
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bioparticles coated with an ionic liquid for the pre-concentration of rare earth elements from microwave-digested tea samples and the subsequent quantification by ETV-ICP-OES†
Sara
Hosseinzadegan‡
a,
Winfried
Nischkauer‡
 a,
Katharina
Bica
b and
Andreas
Limbeck
*a
a,
Katharina
Bica
b and
Andreas
Limbeck
*a
aInstitute of Chemical Technologies and Analytics, TU Wien, Getreidemarkt 9/164-IAC, 1060 Vienna, Austria. E-mail: Andreas.Limbeck@tuwien.ac.at
bInstitute of Applied Synthetic Chemistry, TU Wien, Getreidemarkt 9, 1060 Vienna, Austria
First published on 10th October 2016
Abstract
An analytical procedure for straight-forward quantification of rare earth elements (REEs) in tea was developed. The method consists of three steps: first, dry tea powder is converted into an aqueous sample solution using a microwave-assisted digestion procedure. Then, the REEs are retained on newly designed sorbent particles and are thus effectively extracted from the sample digest. Then, the REE-loaded sorbent material is introduced into the furnace of an electro-thermal vaporization (ETV) unit. There, the core of the particles is pyrolyzed and removed in a first temperature step. Then, the REE analytes are swiftly evaporated at high temperatures, and subsequently analyzed by ICP-OES. The advantages of the method proposed here are as follows: extraction of REEs is accomplished via “dispersed particle extraction”; this avoids typical shortcomings of conventional solid-phase extraction. The analysis via ETV allows separation of the sorbent particles from the analytes in time. Thus, detrimental effects of plasma-loading are circumvented. The method was shown to provide satisfactory detection limits of typically 50 ng g−1 in the dry tea samples (3 s criterion). The method was validated by means of spike recovery experiments, and applied to the analysis of ten different tea samples from China, Japan and India, as sold in European markets.
1. Introduction
Rare earth elements (REEs) are a group of elements which include fourteen lanthanides plus scandium and yttrium. Although they are named rare, their abundance in the environment is comparable to that of other heavy elements such as Cu, Ni, Co and Zn. Recently, there has been increased tendency to apply REEs in several fields which include agriculture,1 catalysts2 and cell phones.3 The increased industrial use of REEs results in pollution of the environment, followed by accumulation of these elements in the biosphere and this accumulation causes an increased risk for human health. Based on reports, accumulation of REEs4 could cause serious problems in different organs of the human body5 especially in bones6 and the nervous system.7 During plant growth, toxic elements present in soil or water can be taken up by plants together with essential elements. Tea made from Camellia sinensis leaves, both black and green, is a common and popular non-alcoholic drink all around the world. Accordingly, it seems that the level of REEs should be monitored in tea. Until now, data about REE levels in tea are very sparse, and there are only a few studies published8–11 that indicate that the level of REEs especially La, Ce, Y, Pr and Nd in tea is considerable.For analysis of trace elements in environmental samples, the most appropriate techniques include inductively coupled plasma mass spectrometry (ICP-MS), inductively coupled plasma optical emission spectrometry (ICP-OES) and electro-thermal atomic absorption spectrometry (ET-AAS).12 However, accurate analysis of REEs in plant samples is challenging – REE levels in the range of a few ng g−1 up to some 100 ng g−1 combined with numerous interferents in the most sensitive analytical techniques are the major difficulties. To overcome limitations in sensitivity and/or selectivity (e.g. due to spectral interferences), the use of sample pre-treatment procedures for pre-concentration and quantitative separation of REEs from interfering sample constituents is recommended. Although several enrichment processes have been reported in the literature, solid phase extraction (SPE) has received the most attention due to easy automation and being inexpensive. In SPE, the analyte-containing matrix is passed through a column of an adsorbent on which the analytes of interest are retained. After a washing step to remove the remaining matrix, the analytes are eluted with an appropriate solvent. Thus, besides analyte enrichment, an effective removal of the potentially interfering matrix can also be achieved. Nevertheless, this way of enrichment has some disadvantages including time demand, column aging and large sample and solvent consumption.13
Regarding the aforementioned disadvantages of SPE, our group has recently introduced an improved approach called dispersed particle extraction (DPE). In this approach, adsorbent particles with surface modified functionalities are suspended in a liquid sample. Targeted analytes are trapped by the particles while the matrix remains in solution. After centrifugation, the supernatant solution is discarded, the sedimented particles are diluted with acid and finally the suspension is measured by a slurry ICP-OES procedure.14 To overcome the main disadvantage of slurry analysis (potential nebulizer clogging), the sorbent particles can also be dissolved with the use of mineral acids, followed by conventional liquid ICP-MS analysis of the derived solution.15,16 However, the dissolved sorbent material still increases the plasma load during ICP-MS measurement, which could affect atomization, excitation and ionization of the target analytes in the plasma.
