
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A fluoride-selective electrode (Fse) for the quantification of fluoride in lithium-ion battery (Lib) electrolytes
A.
Wilken
a,
V.
Kraft
a,
S.
Girod
a,
M.
Winter
ab and
S.
Nowak
*a
aUniversity of Muenster, MEET Battery Research Center, Institute of Physical Chemistry, Corrensstraße 46, 48149 Muenster, Germany. E-mail: sascha.nowak@uni-muenster.de
bHelmholtz Institute Münster, IEK-12 of Forschungszentrum Jülich, Corrensstrasse 46, Münster, Germany
First published on 16th August 2016
Abstract
In this work, a fluoride-selective electrode (FSE) was applied with regard to the analysis of fluoride in lithium hexafluorophosphate-based lithium-ion battery (LIB) electrolytes. The influence of linear organic carbonate solvents dimethyl carbonate (DMC), ethyl methyl carbonate (EMC) and diethyl carbonate (DEC) which are used as co-solvents in battery electrolytes was investigated. The developed FSE method for the analysis of battery electrolytes was comprehensively validated in view of the (1) trueness and recovery rates (nominal vs. actual comparison; influence of different amounts of electrolytes on the performance of the electrode; recovery rates of defined differences in concentration), (2) precision (intra-day precision and inter-day precision), (3) selectivity (influence of the carbonate solvents on different fluoride concentrations; interferents) and (4) linearity and range. Statistical analysis was performed to evaluate the data and to characterize the reproducibility of the method. The determination of the commercially available LP30 (1 mol LiPF6 and ethylene carbonate/dimethyl carbonate (EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DMC, 50:50 wt%)) electrolyte stored over 47 days and at 80 °C, by the FSE technique was compared to the fluoride analysis by ion chromatography (IC). While interferences in the IC method resulted in false-high concentrations, the FSE operated free from interferences, selective and specific. The validation of the method was successfully carried out and enables new areas of application.
:DMC, 50:50 wt%)) electrolyte stored over 47 days and at 80 °C, by the FSE technique was compared to the fluoride analysis by ion chromatography (IC). While interferences in the IC method resulted in false-high concentrations, the FSE operated free from interferences, selective and specific. The validation of the method was successfully carried out and enables new areas of application.
1 Introduction
The fluoride-selective electrode (FSE) came up in the 1960s by Frant & Ross and was successfully applied for fluoride analysis in aqueous solutions.1 The investigation of aging processes in lithium-ion batteries (LIB) is a key point for the further development of rechargeable batteries in the context of research on renewable energies. Therefore, investigations on the changes of the electrolyte composition contribute to the understanding of aging processes that still limit the battery cycling performance. Identification and quantification of compounds and their decomposition products formed in aging processes gain a precise understanding of the aging processes themselves and their influencing factors. Due to the cross-dependence of aging phenomena, they are very complicated to characterize.2 Analysis of electrolytes as the connecting media to all cell compounds (cathode, anode, and separator) provides information about interactions between battery components. Electrolytes based on LiPF6 dissolved in mixtures of organic carbonates such as ethylene carbonate–dimethyl carbonate (EC–DMC), ethylene carbonate–ethyl methyl carbonate (EC–EMC) or ethylene carbonate–diethyl carbonate (EC–DEC) are used most frequently in LIBs.3 The degradation of LiPF6 in organic electrolytes has been investigated intensively.4–6 Campion et al. (2004) proposed a mechanism for the autocatalytic decomposition of LiPF6 in carbonates due to the presence of protic impurities such as traces of alcohol and water.7 In thermally aged electrolytes, the formation of HF was also observed.5,8 The reaction of PF5 with alcohol and water, respectively, forms phosphoryl trifluoride (POF3) and hydrofluoric acid (HF). HF for its part is a weak acid and may catalyze the decomposition reaction of LiPF6 as a further protic compound. The formation of acidic compounds such as HF is known to have negative effects on the stability of the solid electrolyte interphase (SEI) on the anode.9,10 Moreover, Vetter et al. (2005) reported a disproportionation reaction of lithium manganese spinel used as the cathode material. HF dissolved Mn(III) from LiMn2O4 yielding λ-Mn2O4 (Mn IV), Mn2+ (Mn II) and LiF. Mn2+ leaves the spinel and gets dissolved in the electrolyte.4 Mn2+ may be re-precipitated on the cathode as for example, MnxOy, MnF2 or MnCO3. Alternatively, Mn2+ might migrate to the negative electrode and will deposit there. However, while often damaging effects of HF in the context of the electrodes and electrolyte were reported, HF also stabilizes Al current collectors. HF was found to passivate Al used as the current collector due to the formation of chemically stable AlF3 or AlOxFy on the surface.11,34 Overall, the presence of HF has turned out to be decisive for battery life and cyclization performance.Until now, decomposition products of LiPF6-based organic electrolytes were investigated by means of IC as well as IC-electrospray ionization mass spectrometry (IC-ESI-MS),12–16 by electrospray ionization-high resolution mass spectrometry (ESI-HRMS),17 by liquid chromatography-tandem quadrupole mass spectrometry (LC/ESI-MS/MS)18 and by gas chromatography-mass spectrometry (GC-MS).15,19–22 Terborg et al. achieved a baseline separation of the dissociation products by IC of dissolved LiPF6 in water as well as of the components and dissociation products of a commercially available battery electrolyte (1 M LiPF6 in EC:DEC (3:7 w/w)).12 Kraft et al. (2015) monitored the fluoride formation during aging using the IC technique.15 Co-elution of the fluoride peak with the arising ionic organophosphates was observed caused by the low retention of these compounds on a column with a hydrophilic character. Organophosphates are formed unavoidably from the organic carbonates during cyclization of LIBs and indicate aging processes. With increasing cyclization, amounts of organophosphates increase, too. Thus, besides the interesting fluoride concentrations, further aging products arise during ongoing charge and discharge processes impeding the detection of the fluoride. It was noted that fluoride contents of samples with a higher degree in degradation might have given false-high values due to co-elution. A high selectivity in regard to fluoride and organophosphates was achieved on a column with hydrophobic character. However, the analysis time was extremely high caused by a strong interaction of hexafluorophosphate with the stationary phase.14 Furthermore, the two-dimensional IC yielded a successful separation of organophosphates but could not separate fluoride.16 Especially in the case of samples of a higher degree of degradation, the IC analysis capability in terms of fluoride is limited. So far, there is no method reported which can access the fluoride quantification in aged LIB electrolytes. Furthermore, to the best of our knowledge, FSE has not been reported for fluoride quantification in LiPF6-based LIB electrolytes until now. In addition, an analytical validation for the determination of fluoride in this matrix assessing the suitability of the method in this context was not reported in the literature.
