
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparison of ion mobility-mass spectrometry and pulsed-field gradient nuclear magnetic resonance spectroscopy for the differentiation of chondroitin sulfate isomers†
Katharina
Lemmnitzer‡
*a,
Thomas
Riemer‡
a,
Michael
Groessl
b,
Rosmarie
Süβ
a,
Richard
Knochenmuss
b and
Jürgen
Schiller
a
aUniversity of Leipzig, Medical Faculty, Institute of Medical Physics and Biophysics, Härtelstr. 16-18, D-04107 Leipzig, Germany. E-mail: katharina.lemmnitzer@medizin.uni-leipzig.de; Fax: +49-341-9715709; Tel: +49-341-9715741
bTOFWERK, Uttigenstrasse 22, CH-3600 Thun, Switzerland
First published on 9th November 2016
Abstract
Sulfated glycosaminoglycans such as chondroitin sulfate (CS) are important, natural polysaccharides which occur in many biological tissues and possess different biotechnological and medical applications. The analysis of CS is normally based on the evaluation of the unsaturated disaccharides, which can be easily obtained by enzymatic digestion. The 4-sulfate and the 6-sulfate are the typical main products which are usually differentiated and quantified by MS/MS. Here we show that both disaccharide isomers can be readily differentiated by ion mobility-mass spectrometry (IM-MS), with the 6-sulfate isomer having a lower mobility in the gas phase. These findings are further substantiated by pulsed-field gradient (PFG) NMR, which also indicates lower mobility of the 6-sulfate isomer in solution. Based on these already well-studied isomers we provide evidence that the combination of both methods, IM-MS and (PFG) NMR, allows the differentiation of isomers in mixtures, the determination of the relative contributions of both isomers and the structural identification without the absolute need for pure, internal standards.
1. Introduction
Glycosaminoglycans (GAGs) as building blocks of proteoglycans are important constituents of the extracellular matrix (ECM) of connective tissues. These unbranched acidic polysaccharides are omnipresent in mammals and play important roles, not only as structure lending components, for example, in skin or cartilage, but also during the development of the central nervous system, wound healing processes, and biomolecular signaling. These large varieties of functions and the related diseases have been addressed in many recent reviews.1–5One common GAG representative is chondroitin sulfate (CS), consisting of β-1→4-glycosidically linked disaccharides. The repeating units are composed of β-1→3 linked uronic acid and N-acetylgalactosamine (GalNAc) and may vary in the type of the uronic acid, D-glucuronic acid (GlcA) or L-iduronic acid (IdoA), and in the location of the sulfo groups, thus resulting in a high diversity of possible building blocks in the polysaccharide chain. Therefore, the distinction of different CS types is based on the predominant disaccharide repeating unit and the number and position of sulfated hydroxyl groups.
In the old denomination, the most common CS types are referred to as chondroitin sulfate A, B and C. The predominant repeating units of chondroitin sulfate A and C are both composed of GlcA and GalNAc, differing in the position of the sulfation at the C4 and C6 position of the GalNAc, respectively. In type B GlcA is epimerized into IdoA, and sulfation occurs at the C2 position of IdoA, at the C4 and less frequently also at the C6 position of GalNAc.6,7
The detailed CS disaccharide composition varies between species8,9 and between tissues of the same species.10 Furthermore, several diseases are characterized by changed contributions of the CS types.11–13 The homogeneity of the repeating unit sequence and the sulfation pattern of CS can be determined by electrospray ionization (ESI) mass spectrometry (MS)18,19 or matrix-assisted laser desorption and ionization (MALDI) MS.14,15 Since the intact CS polysaccharide with a molecular mass of up to 50 kDa cannot be analyzed directly by its intact mass,1 specific enzymes are used which convert the polysaccharide into defined oligosaccharides which then can be analyzed. Chondroitinase ABC (chondroitin lyase, EC 4.2.2.4) is particularly useful, because this enzyme cleaves all naturally occurring forms of CS and generates nearly exclusively unsaturated disaccharides.16,17
This bottom-up analysis allows the determination of the extent of sulfation (number of sulfo groups per disaccharide unit) simply by mass differences in the mass spectra. In order to differentiate the naturally occurring isomers, particularly the 4- and 6-sulfate (the chemical structures of which are shown schematically in Fig. 1), MS/MS is normally used to determine the positions of the sulfate residues by the yield of characteristic fragment ions.18,19
 | ||
| Fig. 1 Chemical structures of the two most abundant isomeric, unsaturated CS disaccharides which are obtained by enzymatic digestion of the CS polysaccharide. | ||
However, several problems have to be considered during the analysis of sulfated oligosaccharides: even with optimized ESI parameters the in-source loss of sulfo groups can hardly be completely avoided.20 Furthermore, epimerization and sulfation have an impact on the fragmentation process and thus on the abundance of characteristic fragments21 as well as the charge state of the precursor ion.22 Additionally, there are indications of a possible transfer of sulfo groups to another hydroxyl group within the precursor ion during the fragmentation process of oligosaccharides – particularly in ion trap mass spectrometers.23 Achieving quantitative data is more difficult under these conditions in comparison to MS alone and requires at least external calibration with reference compounds, independent of the fragmentation technique used.18,19
To bypass all these sources of error that can occur during the fragmentation process, we have been looking for alternative methods which readily enable the differentiation of CS disaccharide isomers without the need for using sophisticated MSn techniques.