In this work, an innovative approach is presented, which is based on enrichment of REEs using DPE, followed by sensitive measurement of the analyte-containing sorbent particles by means of electrothermal vaporization (ETV) in combination with ICP-OES detection. ETV allows to first pyrolyze the adsorbent material, and only then to introduce the REEs into the ICP. In this way, an effective separation of the particle matrix and analytes in time is enabled, thus plasma load from vaporization of the particles could be avoided during analyte measurement. Since the thermal behavior of REEs and commonly applied silica particles14–16 is quite similar, DPE has been performed with organic sorbent particles in this work for the first time. The applied adsorbent is a natural bio-particle with high porosity, Lycopodium clavatum spore, which was covered with a layer of ionic liquid. The ionic liquid acts as the cation exchanger. These natural plant spores with an average size of 30 microns are an excellent choice as a core material because they are stable in strong acids or bases, inexpensive, easily approachable and ecological.17 Even though the particles are very porous, they do not swell in different solvents.18 The applicability of the method is demonstrated by the analysis of REE levels in different green tea samples.
2. Experimental
2.1 Reagents and standards
High purity water (18 MΩ cm) was prepared with an Easypure purification system (Thermo, USA) and was used in the whole process. Nitric acid 65% (Merck), hydrochloric acid 37% (Merck), hydrofluoric acid 40% (Merck), ethanol 99% (Chem-Lab) and acetone 99.8% (Acros Organics) purchased were of p.a. grade. Acetic acid 96% (Merck) and ammonium acetate (Merck) were used for buffering the samples prior to extraction. Single element ICP standards including La, Eu and Nd (Aristar, 1000 mg L−1), In (Merck, 1000 mg L−1) and a multi-element REE calibration standard (Agilent Technologies, multi-element calibration standard 1, 10 mg L−1, 5% (v/v) HNO3) were diluted to the concentration needed.A set of ten different green tea samples was bought at a local market in Vienna (details on each tea are summarized in Table 1). Prior to chemical digestion, the samples were dried for 24 h at 60 °C.
| Sample ID | Origin | Description |
|---|---|---|
| Tea 1 | China | Gunpowder green tea |
| Tea 2 | India | Organic green tea |
| Tea 3 | Fujian, China | Organic Tiegunyin tea |
| Tea 4 | China | Oolong tea |
| Tea 5 | China | Chun-Mee |
| Tea 6 | China | Sencha |
| Tea 7 | China | “Teekanne” green tea |
| Tea 8 | Japan | Genmaicha |
| Tea 9 | China | Fresh green tea |
| Tea 10 | China | “Grüner Kimono” |
| Tea 11 | China | Fresh green tea |
2.2 Instrumentation
ETV-ICP-OES measurements were performed using radial ICP-OES (Thermo iCAP 6500, Thermo Scientific, USA), coupled to an ETV 4000 unit (Spectral Systems, Germany). The plasma torch contained a corrosion-resistant ceramic injector tube of 1.5 mm inner diameter and was connected to the ETV via 40 cm of PTFE tubing of 4 mm inner diameter.For conventional ICP-OES analysis of liquid samples, a sample introduction kit consisting of a Babbington (V-groove) nebulizer, a glass cyclonic spray chamber with a riser tube and a torch injector tube with 2 mm inner diameter was used.
Digestion of tea leaves was performed by microwave-assisted closed vessel treatment (Multiwave 3000, MF 100 vessels, Anton Paar, Austria).
