HFB gels remained significantly higher than for the LFB formulation (Fig. 5). This 28 day time period is associated with the successful cellular infiltration of the current commercial gold standard dermal scaffold, Intergra, when implanted in vivo.
Quantification of cell infiltration rate: infiltration depth versus time
While the data above indicate the total number of cells within the cryogels, they did not give information regarding the spatial distribution of the cells within. Biochemical assays, such as MTS, also give an estimation of the total number of cells within cryogels (and are less laborious) but also lack spatial cell distribution information. Our images revealed that, in general, appreciably more cells were present on the exterior of all of the cryogel formulations than the interior. Fig. 6 gives an example of the typical numbers of cells observed on the exterior of the cryogels versus the interior. Clearly, more cells were present on the cryogels exterior compared with the interior after 14 days. Thus, it was important to identify those scaffolds that best supported cell infiltration and proliferation within the interior portions – as this would be a requirement for successful integration in vivo. Therefore, we developed a method to quantify cell numbers in defined incremental cross-sections of the cryogels and to reveal the spatial distribution of the cells in the three formulations at specific time intervals. | ||
| Fig. 6 Quantification of the infiltration of dermal fibroblasts into cryogel formulations. (a) LFB, (b) MFB, and (c) HFB, cryogels over time (0–1000 μm). The cryogel with low fibrinogen content (LFB) was infiltrated most rapidly, with cells penetrating as far as 1.0 mm following the seeding process (day 0). For the other two formulations at day 0, cells were limited to the first 200 μm of the scaffold, apart from sporadic cells at greater depths. After 7 days, cell numbers were highest between 0–400 μm with MFB and HFB accommodating the most cells, and the latter accommodating more cells at greater depths than the other two formulations. With all three formulations there were either no apparent increases or minor increases in cell numbers, regardless of depth, between 7 and 14 days. Between 14 and 28 days, large increases in cell numbers were observed with the MFB and HFB formulations at all measured depths. Thus, of importance in a clinical context, after 4 weeks in culture, elevated FB content (MFB, HFB) was associated with increased cell numbers in the interior of the cryogels. Error bars indicate SEM. | ||
The low fibrinogen (LFB) scaffold was infiltrated most rapidly with cells (Fig. 6) penetrating as far as 1.0 mm following the seeding process (day 0). In the other formulations, immediately after the seeding procedure (day 0), cells were limited to the first 200 microns of the scaffold, apart from occasional single cells in the deeper layers. By day 7, cell numbers were still highest between 0–300 μm with MFB and HFB supporting the most cells. With all formulations there were either minor increases or no apparent increases in cell numbers, regardless of depth, between 7 and 14 days. Between 14 and 28 days, large increases in cell numbers were observed with MFB and HFB formulations at all measured depths. This finding is of importance in a clinical context since keratinocytes (required for final wound closure) will only grow in close proximity to underlying dermal fibroblasts. Thus, those cryogels that are able to accommodate cellular infiltration and proliferation within their interiors, in this case those with elevated fibrinogen, would be expected to provide an improved substratum for successful wound healing in vivo.