Using spectroscopic and chromatographic methods for the quantification of fluoride is challenging due to fluoride's heavy excitation because of its high electronegativity and its low retention on ion-exchange columns resulting in co-elution with other compounds having the same chromatographic behavior. A highly element-selective and specific analysis tool was developed by Frant & Ross in 1966.1 A single-crystal of a rare earth fluoride, such as LaF3, NdF3 or PrF3, was used as the electrode-type sensor giving a Nernstian response over several decades of fluoride ion activity. The response of the LaF3 electrode to the concentration of fluoride ions was reported several times to start at 10−6 mol L−1 upwards.1,33 The ISE was applied in the past for fluoride determinations in water, wastewater, pure salts after pre-concentration, electroplating baths, milk, minerals and rocks, plants, phosphoric acid topaz, bones, urine, aliphatic amine solutions, dental tissues, samples containing boron, organic compounds, tea, biological materials, hair and D2O.23 In the present work, the FSE was investigated for its applicability of fluoride analysis in organic electrolytes. Due to the complexity of battery electrolytes, particular attention was paid to the possible adverse reactions of matrix compounds with an FSE. The method was successfully validated considering the following points: (1) trueness (deviation of the measured value due to systematic errors) and recovery rates investigating (a) a nominal actual comparison, (b) the influence of the organic carbonates in various compositions and volumes on fluoride detection and (c) the recovery of defined differences in fluoride concentrations in solutions spiked with different concentrations of a fluoride standard; (2) precision (deviation of analysis values due to random errors) investigated (a) as inter-day precision and intra-day precision; (3) selectivity as (a) a comparison of the analyte in standard solutions with the analyte in the matrix of real samples and (b) the application of TISAB solution for prevention of interferences and achievement of pH stability; (4) linearity and range as a comparison of the linear calibration function with the function of the 2nd degree as well as consideration of the Nernst slope; (5) limits of detection and quantification. After validation, the FSE method was applied to the characterization of thermally aged LiPF6-based LIB electrolytes. The results were compared to the results obtained by analysis using IC.
2 Experimental section
2.1 Chemicals
Sodium carbonate (CertiPUR) and sodium hydrogen carbonate (for analysis) were bought from Merck KGaA (Darmstadt, Germany). Glacial acetic acid (100%, 17.65 mol L−1), sodium chloride and acetonitrile (AcN, for HPLC in gradient grade quality) were purchased from VWR (Darmstadt, Germany). trans-1,2-Diaminocyclohexane-N,N,N′,N′-tetraacetic acid monohydrate (Komplexon IV, ≥99%) and sulphuric acid were obtained from Sigma-Aldrich Chemie GmbH (Steinheim, Germany). Fluoride standard IC (1000 mg L−1 ± 4 mg L−1) was purchased from Fluka Chemie GmbH (Buchs, Switzerland). Dimethyl carbonate (DMC) SelectiLyte™, ethyl methyl carbonate (EMC) SelectiLyte™, diethyl carbonate (DEC) SelectiLyte™ and LP30 SelectiLyte™ (1 M LiPF6 in EC:DMC (50:50, wt%)) were acquired from BASF SE (Ludwigshafen, Germany). All chemicals were of the highest available quality.
Water was purified using a Milli-Q water Advantage A10 system (18.2 MΩ cm, TOC < 3 ppb, Millipore GmbH, Schwalbach, Germany).
2.2 Calibration solutions for method validation
For external calibration, 17 standard solutions of the following fluoride concentrations were diluted in de-ionized water from the fluoride stock solution (1 g L−1): blank, 0. 5 ppm, 1 ppm, 2 ppm, 3 ppm, 4 ppm, 5 ppm, 10 ppm, 20 ppm, 30 ppm, 40 ppm, 50 ppm, 60 ppm, 70 ppm, 80 ppm, 90 ppm and 100 ppm.2.3 FSE instrumentation
The FSE system consisted of an Metrohm 858 Professional Sample Processor, a 905 Titrando connected to an 801 Stirrer, two 800 Dosinos (20 mL for dispensing the sample into the PTFE test vessel and 50 mL for flushing of the PTFE test vessel after the measurement), the FSE type no. 6.0502.15 and the Ag Ag/Cl reference electrode type no. 6.0750.100. The instrument was controlled by Methrom tiamo™ 2.4 and Metrohm MagIC Net™ 3.1. All devices and the software were acquired from Metrohm AG (Herisau, Switzerland). For analysis, 10 mL of TISAB IV solution were filled in the PTFE test vessel and 2 mL of the sample were added.2.4 IC instrumentation
The IC system consisted of a Metrohm 889 IC Sample Center for sample introduction and an 850 Professional IC equipped with a chemical suppression module and a conductivity detector. For the anion separation, the column Metrosep A Supp 4 with the dimensions 250 mm length × 4.0 mm interior diameter and 9 μm particle size was used combined with the matching guard column Metrosep A4/5 guard, 4.0 mm interior diameter. The suppressor applied a 100 mmol L−1 H2SO4 solution for regeneration of the stationary phase of the suppressor. The system was controlled by MagIC Net™ 3.1. All devices and the software were acquired from Metrohm AG (Herisau, Switzerland). The column oven was operated at 40 °C and the flow rate was set to 1 mL min−1. Anions were separated isocratically over 18 min using an aqueous solution of acetonitrile (24 vol%), sodium carbonate (2.52 mmol L−1) and sodium hydrogen carbonate (2.38 mmol L−1) as the eluent.2.5 Statistical analysis
For statistical analysis the open source software R Studio from R Core Team (2015) was used. R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. URL https://www.R-project.org/.2.6 Trueness and recovery rates
:DMC (50:50, wt%), EC:DEC (50:50, wt%) and EC:EMC (50:50, wt%) was investigated in an aqueous solution containing 10 ppm fluoride. 100 μL of the fluoride stock solution (1 mg L−1) was mixed with 0 μL, 10 μL, 50 μL, 100 μL, 500 μL and 1000 μL of the electrolytes, respectively, and filled up with de-ionized water in a 10 mL vial. The samples were analyzed three times and the average of the detected voltages was used for calculations.