Separation of particles during the enrichment process was performed by using a Megafuge 16 Centrifuge (Thermo Scientific, Germany). The centrifuge was equipped with a FIBERLite F15-6 × 100 angular rotor. The sample pretreatment procedure was carried out in 15 mL conical polypropylene centrifugation tubes with a V-bottom (maximum tolerable acceleration: 17![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, VWR, Germany).
000 × g, VWR, Germany).
2.3 Preparation and characterization of sorbent particles
The ionic liquid used for the extraction of REEs consisted of the trihexyl(tetradecyl)phosphonium cation and the di(2-ethylhexyl) phosphate anion (see Fig. S1, ESI†). Throughout this article, the nomenclature [P66614]+[BEHPA]− will be used for the ionic liquid. Synthesis of [P66614]+[BEHPA]− was carried out at the Institute of Applied Synthesic Chemistry of TU Wien, according to the following scheme: trihexyltetradecylphosphonium chloride (5.19 g, 10 mmol), bis(2-ethylhexyl) hydrogenphosphate (3.54 g, 11 mmol) and potassium hydroxide (0.62 g, 11 mmol) were suspended in 100 mL of water:MeOH 1:1 and stirred for 2 h at room temperature. The solution was concentrated to approx. 60 mL and diluted with water. This mixture was repeatedly extracted with dichloromethane, and the combined organic layers were washed with water until no more chloride ions could be detected (checked by addition of an aqueous AgNO3 solution). The organic phase was dried over Na2SO4 and concentrated under reduced pressure. Remaining solvent traces were removed under high vacuum (0.01 mbar, 48 h) with stirring at 50 °C to yield trihexyltetradecylphosphonium bis(2-ethylhexyl)phosphonate as a clear colorless liquid in 94% yield. The derived product was characterized by NMR (see ESI†).
To make a cation exchange adsorbent, 990 mg of Lycopodium clavatum spores were added to the solution of 10 mg of ionic liquid [P66614]+[BEHPA]− in 50 mL ethanol in a 100 mL round bottom flask. The mixture was shaken smoothly on a slow-moving platform shaker for 2 hours at room temperature. Ethanol was subsequently evaporated using rotary pump vacuum and the solid product was dried overnight under high vacuum.
Particles were characterized using scanning electron microscopy (Quanta 200 MK2, FEI, USA) in the initial form and after coating with the ionic liquid. The SEM images of raw Lycopodium clavatum and the treated spores indicate no change in the morphology after treatment. As is shown in Fig. 1, the coated particles have a size of roughly 30 microns.
 | ||
| Fig. 1 SEM images of [P66614]+[BEHPA]−@Lycopodium clavatum, 10 kV acceleration voltage and 100 μm scale-bar. | ||
2.4 Microwave assisted sample digestion