One alternative method is the separation of isomers via LC/HPLC24,25 or electrophoresis.26,27 However, the identification and quantification by these approaches take several minutes per sample, usually involve derivatization of the analytes, and require normally pure reference compounds for calibration and identification of the compounds in the sample. For unknown compounds, the online or offline combination with NMR or MS is needed to obtain structural information.
Solution nuclear magnetic resonance (NMR) spectroscopy is a powerful tool for the determination of the sulfate positions in chondroitin-4- or 6-sulfate,28 but the low sensitivity of the method requires higher sample concentrations and is therefore not suitable for high throughput analysis of diluted samples. Although NMR alone can be used to determine the C4S/C6S ratio in a given sample, the combination with diffusion ordered spectroscopy (DOSY) opens new dimensions.29 It has been used to spot impurities in mixtures, such as oversulfated chondroitin sulfate in heparins30 or to monitor the time-dependent enzymatic depolymerization of heparin.31
Ion mobility-mass spectrometry (IM-MS) is an attractive technique for carbohydrate research due to its ability to separate ions not only by the mass-to-charge ratio but also by their mobility in the gas phase either through space (e.g. by high-field asymmetric waveform ion mobility spectrometry (FAIMS)) or time (e.g. drift tube or travelling wave ion mobility). FAIMS has been used for charge state separation of isobaric chondroitin sulfate oligomers as well as for the separation of synthetic heparin sulfate tetrasaccharide epimers that only differ in the C5 stereochemistry of one uronic acid residue prior to tandem mass spectrometry.32 Another recent study of synthetic trisaccharides using travelling-wave measurements achieved baseline separation between compounds differing in the linkage type and stereoisomers, allowing relative quantification of configurational isomers in a mixture.33
The aim of this work was to test the separation of isomeric CS disaccharides differing only in the position of one sulfate residue by means of IM-MS and DOSY NMR. The utilization of chondroitin sulfate as a thoroughly investigated model substance allowed the comparison with established methods, more precisely MS/MS and 1H NMR, regarding the relative quantification of these isomers in a mixture.
2. Materials and methods
2.1 Chemicals
Polymeric chondroitin sulfate from bovine trachea and all solvents were purchased from Sigma-Aldrich (Deisenhofen, Germany). The used CS oligosaccharides (for comparative purposes) were from Santa Cruz Technology (Heidelberg, Germany). They were used without further purification. Chondroitinase ABC (from proteus vulgaris) was purchased from AMS Biotechnology (Frankfurt, Germany) and the digestion was performed as recently described.342.2 ESI MS and MS/MS
Electrospray ionization mass spectrometry (ESI-MS) in the negative and positive ion mode was carried out on an Amazon SL ion trap mass spectrometer (Bruker Daltonics, Bremen, Germany).The lyophilized digestion sample was dissolved in 50% methanol to obtain a concentration of 5 μg mL−1 and subsequently introduced into the ESI source via a syringe pump at a flow rate of 5 μL min−1. Nitrogen was used as nebulizer gas. ESI-MS conditions were: mass range mode, enhanced resolution; dry temperature, 200 °C; nebulizer, 7.25 psi; dry gas, 4.00 L min−1; HV capillary, 4.5 kV. These parameters enabled the unequivocal identification of the parent ion but provided as well reasonable intensities of fragment ions. Full scan mass spectra were acquired with the ICC (“ion charge control”) target set to 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and a maximum acquisition time of 100 ms. The required accumulation times averaged over 20 spectra were about 85 μs. The MS/MS experiment utilized an end-cap voltage of 0.55 V and an isolation width of 4 Da. Helium was used as collision gas. Data acquisition and analysis were carried out by using the programs TrapControl and DataAnalysis, respectively (Bruker Daltonics, Bremen, Germany).
000 and a maximum acquisition time of 100 ms. The required accumulation times averaged over 20 spectra were about 85 μs. The MS/MS experiment utilized an end-cap voltage of 0.55 V and an isolation width of 4 Da. Helium was used as collision gas. Data acquisition and analysis were carried out by using the programs TrapControl and DataAnalysis, respectively (Bruker Daltonics, Bremen, Germany).