:DMC (50:50, wt%)) from a freshly opened bottle was spiked with 0 μL, 20 μL, 50 μL, 70 μL and 90 μL of the fluoride stock solution (1 mg L−1), respectively, and filled up to 10 mL with de-ionized water resulting in nominal fluoride concentrations of 0 ppm, 20 ppm, 50 ppm, 70 ppm and 90 ppm.
2.7 Precision
For intra-day and inter-day precision, 16 standard solutions of the following fluoride concentrations were diluted in de-ionized water from the fluoride stock solution (1 g L−1): blank, 1 ppm, 2 ppm, 3 ppm, 4 ppm, 5 ppm, 10 ppm, 20 ppm, 30 ppm, 40 ppm, 50 ppm, 60 ppm, 70 ppm, 80 ppm, 90 ppm and 100 ppm. Larger amounts of all concentrations were prepared to use the same stock solutions for all recurring analysis. For intra-day precision, the solutions were analyzed six times successively on the same day. For inter-day analysis precision, the solutions were analyzed for a period of six days consecutively.2.8 Selectivity
:DEC (50:50, wt%) containing the following fluoride concentrations were prepared: 0.5 ppm, 1 ppm, 2 ppm, 5 ppm, 10 ppm, 15 ppm, 25 ppm, 50 ppm, and 100 ppm. In the first batch, the samples are just filled up with de-ionized water. In the second batch, 100 μL of EC:DEC (50:50, wt%) were added to each solution and prior to filling up with deionized water. Each batch was analyzed three times. The average of the detected voltages for each concentration step was used for calculations.
2.9 Linearity and range
For investigation of the linearity, data of the calibration curve were used. Linearity was determined by comparing the linear calibration function to a function of the 2nd degree. The adjustment of the two functions was compared by an F-test.2.10 Sample preparation and hydrolysis stability
Since traces of water or moisture lead to the rapid decomposition of the conducting salt, the electrolytes were handled in a dry room or glovebox to prevent contamination. However, water can be used as a solvent during sample preparation. Terborg et al. showed that lithium hexafluorophosphate is stable in aqueous solution for several days.12 The same was reported by Metrohm who investigated the stability of PF6 in water as well as the stability of PF6 in electrolytes when diluted with water.35 Regarding the hydrolysis of the organophosphates, Kraft et al. demonstrated that these compounds are stable against water and therefore water is an appropriate solvent during sample preparation.162.11 Sample preparation for aging experiments of a battery electrolyte at 80 °C
For the investigation of the formation of fluoride in the electrolytes, 2 mL of commercially available LP30 was filled in aluminum vials closed with a screw cap. In total 40 sample containers were stored at 80 °C for 47 days. The samples were analyzed on days 1, 3, 5, 9, 14, 16, 20, 28, 35 and 47. At each time, four sample vials were analyzed and each sample was analyzed threefold by IC combined with a conductivity detector as well as with a FSE. Before analysis by means of FSE, all samples were diluted 1:100 with de-ionized water. For analysis via IC, all samples were diluted further 1:1 with de-ionized water. The average of each time was used for calculations.
2.12 Sample preparation for aging experiments of a battery electrolyte at several temperatures
For the investigation of fluoride formation depending on the temperature, 2 mL of commercially available LP30 was filled in aluminum vials closed with a screw cap. Groups of four samples were stored at −16 °C, 6 °C, 20 °C, 40 °C, 60 °C and 80 °C, respectively, for the duration of 47 days. For analysis by means of FSE, all samples with the exception of the samples stored at −16 °C, 6 °C and 20 °C were diluted 1:100 with de-ionized water. The samples stored at −16 °C, 6 °C and 20 °C were diluted 1:10 and analyzed in this dilution by FSE as well as by IC due to the expected very low fluoride concentrations. For analysis via IC, all remaining samples were subsequently diluted 1:1 with de-ionized water. All samples were analyzed threefold by IC as well as by FSE. The average of each temperature was used for calculations.
2.13 External calibration for analysis by means of IC and FSE
For external calibration, ten standard solutions of the following fluoride concentrations were diluted in de-ionized water from the fluoride stock solution (1 g L−1): blank, 0. 5 ppm, 1 ppm, 2 ppm, 5 ppm, 10 ppm, 15 ppm, 25 ppm, 50 ppm and 100 ppm. The external calibration was performed daily before sample analyses by FSE and IC.3 Results & discussion
3.1 Trueness and recovery rates
:DMC, EC:DEC and EC:EMC, each in a ratio of (50:50, wt%), were spiked to an aqueous solution containing 10 ppm fluoride. All samples were examined by a three-fold determination procedure. For external calibration, electrolyte-free calibration solutions were applied to make the influence of the electrolytes apparent. An electrolyte-free solution (0; 0%) containing 10 ppm fluoride was analyzed as reference. The recovery rates of fluoride observed for different contents of carbonates are between 95% and 109% as summarized in Table 1.