Immunofluorescent staining of the cell seeded cryogels
Fibroblasts staining positively for α-sma were predominantly found on, or close, to the outer surface of all three cryogel formulations, with cells in the interior of the scaffolds predominantly α-sma negative and F-actin positive. Fig. 7a shows the presence of elongated cells with stress fibres positively expressing α-sma, on the flat seeded surface and the curved outer regions of the cryogels, corresponding to the myofibroblast phenotype. The presence of these cells indicates contractile behaviour such as is typically observed with fibroblasts in late stage granulation tissue in vivo,26 these α-sma positive fibres exerting more contractile activity than stress fibres positive for F-actin.27 Such cells are known to be immotile,28 therefore the α-sma positive cells on the curved outer portion of the cryogels will have migrated there from either the TC surface, or from progeny cells within the cryogels, whilst not expressing α-sma. Those present on the seeded surface that were found to be α-sma positive may have been directly transferred from the TC plastic by simple adhesion to the cryogels, without active migration, and thus may have been α-sma positive before transfer. Cells that had migrated into the scaffold interiors were found to predominantly express F-actin (rather than α-sma) which would be expected for motile cells. Thus, at the curved periphery of the cryogels and possibly at the seeded surfaces, the fibroblasts responded to the prevailing conditions by applying contractile forces to the scaffolds. | ||
| Fig. 7 The differential expression of F-actin and α-smooth muscle actin of dermal fibroblasts growing in cryogels and on glass. F-actin is shown in blue and α-smooth muscle actin shown in green. Cell nuclei are shown in gold (a) or pink (b, c, d). Cryogel is (a) purple or (b) magenta. (a) Shows fibroblasts in a cryogel (formulation Low FB) in cross-section. The yellow line marks a minor discontinuity between merged images from the same sample. Note the increased expression of α-sma on the periphery of the cryogel. This is indicative of contractile forces at the liquid (curved portion) and TC (base) interfaces. However, α-sma expression is largely absent in the cells in the cryogel interior, where the F-actin expressing phenotype dominates. (b) A single fibroblast in a cryogel expressing both α-sma and F-actin (Low FB) (c and d) the differential expression of α-sma and F-actin in fibroblasts growing on glass slides shown at (c) high and (d) low magnification. Note in (d) that α-sma was expressed in the cells that were on the periphery of an acellular zone (bottom mid-left to top mid-right) similar to those in (a) at the cryogel periphery. | ||
The cryogels used in this study were denser than the surrounding growth medium and sunk to the bottom of the tissue plate wells. Previous studies have found a lack of α-sma expression in cells in free floating collagen based lattices.29 Since α-sma expression was noted in the fibroblasts around the outer portions of the cryogels (Fig. 7a), these scaffolds may have been subject to tension from the well base, perhaps by means of cells bridging the TC plastic and the cryogels, which would have favoured α-sma expression.
Some cells were noted to express both α-sma and F-actin and were generally observed sandwiched between the F-actin positive cells in the scaffold centre and the α-sma positive cells on the outer surface (Fig. 7a). It was notable the α-sma staining tended to be centred close to the cell nucleus with F-actin predominating at the outer extremities of the cell fibrils (Fig. 7b). Either α-sma or F-actin filaments were observed in discrete cytoplasmic processes extending from the main body of the cells, where they intimately associated with the cryogel pore walls (Fig. 7b). The same fibroblasts grown on glass slides (Fig. 7c and d) were found to express these proteins in a similar manner – Fig. 7c shows a lone cell staining strongly positively for α-sma amongst F-actin positive cells (validating the images produced of cells within the cryogels). In Fig. 7d an apparent discontinuity is present in the centre of the image (top to bottom) where cell to cell contact is incomplete in one plane. Many of these cells stained positively for α-sma and were thus comparable with those α-sma positive cells on the periphery of the cryogels that lacked cell to cell contact in one plain at the liquid or TC plastic interface.
Primary human dermal fibroblasts were used in this study to model the initial phase of dermal regeneration i.e. cell infiltration of the cryogels and the formation of nascent ECM (comparable with the formation of dermal granulation tissue in vivo following injury) of which, collagen and fibrinogen are both key components. FB has been shown to be a potent chemoattractant for fibroblasts and to reduce the contraction of fibroblast populated collagen lattices in vitro in a concentration dependent manner.30 Thus, the application of such FB-containing matrices in vivo may readily recruit fibroblasts necessary for ECM production and remodelling, yet discourage the formation of unsightly, and mechanically weak, scar tissue. This hypothesis is partially backed by the findings of this study. The interior portions of the cryogels were largely lacking in cells of the myofibroblast phenotype, with the non-scarring F-actin phenotype predominating, suggesting that the internal portions of the cryogels may provide a suitable microenvironment for the scar-free remodelling of the materials in vivo. This may have been partially due to the relatively high content of locally available fibrinogen surrounding the cells in three dimensions, unlike at the cryogel surface. Where applied clinically, a cryogel dermal regeneration scaffold would be situated proximal to the wound bed, with a potential for recruitment from a ready supply of a variety of host cells, and would be subject to pervasion by the associated myriad of chemical signals and gradients these elicit. Moreover, there would not be a large cell-free liquid interface around the periphery of the cryogels as was the case in these in vitro experiments. Thus, the presence of α-sma positive cells on the periphery of the cryogels observed in this study, indicative of potential scarring, would not necessarily be replicated in vivo. In successfully re-epithelialised wounds, myofibroblasts undergo apoptosis and are thus cleared from the site.31 With the absence of epithelial cells in the experiments described herein, such a transition may not occur, regardless of the apparent compatibility of the material with the fibroblasts (as evidenced by F-actin expression in the cryogel interiors).