2.3 Ion mobility-mass spectrometry (IM-MS)
All IM-MS spectra were recorded on a TOFWERK IMS-TOF instrument, which has been comprehensively described before.35 The dynamic range of IM-MS in combination with a TOF mass analyzer is approximately 1.000.36 Analytes were ionized by ESI and nitrogen was used as drift gas at atmospheric pressure and 30 °C in the enclosed IMS cell. Samples were diluted in methanol (final concentration 1 μg mL−1), and sodium acetate was added to enhance the formation of completely sodiated ions in the positive ion spectra. This warrants the optimum separation of both CS isomers (vide infra).The theoretical background of IMS has been discussed in detail elsewhere37 and can be briefly summarized as follows: ions traveling through a drift region of the length L filled with a neutral collision gas under the influence of a weak homogeneous electric field (E = V/L) are separated on the basis of their average ion velocity vd, which depends on the ion mobility K. The measured drift time td depends on the length of the drift region and the ion velocity:

Since ion mobility depends on the applied experimental conditions such as temperature T, pressure P and the strength of the electric field, the reduced mobility value K0 is normally calculated to allow comparisons of different experimental setups:
Reduced mobilities (K0) and collision cross-sections (σ) can be calculated by normalizing the ion mobility drift time to drift length, potential, pressure and temperature and a modified zero field (so-called Mason–Schamp) equation. This momentum transfer scan law includes field-dependent corrections for both collisional momentum transfer and collision frequency (α and β terms, respectively). For more detailed information see Siems et al.38

K0 = reduced mobility [cm2 V−1 s−1], L = drift length [cm], V = drift potential [V], td = drift time [s], P = pressure in the drift cell [mbar], T = temperature of the drift cell [K], σ = integrated collision cross-section, μ = reduced mass of the analyte and the drift gas, k = Boltzmann's constant, q = elementary charge, z = charge number, E = electric field, vd = drift velocity, N = neutral gas number density, βMT = correction coefficient for momentum transfer, αMT = correction coefficient for collision frequency, and vT = thermal velocity.

2.4 Proton (1H) nuclear magnetic resonance (NMR) spectroscopy
Samples were investigated as 5 mg ml−1 solutions in D2O. Apart from a reduced solvent signal, the use of D2O has the additional advantage that it helps to obtain deuterium frequency lock stabilization, which improves the spectral quality. The proton NMR spectrum is displayed in Fig. 4; trace A was recorded at 25 °C using an inverse 5 mm probe on a Bruker 600 MHz NMR spectrometer.The relative moieties of C4S and C6S in the mixture were determined by comparing the area of the resonances assigned to the N-acetyl groups (NAc) and the double bond in the uronic acids (UA H4) of the two isomers for three separate measurements at different temperatures: 303.15 K/298.15 K/310.15 K.
2.5 Diffusion-ordered spectroscopy (DOSY) pulsed-field gradient (PFG) NMR
Diffusion-ordered spectroscopy (DOSY) was performed at 30 °C (303 ± 1 K) by applying magnetic field gradients g and measuring the intensity I in dependence on the applied field gradients.39The attenuation of the observed signal intensity I due to the applied gradient strength g depends – besides the apparent self-diffusion coefficient D of the molecule of interest – on the non-attenuated signal intensity I0 (i.e. without gradients), the gyromagnetic ratio γ of the observed nucleus (γ = 4.26 × 107 Hz T−1), the diffusion time Δ (0.05 s) and the duration of the applied gradient δ (0.004 s), first described by Steijskal and Tanner in 1965:40
| I = I0exp[−Dγ2g2δ2(Δ − δ/3)] |
Consequently, the slope of the logarithmic plot of the decreasing signal intensity with increasing field gradient is proportional to the apparent self-diffusion coefficient D of the corresponding molecules:
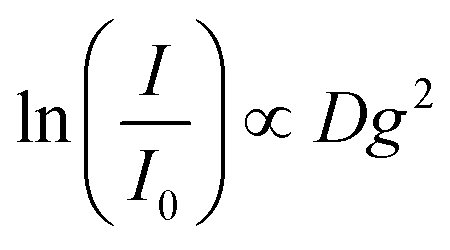
The calibration of the magnetic field gradient strength was performed as described by Holz et al.41 The temperature control was verified by measuring the temperature dependent chemical shift of ethylene glycol according to Ammann et al.42
3. Results and discussion
3.1 ESI MS and MS/MS
Fig. 2 shows the ESI mass spectra of chondroitin sulfate from bovine trachea after enzymatic digestion with chondroitinase ABC in the positive (A) and negative ion mode (B). The assignment of all signals can be found in Table 1 including the observed and calculated m/z values. | ||
| Fig. 2 ESI-MS of chondroitin sulfate from bovine trachea after enzymatic digestion with chondroitinase ABC, in the positive (A) and negative (B) ion mode. Trace (C) shows the negative ion mode mass spectrum of a mixture of the two isomeric standards, C4S and C6S (1:1, v/v). | ||
| m/z | z | Assignment | Hill notation | Calculated m/z (monoisotopic) |
|---|---|---|---|---|
| A | ||||
| 526.00 | 1 | [M − 2H + 3Na]+ | C14H19NNa3O14S | 526.02 |
| 504.02 | 1 | [M − H + 2Na]+ | C14H20NNa2O14S | 504.04 |
| 424.08 | 1 | [M − SO3 − H + 2Na]+ | C14H20NNa2O11 | 424.08 |
| 475.03 | 2 | [2M − SO3 − 3H + 5Na]2+ | C28H39N2Na5O25S | 475.05 |
|
||||
| B | ||||
| 458.06 | 1 | [M − H]− | C14H20NO14S | 458.06 |
| 480.02 | 1 | [M + Na − 2H]− | C14H19NNaO14S | 480.04 |
| 378.09 | 1 | [M − SO3 − H]− | C14H20NO11 | 378.10 |
In the positive ion mode (Fig. 2, trace A) the most abundant species with m/z 526.00 corresponds to singly charged ions containing three sodium ions. The negative ion spectrum shows the singly charged deprotonated form (m/z 458.06) and a sodiated species (m/z 480.02). In both spectra (A) and (B) low intense ions (m/z 424.06 and 378.11) were observed, representing unsaturated disaccharides without sulfate groups. This might be due to a sulfate loss during measurement; however, optimization of the experimental conditions could not minimize this effect and the same measurement of a mixture of the chondroitin sulfate disaccharide isomers C4S and C6S in the negative ion mode (shown in Fig. 2, trace C) also provided no evidence of the loss of sulfate groups under these experimental conditions. This indicates that these ions correspond to naturally occurring (low abundant) non-sulfated repeating units within the CS polysaccharide chains from bovine trachea.