:DMC (50:50, wt%), EC:DEC (50:50, wt%) and EC:EMC (50:50, wt%)
| Added carbonate [μL, %] | Recovery rate of 10 ppm fluoride [%] | ||
|---|---|---|---|
| EC:DMC |
EC:DEC |
EC:EMC |
|
| 0; 0% | 101 | 96 | 96 |
| 10; 0.1% | 101 | 95 | 100 |
| 50; 0.5% | 101 | 95 | 100 |
| 100; 1% | 102 | 96 | 101 |
| 500; 5% | 106 | 98 | 103 |
| 1000; 10% | 109 | 102 | 108 |
A slightly positive trend becomes visible: with the increasing amount of the electrolyte, increasing recovery rates resulted. However, up to the 500 μL (5%) amount of the electrolyte the recovery rates (95–106%) are in good accordance to recovery rates determined by repeated injections of fluoride standard solution containing no electrolyte (10 ppm: 97–103% and 70 ppm: 98–103%; Chapter 3.1.1). Therefore, samples containing 10–500 μL (0.1–5%) of the electrolyte, which corresponds to the volume of the electrolyte that is typically recovered from cycled cells, can be analyzed by means of the fluoride selective electrode method without any compensation of matrix effects.
| Expected concentration [ppm] | Determined concentration [ppm] | Expected differences [ppm] | Determined differences [ppm] |
|---|---|---|---|
| 0 | 2.1 | ||
| 20 | 21.8 | 20 − 0 = 20 | 21.8 − 2.1 = 19.7 |
| 50 | 52.3 | 50 − 20 = 30 | 52.3 − 21.8 = 30.5 |
| 70 | 71.8 | 70 − 50 = 20 | 71.8 − 52.3 = 19.5 |
| 90 | 93.1 | 90 − 70 = 20 | 93.1 − 71.8 = 21.3 |
The fluoride concentration in a freshly opened bottle of LP30 has already been determined to be 2.1 ppm. The initial amount was determined in all samples beyond the spiked amount and led to increased fluoride concentrations. The calculated theoretical concentrations were recovered plus about 2 ppm. For example, after spiking 20 ppm fluoride, a total content of 21.8 ppm was determined. Differences in fluoride concentrations between the samples of about 20 ppm and 30 ppm, caused by spiked fluoride concentrations were observed as expected. Differences between samples could be recovered accurately throughout the concentration range.
3.2 Precision
The precision, i.e., the accuracy of the measurement, specifies scattering of analysis values due to random errors. The scattering of analysis values can be expressed as the variation coefficient of repeated analysis. Variation in the analysis value due to the analysis instrument itself is referred to as ‘repeatability of the instrument’. It was specified by a sixfold analysis of the same fluoride containing standard solutions. Here, the calibration curve consisting of ten concentration points between 1 ppm and 100 ppm was used to observe variations over the whole calibration range.The intra-day and inter-day precision, moreover, proved process stability, repeatability and comparability of analysis pointing to a high robustness of the method.
3.3 Selectivity
:DEC mixture in Fig. 1.
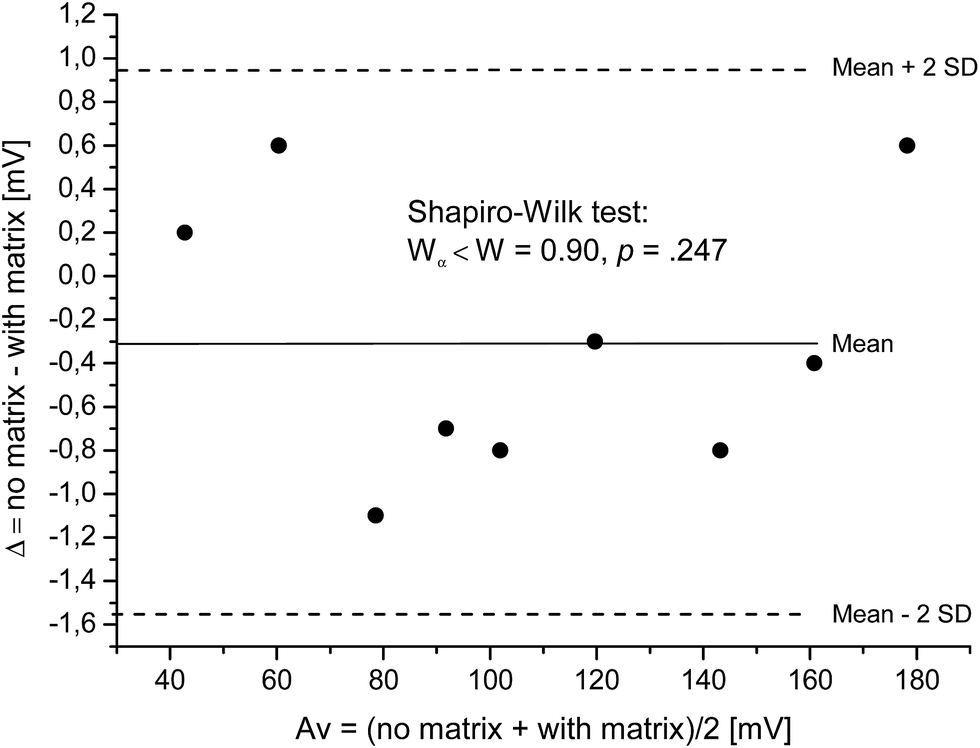 | ||
| Fig. 1 Bland–Altman-plot. Difference vs. average of detected voltages for calibration curves representing “no matrix” and “with matrix”; with 95% limits of agreement (broken lines) and regression line (y = −0.287 to 1.191 × 10−4) overlying with the mean (−0.30), both solid lines. The Shapiro–Wilk test confirms the normal distribution of data values. | ||
The differences follow a normal distribution as confirmed by the Shapiro–Wilk test, Wα=0.05 = 0.829 < W = 0.899 and p = 0.247. The differences are distributed normally around the mean value. Thus, the mean value of the differences (−0.30 mV) gives the constant bias between the two methods “no matrix” and “with matrix”. The measured voltage does slightly and constantly increase over the whole concentration range, if standard solutions contain matrix compounds.