Conclusion
The infiltration of human dermal fibroblasts into fibrinogen-gelatin cryogel matrices was quantitatively assessed in vitro using a combination of cell culture and confocal laser scanning microscopy. In all formulations, more cells were present on the outer 100 μm portion that in the interior. In the two cryogel formulations with higher FB concentrations (MFB, HFB), we observed both a greater total cell number throughout the scaffold and a larger number of fibroblasts penetrating the interior. The extent of cellular infiltration, as measured by the number of cells per distance travelled versus time, was found to be positively correlated with the fibrinogen concentration of the cryogel scaffolds – a known potentiator of cell migration and angiogenesis within regenerating tissue. This is a critical finding in a clinical context since keratinocytes will only grow in close proximity to underlying dermal fibroblasts. Cryogels with elevated fibrinogen concentrations would be expected to provide an improved substratum for successful wound healing in vivo if they are more readily infiltrated with fibroblasts that are required for subsequent epithelial growth on the cryogel exterior.An analysis of the proteins expressed by infiltrating fibroblasts revealed that the cells that had migrated into the interior portion of the scaffolds expressed predominantly F-actin, whereas those present on or near the surface of the cryogels expressed predominantly α-smooth muscle actin, indicative of a nonmotile, myofibroblast phenotype associated with wound contraction. Positive staining for F-actin in the central regions of the cryogels indicated that fibroblast contraction in vivo could be expected to be minimised.
For the successful integration of a dermal replacement material it must be adequately vascularised, achieved through the action of endothelial cells. Therefore, the next objective would be to co-culture fibroblasts and endothelial cells on FB-gelatin cryogels in vitro, to examine the rate of cell infiltration, ECM deposition, and vascularisation. In addition, work is now ongoing in producing ECM-based cryogels in the form of easily manipulated flat sheets, as the next step towards clinical use.
Abbreviations
| CLSM | Confocal laser scanning microscopy |
| DMEM | Dulbecco's Modified Eagle Medium |
| ECM | Extracellular matrix |
| FB | Fibrinogen |
| FCS | Foetal calf serum |
| GLM | Generalised linear model |
| GTA | Glutaraldehyde |
| HFB | High fibrinogen |
| LFB | Low fibrinogen |
| MFB | Middle fibrinogen |
| NFB | Neutral buffered formalin |
| PBS | Phosphate buffered saline |
| PI | Propidium iodide |
| RH | Relative humidity |
| SEM | Standard Error of the Mean |
| TC | Tissue culture |
References
- J. E. Jones, E. A. Nelson and A. Al-Hity, Cochrane Database Syst. Rev., 2013, 1, CD001737 Search PubMed.
- S. MacNeil, Nature, 2007, 445, 874–880 CrossRef CAS PubMed.
- R. E. Horch, J. Kopp, U. Kneser, J. Beier and A. D. Bach, J. Cell. Mol. Med., 2005, 9, 592–608 CrossRef PubMed.
- Y. Ikada, J. R. Soc., Interface, 2006, 3, 589–601 CrossRef CAS PubMed.
- R. V. Shevchenko, S. L. James and S. E. James, J. R. Soc., Interface, 2010, 7, 229–258 CrossRef CAS PubMed.
- S. T. Boyce, Burns, 2001, 27, 523–533 CrossRef CAS PubMed.
- A. El Ghalbzouri, P. Hensbergen, S. Gibbs, J. Kempenaar, R. van der Schors and M. Ponec, Lab. Invest., 2004, 84, 102–112 CrossRef CAS PubMed.