The determination of the relative moieties of C4S and C6S was performed as essentially described by Desaire and Leary:18 the relative intensities of the two fragment ions m/z 282 and m/z 300 of the deprotonated ion (m/z 458.06) determined by MS/MS measurements of the indicated disaccharide standards were used in a given system of equations, and normalized (among other influencing factors) to differences in the ionization efficiencies of both isomers. In the case of the here analysed chondroitin sulfate from bovine trachea, the determined moiety of C4S was about (64 ± 4)% (detailed information and the used data can be found in the ESI†).
3.2 Ion mobility-mass spectrometry (IM-MS)
Negative and positive IM spectra of two commercially available CS disaccharide standards and one CS sample subsequent to exhaustive digestion with chondroitinase ABC are shown in Fig. 3. | ||
| Fig. 3 IM-MS of the positively charged triply sodiated disaccharide (m/z 526; trace A) and the singly negatively charged deprotonated species (m/z 458; trace B). In addition to the results of the enzymatic digestion of native CS (from bovine trachea) with chondroitinase ABC, the isolated unsaturated CS disaccharides (commercially available C4S and C6S standards) are also shown. | ||
In both detection modes, the positive (A) and the negative (B) ion mode, two peaks corresponding to the 4- and 6-sulfate are detected (baseline separation only in positive ion mode). There were no indications for different conformations of the disaccharides in the gas phase as observed for sodiated disaccharides in IM-MS measurements done before.43
Experiments with the purified C4S and C6S disaccharides enable the unequivocal assignments of both isomers: the first peak, characterized by a shorter drift time and therefore a smaller collision cross-section, represents the 4-sulfate and the second peak the 6-sulfate. The observed drift times, the corresponding calculated collision cross-sections and the reduced ion mobility data are given in Table 2. The observed drift time differences are caused by different collision cross-sections of the ions: the C4 sulfo group close to the helical axis of the disaccharide results in a more compact form, whereas the sulfate residue in the C6 position is more space-demanding. This difference is increased by the attachment of sodium ions, thus allowing baseline separation of the two signals in the positive ion mode. This is a significant advantage of the positive ion mode although sulfated oligosaccharides are normally more sensitively detected as negative ions.
| A | ||
|---|---|---|
| [M − 2H + 3Na]+ | C4S | C6S |
| t d (ms) | 48.4 | 49.4 |
| σ (Å2) | 223.8 | 227.9 |
| K 0 (cm2 V−1 s−1) | 1.084 | 1.064 |
| Relative area (%) | 67 ± 3 | 33 ± 3 |
| B | ||
|---|---|---|
| [M − H]− | C4S | C6S |
| t d (ms) | 45.4 | 45.9 |
| σ (Å2) | 211.0 | 213.3 |
| K 0 (cm2 V−1 s−1) | 0.980 | 0.969 |
| Relative area (%) | 78 ± 4 | 22 ± 4 |
Integration of the peak areas resulted in different percentages of C4S in the positive (67 ± 3)% and the negative ion mode (78 ± 4)% (also included in Table 2). The pronounced difference in the relative contribution of the C4S isomer as the fully sodiated ion (positive ion mode) and deprotonated ion (negative ion mode) indicates that the CS isomers exhibit different ionization and/or adduct formation efficiencies during ESI.
The normalization factor for the different ionization efficiencies in the negative ion mode (that was determined during MS2 quantification) was used to take these differences into account. The percentage of C4S determined in this manner in the negative ion mode of (62 ± 5)% is in good agreement with the result of the 1H NMR measurement (60 ± 4%).