Due to the normal distribution, 95% of differences lie between the mean difference ![[d with combining macron]](https://www.rsc.org/images/entities/i_char_0064_0304.gif) and twice the standard deviation of the differences s. Here, differences are expected between ± 2s resulting in [−1.54 and 0.94]. These values are referred to as “limits of agreement” and are estimates of values applying to the whole population. To estimate the precision of the limits of agreement, confidence intervals (CI) for both limits are calculated. The 95%-CI for the lower limit is [−2.02 and −1.05] and for the upper limit [0.45 and 1.42]. Even though the number of samples was low and, thus, CI are wide, most data points are between the higher limit of the lower limit of agreement [−1.05] as well as the lower limit of the higher limit of agreement [0.45].
and twice the standard deviation of the differences s. Here, differences are expected between ± 2s resulting in [−1.54 and 0.94]. These values are referred to as “limits of agreement” and are estimates of values applying to the whole population. To estimate the precision of the limits of agreement, confidence intervals (CI) for both limits are calculated. The 95%-CI for the lower limit is [−2.02 and −1.05] and for the upper limit [0.45 and 1.42]. Even though the number of samples was low and, thus, CI are wide, most data points are between the higher limit of the lower limit of agreement [−1.05] as well as the lower limit of the higher limit of agreement [0.45].
The constant bias of −0.30 mV corresponds to a deviation of 0.23% at 1 ppm and 1.79% at 100 ppm. These variations are smaller than the variation due to random errors (intra-day: 2.5%; inter-day: 3.8%) for precision variation and will not be considered any further. Corresponding analysis were performed with EC:DMC and EC:EMC resulting in comparable data.
3.4 Linearity and range
A look on the calibration curve providing the voltage in mV plotted against log(c) in log(ppm) yielding a regression coefficient of R = 0.999 confirms that a linear relation between the electrode response and the fluoride concentration is given. However, the examination of linearity is performed for the sake of completeness. As data of the analysis of precision (Chapter 3.2) show homogeneous variances (homoscedasticity), the criteria for linear regression are fulfilled. The test of linearity is done by comparing the linear calibration function with the calibration function of the 2nd degree. The reduction of the residual variance resulting due to the regression model of higher order is checked for statistical significance by an F-test, F(14, 13) = 20810 and p < 0.01. As expected, data values fit perfectly to a linear model. The comparison of linear calibration functions with calibration functions of the 2nd degree has been proposed by Mandel in the 1930s and is known as Mandel's fitting test in the field of method validation in analytical chemistry.24 The range for the determination of fluoride here is 0.5–100 ppm with a Nernst slope of −58.5 mV at 20 °C in ambient air.
3.5 Limits of detection and quantification
The response of the LaF3 electrode to the concentration of fluoride ions was reported in the literature several times to start at 10−6 mol L−1 upwards.1,33 The working range here was starting at 2.6 × 10−2 mol L−1 = 0.5 ppm and, thus, the lowest concentration is clearly above the limits of detection and quantification and was not determined here.3.6 Analysis of a battery electrolyte thermally aged at 80 °C
A central issue for the understanding of degradation processes in LIBs is the understanding and monitoring of fluoride formation from the conducting salt LiPF6. Fluoride formation is influenced by temperature and/or by the aging process.1,6,14After validation of the FSE method in the context of battery electrolytes, the newly evaluated FSE method is compared to conventional fluoride analysis by means of IC. The commercially available LP30 battery electrolyte was aged at 80 °C and analyzed at various times with a maximum of 47 days. After dilution the samples were analyzed by means of FSE and IC. The resulting fluoride contents are depicted in Fig. 2.
 | ||
| Fig. 2 Fluoride contents in a LP30 electrolyte aged at 80 °C for 1 day up to 47 days. Samples were analyzed by means of IC and FSE. The fluoride concentrations determined by IC are depicted in blue and the fluoride concentrations analyzed by FSE are shown in orange. Simple standard deviations are given as error bars in black. Concentrations observed by FSE are lower than the concentrations observed by IC analysis. Apparently false-high values in IC are due to the co-elution of fluoride and organophosphates. Results of FSE remain unaffected by the formation of aging products such as organophosphates. | ||
Data from FSE as well as IC analysis show increased fluoride concentrations with passing time from the first day to the 47th day. In FSE analysis, fluoride concentrations increased from 224.6 ppm to 2413.1 ppm with standard deviations (SD) between 35.1 ppm and 295.0 ppm, which corresponds to SD = 3–19%. In IC analysis, fluoride concentrations increased from 238.0 ppm to 2639.2 ppm with approximately SD = 3–17%. The standard deviations varied between samples. For most samples, SDs are in a similar range except for the 9th, the 14th, 20th and 47th day exhibiting a higher SD. In total, SDs of both techniques overlap for the most days. Both techniques led to fluoride concentrations with real values in similar ranges.