- M. B. Dainiak, I. U. Allan, I. N. Savina, L. Cornelio, E. S. James, S. L. James, S. V. Mikhalovsky, H. Jungvid and I. Y. Galaev, Biomaterials, 2010, 31, 67–76 CrossRef CAS PubMed.
- M. W. Mosesson, J. Thromb. Haemostasis, 2005, 3, 1894–1904 CrossRef CAS PubMed.
- D. Greiling and R. A. Clark, J. Cell Sci., 1997, 110(Pt 7), 861–870 CAS.
- B. J. Rybarczyk, S. O. Lawrence and P. J. Simpson-Haidaris, Blood, 2003, 102, 4035–4043 CrossRef CAS PubMed.
- W. F. Skogen, R. M. Senior, G. L. Griffin and G. D. Wilner, Blood, 1988, 71, 1475–1479 CAS.
- L. A. Sporn, L. A. Bunce and C. W. Francis, Blood, 1995, 86, 1802–1810 CAS.
- R. A. A. Muzzarelli, Carbohydr. Polym., 1993, 20, 7–16 CrossRef CAS.
- M. Barczyk, S. Carracedo and D. Gullberg, Cell Tissue Res., 2010, 339, 269–280 CrossRef CAS PubMed.
- V. Ottani, M. Raspanti and A. Ruggeri, Micron, 2001, 32, 251–260 CrossRef CAS PubMed.
- S. P. Palecek, J. C. Loftus, M. H. Ginsberg, D. A. Lauffenburger and A. F. Horwitz, Nature, 1997, 385, 537–540 CrossRef CAS PubMed.
- I. U. Allan, R. V. Shevchenko, B. Rowshanravan, B. Kara, C. A. Jahoda and S. E. James, Soft Matter, 2009, 7, 319–341 CrossRef CAS.
- J. Solon, I. Levental, K. Sengupta, P. C. Georges and P. A. Janmey, Biophys. J., 2007, 93, 4453–4461 CrossRef CAS PubMed.
- Y. Mao and J. E. Schwarzbauer, Matrix Biol., 2005, 24, 389–399 CrossRef CAS PubMed.
- H. Bannasch, T. Unterberg, M. Fohn, B. Weyand, R. E. Horch and G. B. Stark, Burns, 2008, 34, 1015–1021 CrossRef PubMed.
- M. H. Desai, J. M. Mlakar, R. L. McCauley, K. M. Abdullah, R. L. Rutan, J. P. Waymack, M. C. Robson and D. N. Herndon, J. Burn Care Rehabil., 1991, 12, 540–545 CrossRef CAS PubMed.
- J. F. Hansbrough, C. Dore and W. B. Hansbrough, J. Burn Care Rehabil., 1992, 13, 519–529 CrossRef CAS PubMed.
- N. L. Parenteau, P. Bilbo, C. J. Nolte, V. S. Mason and M. Rosenberg, Cytotechnology, 1992, 9, 163–171 CrossRef CAS PubMed.
- L. Almany and D. Seliktar, Biomaterials, 2005, 26, 2467–2477 CrossRef CAS PubMed.
- G. Gabbiani, J. Pathol., 2003, 200, 500–503 CrossRef CAS PubMed.
- L. K. Wrobel, T. R. Fray, J. E. Molloy, J. J. Adams, M. P. Armitage and J. C. Sparrow, Cell Motil. Cytoskeleton, 2002, 52, 82–90 CrossRef PubMed.
- L. Ronnov-Jessen and O. W. Petersen, J. Cell Biol., 1996, 134, 67–80 CrossRef CAS PubMed.
- H. P. Ehrlich and T. Rittenberg, J. Cell. Physiol., 2000, 185, 432–439 CrossRef CAS PubMed.
- Y. D. Nien, Y. P. Han, B. Tawil, L. S. Chan, T. L. Tuan and W. L. Garner, Wound Repair Regen., 2003, 11, 380–385 CrossRef PubMed.
- A. Desmouliere, M. Redard, I. Darby and G. Gabbiani, Am. J. Pathol., 1995, 146, 56–66 CAS.
| This journal is © The Royal Society of Chemistry 2016 |