However, in the case of the positive ion mode the obtained results differed only slightly from the values determined by 1H NMR. This indicates that the ionization efficiencies of the fully sodiated isomers in the positive ion mode are reduced or compensated by other effects such as differences in the adduct formation tendencies of both isomers. A more detailed evaluation of these aspects, however, was beyond the scope of this study.
3.3 Proton (1H) nuclear magnetic resonance (NMR) spectroscopy and diffusion-ordered spectroscopy (DOSY) pulsed-field gradient (PFG) NMR
Even if basically all resonances of the unsaturated disaccharides can be used to perform DOSY experiments, the resonances of the N-acetyl groups (NAc) are most useful for three reasons: (i) the N-acetyl resonances are well separated from all remaining carbohydrate resonances, i.e. there is no overlap. (ii) There are three equivalent protons in each N-acetyl group and this (in combination with the aspect that there is no coupling which would lead to peak splitting) confers high sensitivity even if diluted samples (in comparison to the rather concentrated solutions used here) are investigated. (iii) The characteristic chemical shifts enable the assignment to the different unsaturated disaccharides,28 while the α- and β-anomers cannot be differentiated because they result in exactly the same chemical shifts.Of course, the resonances of the H4 protons located at the double bond within the uronic acid can also be used because there is a clear assignment to the two isomers and a good separation from all other signals: although the intensities of these signals are much lower, nearly identical data (as in the case of the N-acetyl groups) can be obtained if these resonances are used. Although NMR is by far less sensitive in comparison to MS it should be noted that the here used considerable concentrations (5 mg ml−1) are not a must but were used to save instrument time. Reasonable NMR spectra can also be obtained for diluted samples such as synovial fluids or other body fluids.44
All in all, this results in a set of two signals on each “end” of the 1H NMR spectrum as illustrated in Fig. 4(A). During PFG NMR measurements, the intensities of these signals decrease with increasing gradient strength. This is exemplary shown for the NAc resonances in Fig. 4(B).
 | ||
| Fig. 4 600 MHz high resolution 1H NMR spectrum of the enzymatic digest of native CS from bovine trachea with chondroitinase ABC (A). Two resonances are very characteristic and well separated from the remaining carbohydrate signals: the resonance at 2.088 ppm can be assigned to the N-acetyl group (NAc) of C4S, while the N-acetyl group at 2.050 ppm stems from C6S.28 In the same way, the resonances of the double bond in the uronic acid (UA) can be assigned to C4S (5.977 ppm) and C6S (5.883 ppm).28 These selected resonances are explicitly labeled in the spectrum. In trace (B) the attenuation of the N-acetyl resonances in dependence on the applied gradient strength is shown to illustrate the effect of 1% and 95% gradient strength on the signal intensities. | ||
Fig. 5 shows the DOSY logarithmic plot of the NAc resonances in (A) as a function of the square of the applied field gradient strength. The straight lines emphasize (i) that there is only one single apparent self-diffusion coefficient D per compound and (ii) that the two isomers can be differentiated due to their diffusion behavior. The differences in the diffusion behavior are only based on the different spatial extensions of the related ions: the introduction of a sulfate residue at the C6 position is obviously more space-demanding in comparison to the same modification at the C4 position and results in smaller diffusion coefficients, respectively.
 | ||
| Fig. 5 DOSY plot of the N-acetyl resonances of the two CS isomers (A): the attenuation of the relative signal intensity I/I0 of C4S is slightly higher than that of the C6S corresponding to a higher diffusion coefficient of the 4-sulfate. The individual plots for the NAc and UA H4 resonances for both isomers and the corresponding linear regressions are given in the supplement (S1). Trace (B) shows the determined diffusion coefficients for all discussed resonances and the error bars from the linear regression. | ||
The diffusion coefficients calculated from each selected resonance are given in Table 3. Detailed information on the linear regression, R2 values and errors is also available in the electronic supplement. The comparison of these values allows an estimation of the error and thus the validity range of the method: the relative error of the fit itself is 0.2% and within one isomer species (NAc C4S and UA H4 C4S or NAc C6S and UA H4 C6S) the difference amounts to about 1%. However, the difference between the two isomers is at least 2%. This is graphically illustrated in Fig. 5(B): considering the different diffusion coefficients a clear distinction between the two isomers can be made.