Even when a similar trend in concentrations was observed, it is noteworthy that concentrations observed by FSE are always lower than the concentrations observed by IC analysis. Fig. 3 shows the ion chromatograms of LP30 analyzed after the first day in the oven at 80 °C and LP30 after the 28th day at 80 °C. While after the 1st day only one signal according to fluoride is visible, the chromatogram shows two signals after the 28th day. The second signal and the apparent peak shoulder of the fluoride peak can be assigned to organophosphates, a group of electrolyte degradation products that could not be baseline-separated using all conceivable possibilities. Co-eluting the organophosphate compounds does change the results towards “dust” findings which explains the higher “fluoride” concentrations found in IC analysis. False-high values increase with the number of days. The detected difference in concentration between the FSE and IC analysis Δc (F−) was 13.4 ppm for the first day with low degradation and is successively increased until the 47th day with high degradation, revealing a difference of 226.0 ppm. Fluoride analysis by IC does not only determine the analyte of interest and, thus, cannot be considered as a valid technique for this purpose.
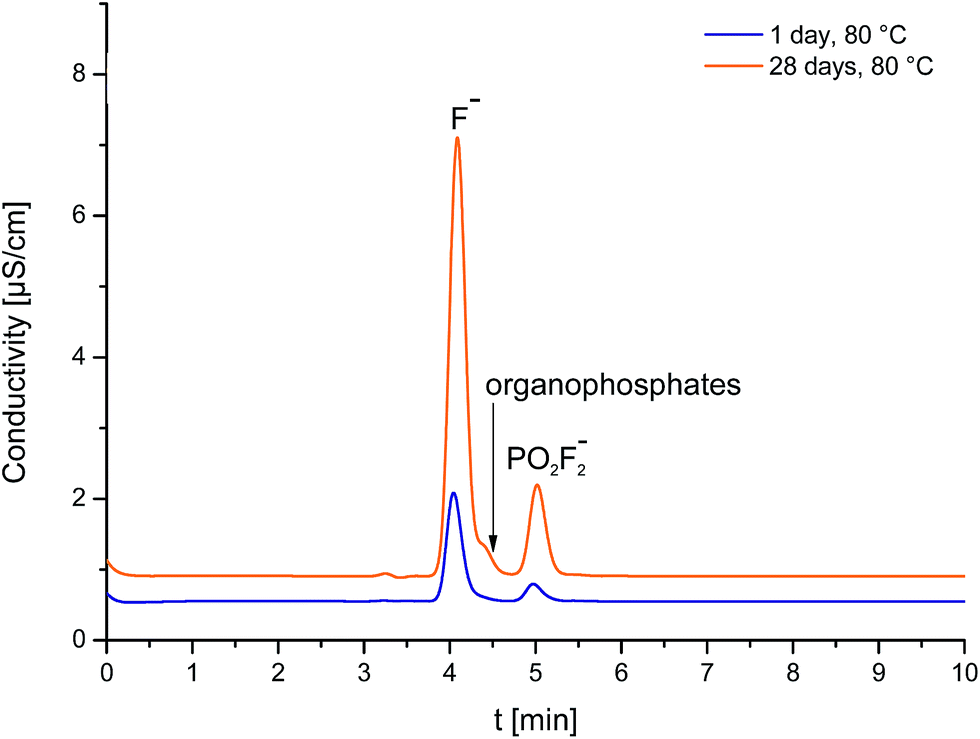 | ||
| Fig. 3 Ion chromatogram of the aged LP30 electrolyte after one day (blue line) and after 28 days (orange line) at 80 °C. After 28 days of aging, organophosphates were formed and co-eluted with the fluoride peak resulting in an overestimation of fluoride contents. | ||
The observed co-elution of fluoride and organophosphates is in good accordance with previous results reported from our working group.14,15 Kraft et al. (2015) identified organophosphates by means of IC-ESI-MS and discussed the influence of the co-eluting organophosphates.15 They assumed that the influence of the co-elution might be negligible if the content of ionic organophosphates is very low. For samples of high degradation degree, co-eluting compounds were expected to lead in false-high values.14,15 However, data shown here confirmed their assumptions: analysis of battery electrolytes by means of FSE prevents false-high concentrations especially in strongly aged samples.
The influence of water was proven to have no influence on the degradation of NaPF6 and the formation of fluoride as shown elsewhere.12 Compared to LiPF6, the dissociation of NaPF6 is advantageous due to entropic effects. Nevertheless, an aqueous solution (1% EC/DMC, v/v) of NaPF6 containing 22.5 ppm Na/P and 22.5–135 ppm fluoride, respectively, were analyzed repetitively for four hours. About 20 minutes after NaPF6 was dissolved, a fluoride concentration of 0.3 ppm was detected by means of FSE. After four hours, the fluoride concentration increased marginally to 0.7 ppm and, thus, the increase in fluoride concentration due to hydrolysis of NaPF6 is negligible. The sample preparation in water was proven to have no influence on detected fluoride concentrations.
To investigate the influence of different temperatures, commercially available LP30 was stored at −16 °C, 6 °C, 20 °C, 40 °C, 60 °C and 80 °C for 47 days. The samples were analyzed again by means of the FSE and IC techniques. In Fig. 4 the fluoride contents are shown as a comparison of both methods. While detected fluoride concentrations were low when the sample was kept at low temperatures (−16 °C: 42.3 ppm/45.1 ppm (FSE/IC), 6 °C: 94.1 ppm/99.8 ppm; 20 °C: 115.3 ppm/114.9 ppm), fluoride concentrations increased when the sample was kept at higher temperatures (40 °C: 484.7 ppm/538.5 ppm (FSE/IC); 60 °C: 1497.8 ppm/1795.6 ppm; 80 °C: 3032.6 ppm/3321.1 ppm).
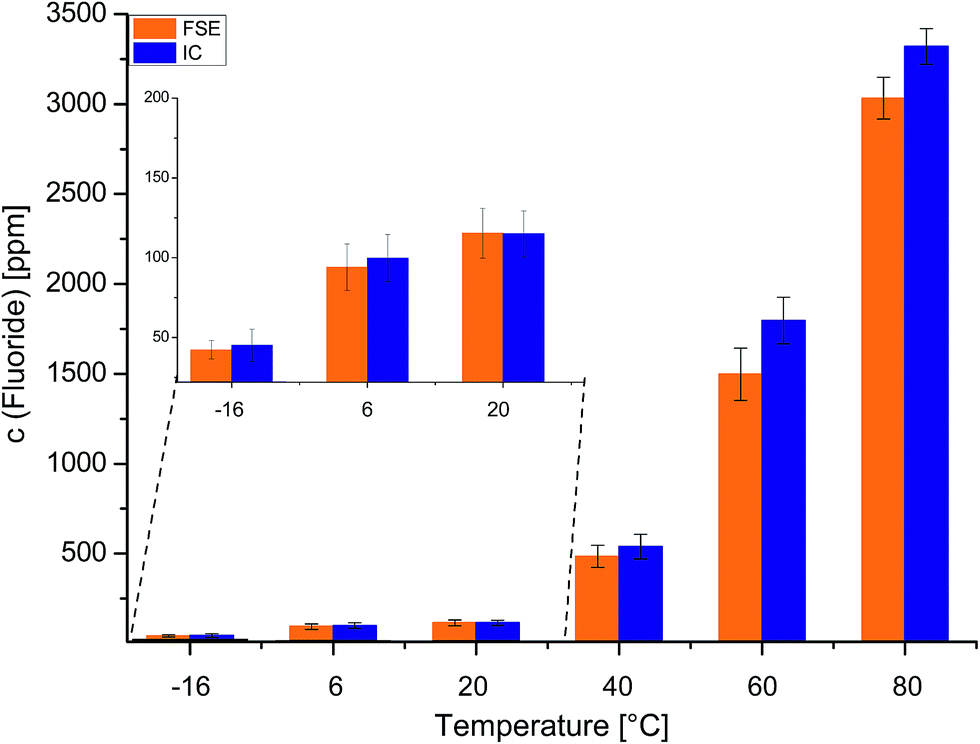 | ||
| Fig. 4 Fluoride contents in the LP30 aged electrolyte at −16 °C, 6 °C, 20 °C, 40 °C, 60 °C and 80 °C, respectively, for 47 days. The samples were analyzed by means of IC and FSE. Concentrations observed by FSE are lower than concentrations observed by IC analysis. | ||
With the exception of the sample stored at 20 °C, fluoride concentrations were higher when samples were analyzed by IC. The differences in detected fluoride concentrations Δc (F−) of FSE and IC analysis yielded again higher differences for samples that were strongly aged. The more a sample was aged and the more LP30 electrolyte degradation products were present, the more results differed for FSE and IC analysis. Especially for strongly aged electrolyte samples the FSE as the fluoride selective method was able to provide a precise analysis, free from interferences with matrix compounds.
4 Conclusions
For the first time, a fluoride selective electrode (FSE) has been applied for the analysis of fluoride in organic carbonates (EC:DEC, EC:EMC, EC:DEC, each (50:50, wt%)) as compounds of LiPF6-based LIBs electrolytes as well as in LP30 (1 M LiPF6 in EC:DMC) as a representative of commercially available battery electrolytes. Method validation of FSE for the analysis of fluoride in LiPF6-based LIB electrolytes was performed: experimentally achieved concentrations were proven not to differ from nominal concentrations within a 95% confidence level. Samples containing 0.1–5% of the electrolyte can be analyzed by FSE without further compensation of matrix effects. Defined fluoride concentrations spiked to an electrolyte from a freshly opened bottle were recovered as expected over the whole concentration range (0.5–100 ppm). Scattering of analysis values in repeated analysis of samples were proven to be caused by random errors (0.2–2.5% in sixfold analysis on one day; 0.5–3.8% in sixfold analysis for a period of six days consecutively) and are not based on any systematic effects.
Samples containing increasing fluoride concentrations and a typical amount of the electrolyte recovered back from cycled battery cells yielding a 1% electrolyte solution showed no substantial differences in detected voltages to samples containing the fluoride and no electrolyte. The application of TISAB IV keeps the pH constant avoiding cross-sensitivity with hydroxide ions and fluoride loss due to the formation of HF and HF2−. The constant ionic strength allows drawing the desired conclusions from the analyte concentration instead of from the analyte ionic activity. Moreover, reactions of fluoride with metal ions, such as with Al3+, that form complexes, which make analysis more difficult, are avoided. The calibration curve (0.5–100 ppm) yielded a regression coefficient of R = 0.999 and a Nernst slope of −58.5 mV at 20 °C. LP30 samples were thermally aged at 80 °C for various durations and for 47 days at various temperatures. Fluoride contents were analyzed by means of IC combined with a conductivity detector and by means of FSE. The comparison of the observed fluoride contents yielded an overestimation of fluoride analyzed by IC in strongly aged electrolytes due to co-elution of aging products with the fluoride peak. The FSE method worked free from interferences, selective and specific and thus could be introduced here as a robust tool for fluoride analysis in the complex matrix of battery electrolytes.
Acknowledgements
The authors wish to thank the German Federal Ministry of Education and Research (BMBF) for funding this work in the project ‘Elektrolytlabor-4E’ (03X4632).References
- M. S. Frant and J. W. Ross, Science, 1966, 154(3756), 1553–1555 CAS.
- A. Barrè, B. Deguilhem, S. Grolleau, M. Gérad, F. Suard and D. Riu, J. Power Sources, 2013, 241, 680–689 CrossRef.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- J. Vetter, P. Novák, M. R. Wagner, C. Veit, K. C. Möller, J. O. Besenhard, M. Winter, M. Wohlfahrt-Mehrens, C. Vogler and A. Hammouche, J. Power Sources, 2005, 147, 269–281 CrossRef CAS.
- C. L. Campion, W. Li and B. L. Lucht, J. Electrochem. Soc., 2005, 152(12), A2327–A2334 CrossRef CAS.