| Resonance [ppm] | Assignment | D [10−10 m2 s−1] | Integration limits [ppm] | Rel. intensitiesa |
|---|---|---|---|---|
| a Values from three separate measurements at different temperatures: 303.15 K/298.15 K/310.15 K. | ||||
| 2.050 | NAc C6S | (4.776 ± 0.010) | 2.04–2.06 | 0.40/0.42/0.42 |
| 2.088 | NAc C4S | (4.876 ± 0.004) | 2.07–2.10 | 0.60/0.58/0.58 |
| 5.883 | UA H4 C6S | (4.744 ± 0.006) | 5.86–5.91 | 0.41/0.36/0.39 |
| 5.977 | UA H4 C4S | (4.897 ± 0.005) | 5.96–5.99 | 0.59/0.64/0.61 |
4. Conclusions
The differentiation of the two most common chondroitin sulfate disaccharide isomers with the sulfo group either on the C4 or the C6 position of the N-acetylgalactosamine could be realized by means of IM-MS measurements. The unequivocal assignment of the two isomers to the respective signal in the ion mobility spectrum was obtained by the simultaneous investigation of both disaccharide standards. The collision cross-sections (σ) with nitrogen as collision gas and the reduced ion mobility (K0) of the two disaccharides as deprotonated [M − H]− as well as fully sodiated [M − 2H + 3Na]+ ions were determined, and baseline separation of both isomers in a crude digestion sample of the CS polysaccharide from a biological sample was achieved.Further NMR DOSY experiments confirmed the MS results: under both conditions, i.e. in solution and in the gas phase, the small differences in the structures of the two isomers led to observable differences. In both, solution (NMR) and gas phase (IM), the C4S, with its more “compact form” due to the position of the sulfo group near to the helical axis of the disaccharide, showed a higher mobility than the C6S isomer.
The comparison of the obtained C4S and C6S relative moieties by means of the different methods is given in Table 4. The results determined by IM-MS in the positive ion mode and in the negative ion mode subsequent to normalization are in excellent agreement with the results of the MS2 and 1H NMR experiments.
| % | MS2 | IM-MS | 1H NMR | ||
|---|---|---|---|---|---|
| Pos. IM | Neg. IM | Neg. IM × Norm 1 | |||
| C4S | (64 ± 4)% | (67 ± 3)% | (78 ± 4)% | (62 ± 5)% | (60 ± 4)% |
| C6S | (37 ± 2)% | (33 ± 3)% | (22 ± 4)% | (38 ± 5)% | (40 ± 4)% |
In a nutshell, a rapid separation (a measurement takes only a few seconds) of both CS isomers in a mixture can be performed by means of IM-MS in the positive ion mode without the need for any pre-separation or derivatization. This can be regarded as a milestone regarding the fast elucidation of the CS compositions of a large number of biological samples. Additionally, the IM-MS method exhibits an about 5000 times higher sensitivity compared to the NMR method.
Finally, the use of the two methods allows the characterization of (at least) carbohydrate isomers in mixtures without the absolute need for purified standard substances: the agreement between the behavior of both isomers in solution, determined by PFG NMR, as well as in the gas phase shown by IM-MS serves as a bridge between NMR structure elucidation and the identification of characteristic MS/MS fragments which will be the focus of our future research.
Acknowledgements
This work was supported by the German Research Council (Transregio 67, projects A2 and A8).References
- J. Schiller and D. Huster, New methods to study the composition and structure of the extracellular matrix in natural and bioengineered tissues, Biomatter, 2012, 2, 115–131 CrossRef PubMed.
- K. Sugahara, T. Mikami, T. Uyama, S. Mizuguchi, K. Nomura and H. Kitagawa, Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate, Curr. Opin. Struct. Biol., 2003, 13, 612–620 CrossRef CAS PubMed.
- K. Sugahara and T. Mikami, Chondroitin/dermatan sulfate in the central nervous system, Curr. Opin. Struct. Biol., 2007, 17, 536–545 CrossRef CAS PubMed.
- K. Gulati and K. M. Poluri, Mechanistic and therapeutic overview of glycosaminoglycans: the unsung heros of biomolecular signaling, Glycoconjugate J., 2016, 33, 1–17 CrossRef CAS PubMed.
- F. Sasarman, C. Maftei, P. M. Campeau, C. Brunel-Guitton, G. A. Mitchell and P. Allard, Biosynthesis of glycosaminoglycans: associated disorders and biochemical tests, J. Inherited Metab. Dis., 2016, 39, 173–188 CrossRef CAS PubMed.
- D. M. Toleffsen and L. Zhang, in Hemostasis and Thrombosis, ed. R. W. Colman, Lippincott Williams & Wilkins, 2006, pp. 271–283 Search PubMed.
- N. Volpi, Dermatan sulfate: recent structural and activity data, Carbohydr. Polym., 2010, 82, 233–239 CrossRef CAS.
- A. V. Noulas, S. S. Skandalis, E. Feretis, D. A. Theocharis and N. K. Karamanos, Variations in content and structure of glycosaminoglycans of the vitreous gel from different mammalian species, Biomed. Chromatogr., 2004, 18, 457–461 CrossRef CAS PubMed.
- N. Volpi, Analytical aspects of pharmaceutical grade chondroitin sulfates, J. Pharm. Sci., 2007, 96, 3168–3180 CrossRef CAS PubMed.
- F. Cheng, D. Heinegård, A. Malmström, A. Schmidtchen, K. Yoshida and L.-A. Fransson, Patterns of uronosyl epimerization and 4-/6–0-sulphation in chondroitin/dermatan sulphate from decorin and biglycan of various bovine tissues, Glycobiology, 1994, 4, 685–696 CrossRef CAS PubMed.