- H. Yang, G. V. Zhuang and P. N. Ross Jr, J. Power Sources, 2006, 161, 573–579 CrossRef CAS.
- C. L. Campion, W. Li, W. B. Euler, B. L. Lucht, B. Ravdel, J. F. DiCarlo, R. Gitzendanner and K. M. Abraham, Electrochem. Solid-State Lett., 2004, 7(7), A194–A197 CrossRef CAS.
- P. Handel, G. Fauler, K. Kapper, M. Schmuck, C. Stangl, R. Fischer, F. Uhlig and S. Koller, J. Power Sources, 2014, 267, 255–259 CrossRef CAS.
- T. Kawamura, A. Kimura, M. Egashira, S. Okada and J.-I. Yamaki, J. Power Sources, 2002, 104, 260–264 CrossRef CAS.
- S. S. Zhang, J. Power Sources, 2006, 162, 1379–1394 CrossRef CAS.
- S.-T. Myung, Y. Hitoshi and Y.-K. Sun, J. Mater. Chem., 2011, 21, 9891–9911 RSC.
- L. Terborg, S. Nowak, S. Passerini, M. Winter, U. Karst, P. R. Haddad and P. N. Nesterenko, Anal. Chim. Acta, 2012, 714, 121–126 CrossRef CAS PubMed.
- L. Terborg, S. Nowak, F. Blaske, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2013, 242, 832–837 CrossRef CAS.
- V. Kraft, M. Grützke, W. Weber, M. Winter and S. Nowak, J. Chromatogr. A, 2014, 1354, 92–100 CrossRef CAS PubMed.
- V. Kraft, W. Weber, M. Grützke, M. Winter and S. Nowak, RSC Adv, 2015, 5, 80150–80157 RSC.
- V. Kraft, M. Grützke, W. Weber, J. Menzel, S. Wiemers-Meyer, M. Winter and S. Nowak, J. Chromatogr. A, 2015, 1409, 201–209 CrossRef CAS PubMed.
- G. Gachot, S. Grugeon, M. Armand, S. Pilard, P. Guenot, J.-M. Tarascon and S. Laruelle, J. Power Sources, 2008, 178, 409–421 CrossRef CAS.
- V. Kraft, W. Weber, B. Streipert, R. Wagner, C. Schulz, M. Winter and S. Nowak, RSC Adv., 2016, 6, 8–17 RSC.
- G. Gachot, P. Ribière, D. Mathiron, S. Grugeon, M. Armand, J.-B. Leriche, S. Pilard and S. Laruelle, Anal. Chem., 2011, 83, 478–485 CrossRef CAS PubMed.
- G. Gachot, S. Grugeon, G. G. Eshetu, D. Mathiron, P. Ribière, M. Armand and S. Laruelle, Electrochim. Acta, 2012, 83, 402–409 CrossRef CAS.
- L. Terborg, S. Weber, S. Passerini, M. Winter, U. Karst and S. Nowak, J. Power Sources, 2014, 245, 836–840 CrossRef CAS.
- W. Weber, V. Kraft, M. Grützke, R. Wagner, M. Winter and S. Nowak, J. Chromatogr. A, 2015, 1394, 128–136 CrossRef CAS PubMed.
- J. Koryta, Anal. Chim. Acta, 1984, 159, 1–46 CrossRef CAS.
- S. Kromidas, Handbuch Validierung in der Analytik, Wiley-VCH Verlag & Co. KGaA, Weinheim, Deutschland, 2011 Search PubMed.
- WTW – Die ionenselektive Messtechnik: https://www.omnilab.de/tl_files/Omnilab_2011/Downloads/Infomaterial_Anwenderberichte/Elektrochemie/ISE_D.pdf, accessed May 2016.
- Umweltbundesamt – Qualitätssicherungsstelle im Bund/Länder-Messprogramm Nord- und Ostsee (BLMP), Leitlinie zur Methodenvalidierung 01/05, J. Wellmitz and M. Gluschke; https://www.umweltbundesamt.de/sites/default/files/medien/publikation/long/2832.pdf, accessed May 2016.
- E. Lindner and B. D. Pendley, Anal. Chim. Acta, 2013, 762, 1–13 CrossRef CAS PubMed.
- D. G. Altman and J. M. Bland, The Statistician, 1983, 32, 307–317 CrossRef.
- J. M. Bland and D. G. Altman, The Lancet, 1986, 30, 307–310 CrossRef.
- J. M. Bland and D. G. Altman, The Lancet, 1995, 346(8982), 1085–1087 CrossRef CAS.
- Metrohm – Application Bulletin Nr. 82/3 d-Bestimmung von Fluorid mit der ionenselectiven Elelektrde, https://www.google.de/url?sa=t%26rct=j%26q=%26esrc=s%26source=web%26cd=1%26cad=rja%26uact=8%26ved=0ahUKEwinqvHJltfMAhXHORQKHSMyCUkQFggdMAA%26url=http%3A%2F%2Fpartners.metrohm.com%2FGetDocument%3Faction%3Dget_dms_document%26docid%3D692912%26usg=AFQjCNH2B9Q4sCUI8pXaYNmhWspOlcyJ1g%26sig2=lBOEzdoKhOiWv5sEpX_ZSQ, accessed May 2016.
- M. S. Frant and J. W. Ross, Anal. Chem., 1968, 40(7), 1169–1171 CrossRef CAS PubMed.
- J. E. Harwood, Water Res., 1969, 3, 273–280 CrossRef CAS.
- E. Krämer, T. Schedlbauer, B. Hoffmann, L. Terborg, S. Nowak, H. J. Gores, S. Passerini and M. Winter, J. Electrochem. Soc., 2013, 160(2), A356–A360 CrossRef.
- IC Application Work: AW DE8-0768-122010: Determination of hexafluorophosphate and lithium in battery electrolyte.
| This journal is © The Royal Society of Chemistry 2016 |