- M. Shinmei, S. Miyauchi, A. Machida and K. Miyazaki, Quantitation of chondroitin 4-sulfate and chondroitin 6-sulfate in pathologic joint fluid, Arthritis Rheum., 1992, 35, 1304–1308 CrossRef CAS PubMed.
- A. H. K. Plaas, L. A. West, S. Wong-Palms and F. R. T. Nelson, Glycosaminoglycan sulfation in human osteoarthritis: disease-related alterations at the non-reducing termini of chondroitin and dermatan sulfate, J. Biol. Chem., 1998, 273, 12642–12649 CrossRef CAS PubMed.
- F. Mannello, F. Maccari, D. Ligi, M. Canale, F. Galeotti and N. Volpi, Characterization of oversulfated chondroitin sulfate rich in 4,6-O-disulfated disaccharides in breast cyst fluids collected from human breast gross cysts, Cell Biochem. Funct., 2014, 32, 344–350 CrossRef CAS PubMed.
- T. N. Laremore and R. J. Linhardt, Improved matrix-assisted laser desorption/ionization mass spectrometrc detection of glycosaminoglycan disaccharides as cesium salts, Rapid Commun. Mass Spectrom., 2007, 21, 1315–1320 CrossRef CAS PubMed.
- C. C. Hsieh, J. Y. Guo, S. U. Hung, R. Chen, Z. Nie, H.-C. Chang and C.-C. Wu, Quantitative analysis of oligosaccharides derived from sulfated glycosaminoglycans by nanodiamond-based affinity purification and matrix-assisted laser desorption/ionization mass spectrometry, Anal. Chem., 2013, 85, 4342–4349 CrossRef CAS PubMed.
- K. A. Jandik, K. Gu and R. J. Linhardt, Action pattern of polysaccharide lyases on glycosaminoglycans, Glycobiology, 1994, 4, 289–296 CrossRef CAS PubMed.
- A. Nimptsch, S. Schibur, M. Schnabelrauch, B. Fuchs, D. Huster and J. Schiller, Characterization of the quantitative relationship between signal-to-noise (S/N) ratio and sample amount on-target by MALDI-TOF MS: determination of chondroitin sulfate subsequent to enzymatic digestion, Anal. Chim. Acta, 2009, 635, 175–182 CrossRef CAS PubMed.
- H. Desaire and J. A. Leary, Detection and quantification of the sulfated disaccharides in chondroitin sulfate by electrospray tandem mass spectrometry, J. Am. Soc. Mass Spectrom., 2000, 11, 916–920 CrossRef CAS PubMed.
- J. Zaia and C. E. Costello, Compositional analysis of glycosaminoglycans by electrospray mass spectrometry, Anal. Chem., 2001, 73, 233–239 CrossRef CAS PubMed.
- P. Hu, L. Fang and E. K. Chess, Source-induced fragmentation of heparin, heparan, and galactosaminoglycans and application, Anal. Chem., 2009, 81, 2332–2343 CrossRef CAS PubMed.
- A. Hitchcock, K. E. Yates, C. Costello and J. Zaia, Comparative glycomics of connective tissue glycosaminoglycans, Proteomics, 2008, 8, 1384–1397 CrossRef CAS PubMed.
- J. E. McClellan, C. E. Costello, P. B. O'Connor and J. Zaia, Influence of charge state on product ion mass spectra and the determination of 4S/6S sulfation sequence of chondroitin sulfate oligosaccharides, Anal. Chem., 2002, 74, 3760–3771 CrossRef CAS PubMed.
- D. T. Kenny, S. M. A. Issa and N. G. Karlsson, Sulfate migration in oligosaccharides induced by negative ion mode ion trap collision-induced dissociation, Rapid Commun. Mass Spectrom., 2011, 25, 2611–2618 CrossRef CAS PubMed.
- G. J. Lee, J. E. Evans and H. Tieckelmann, Rapid and sensitive determination of enzymatic degradation products of isomeric chondroitin sulfates by high-performance liquid chromatography, J. Chromatogr., Biomed. Appl., 1978, 146, 439–448 CrossRef CAS.
- N. Volpi, F. Galeotti, B. Yang and R. J. Linhardt, Analysis of glycosaminoglycan-derived precolumn 2-aminoacridone-labeled disaccharides with LC-fluorescence and LC-MS detection, Nat. Protoc., 2014, 9, 541–558 CrossRef CAS PubMed.
- A. Zinellu, S. Pisanu, E. Zinellu, A. J. Lepedda, G. M. Cherchi, S. Sotgia, C. Carru, L. Deiana and M. Zormato, A novel LIF-CE method for the separation of hyaluronan- and chondroitin sulfate-derived disaccharides: application to structural and quantitative analyses of human plasma low- and high-charged chondroitin sulfate isomers, Electrophoresis, 2007, 28, 2439–2447 CrossRef CAS PubMed.
- E. Ucakturk, C. Cai, L. Li, G. Li, F. Zhang and R. J. Linhardt, Capillary electrophoresis for total glycosaminoglycan analysis, Anal. Bioanal. Chem., 2014, 406, 4617–4626 CrossRef CAS PubMed.
- S. Yamada, K. Yoshida, M. Sugiura and K. Sugahara, One- and two-dimensional 1H-NMR characterization of two series of sulfated disaccharides prepared from chondroitin sulfate and heparan sulfate/heparin by bacterial eliminase digestion, J. Biochem., 1992, 112, 440–447 CAS.
- D. Jeannerat and J. Furrer, NMR experiments for the analysis of mixtures: beyond 1D 1H spectra, Comb. Chem. High Throughput Screening, 2012, 15, 15–35 CrossRef CAS PubMed.
- J. Sitkowski, E. Bednarek, W. Bocian and L. Kozerski, Asessment of oversulfated chondroitin sulfate in low molecular weight and unfractioned heparins diffusion ordered nuclear magnetic resonance spectroscopy method, J. Med. Chem., 2008, 51, 7663–7665 CrossRef CAS PubMed.
- J. F. K. Limtiaco, S. Beni, C. J. Jones, D. J. Langeslay and C. K. Larive, NMR methods to monitor the enzymatic depolymerization of heparin, Anal. Bioanal. Chem., 2011, 399, 593–603 CrossRef CAS PubMed.
- M. J. Kailemia, M. Park, D. A. Kaplan, A. Venot, G.-J. Boons, L. Li, R. J. Linhardt and I. A. Amster, High-field asymmetric-waveform ion mobility spectroscopy and electron detachment dissociation of isobaric mixtures of glycosaminoglycans, J. Am. Soc. Mass Spectrom., 2014, 25, 258–268 CrossRef CAS PubMed.
- J. Hofmann, H. S. Hahm, P. H. Seeberger and K. Pagel, Identification of carbohydrate anomers using ion-mobility-mass spectrometry, Nature, 2015, 526, 241–244 CrossRef CAS PubMed.
- J. Schiller, J. Arnhold, S. Benard, S. Reichl and K. Arnold, Cartilage degradation by hyaluronate lyase and chondroitin ABC lyase: a MALDI-TOF mass spectrometric study, Carbohydr. Res., 1999, 318, 116–122 CrossRef CAS PubMed.
- K. Kaplan, S. Graf, C. Tanner, M. Gonin, K. Fuhrer, R. Knochenmuss, P. Dwivedi and H. H. Hill, Resistive glass IM-TOF MS, Anal. Chem., 2010, 82, 9336–9343 CrossRef CAS PubMed.
- M. Groessl, S. Graf and R. Knochenmuss, High resolution ion mobility-mass spectrometry for separation and identification of isomeric lipids, Analyst, 2015, 140, 6904–6911 RSC.
- G. A. Eiceman and Z. Karpas, Ion Mobility Spectrometry, CRC Press, Taylor and Francis Group, Boca Raton, FL. 2nd edn, 2005 Search PubMed.
- W. F. Siems, L. A. Viehland and H. H. Hill Jr, Improved momentum-transfer theory for ion mobility. 1. Derivation of the fundamental equation, Anal. Chem., 2012, 84, 9782–9791 CrossRef CAS PubMed.
- J. Schiller, L. Naji, R. Trampel, W. Ngwa, R. Knauss and K. Arnold, Pulsed-field gradient-nuclear magnetic resonance (PFG NMR) to measure the diffusion of ions and polymers in cartilage: applications in joint diseases, Methods Mol. Med., 2004, 101, 287–302 CAS.
- E. O. Stejskal and J. E. Tanner, Spin diffusion measurements: spin echoes in the presence of a time dependent field gradient, J. Chem. Phys., 1965, 42, 288–292 CrossRef CAS.
- H. Holz, S. R. Heil and A. Sacco, Temperature self-diffusion of water and six selected molecular liquids for calibration in accurate 1H NMR PFG measurements, Phys. Chem. Chem. Phys., 2000, 2, 4740–4742 RSC.
- C. Ammann, P. Meier and A. E. Merbach, A simple multinuclear NMR thermometer, J. Magn. Reson., 1982, 46, 319–321 CAS.
- P. Dwivedi, B. Bendiak, B. H. Clowers and H. H. Hill Jr, Rapid resolution of carbohydrate isomers by electrospray ionization ambient pressure ion mobility spectrometry-time-of-flight mass spectrometry (ESI-APIMS-TOFMS), J. Am. Soc. Mass Spectrom., 2007, 18, 1163–1175 CrossRef CAS PubMed.
- B. Mickiewicz, J. J. Kelly, T. E. Ludwig, A. M. Weljie, J. P. Wiley, T. A. Schmidt and H. J. Vogel, Metabolic analysis of knee synovial fluid as a potential diagnostic approach for osteoarthritis, J. Orthop. Res., 2015, 33, 1631–1638 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay02531e |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2016 |