For complete decomposition of tea, 0.6 g of the dried leaves were transferred to clean microwave vessels and digested in a mixture of 3 mL HNO3 (65 wt%), 2 mL HCl (37 wt%), 1 mL H2O2 (30 wt%) and 0.1 mL HF (40 wt%) under elevated pressure and temperature: 40 min heating ramp, 30 min holding time at 800 W power, a maximum temperature of 190 °C and a maximum pressure of 20 bar. The obtained clear solutions were quantitatively transferred into PFA beakers (Nalgene, USA) and evaporated to dryness on a heating block at 140 °C. The dried residues were treated for another 30 minutes with a mixture of 2 mL HNO3 (65 wt%) and 2 mL H2O2 (30 wt%) at 90 °C with closed caps. Then, the solutions were again evaporated to dryness to remove any remaining HF.19,20 The residues were dissolved with 50 μL of 0.07 M HNO3 in an ultrasonic bath (5 min) and then filled up to 35 mL with high purity water.2.5 Dispersed particle extraction procedure
A graphical illustration of the developed sample pre-treatment procedure is presented in Fig. 2. For analysis 10 mL of sample solution was pipetted into 15 mL conical tubes, and ammonium buffer was added for pH adjustment. Dry sorbent particles were added to the sample solution (step I). After a reaction time of 5 minutes, necessary to retain the REE ions on the sorbent particles, the sample solution was centrifuged for the separation of solution and analyte-containing particles. After removal of the supernatant solution (step II), the precipitated sorbent particles were re-suspended in dilute nitric acid (1% v/v) to obtain a final volume of 500 μL in the case of ETV experiments (step III). For conventional slurry ICP-OES measurement, the volume was adjusted to a final volume of 2 mL. During step III, indium was added as the internal standard with a final concentration of 6 mg L−1.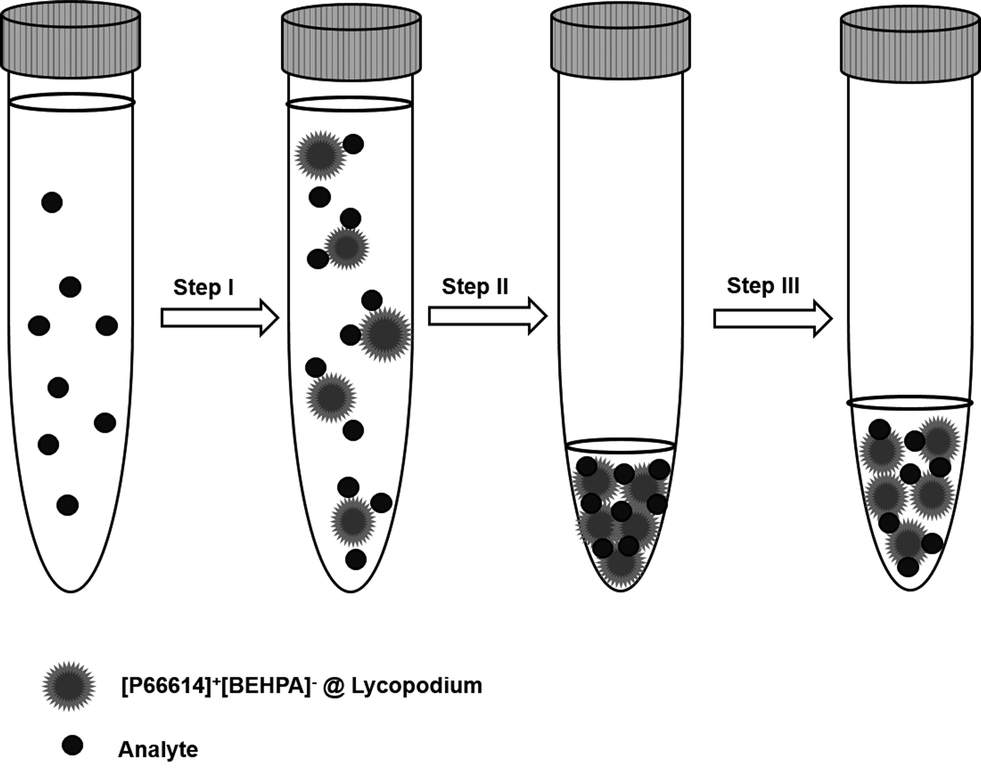 | ||
| Fig. 2 Illustration of the dispersed particle extraction (DPE) enrichment process. | ||
2.6 Sample analysis
During method development, the slurry obtained after DPE extraction was analyzed via slurry nebulization ICP-OES. Measurement was done with the use of a Babington nebulizer with a sample flow rate of 0.6 mL min−1. Background-corrected signals were recorded in 5-fold replicates using an analysis time of 5 s per replicate.For the final ETV-ICP-OES method, 30 μL of the suspension obtained via DPE was transferred into a cleaned graphite boat. Before measurement, the solvent was evaporated by gentle heating with an infrared lamp. Then, the boat was placed in the ETV oven and heated following a defined program for 90 s. The boat was first heated to 400 °C by a slow ramp in 40 s and was then held at that temperature for 30 s. Then, it was swiftly heated up to 2150 °C and was held for 20 s at that temperature, followed by a cooling phase. The instrumental parameters of the applied analytical procedures are summarized in Table 2. Since the resolution of the time-axis is inversely proportional to the number of observed emission lines, only one emission line per element was selected. With those settings, “Timescan” mode was used to record the derived transient signals by collecting data points every 0.4 s.
| ETV sample introduction | Liquid sample introduction | ||
|---|---|---|---|
| Plasma power | W | 1200 | 1200 |
| Radial observation height | mm | 11 | 11 |
| Plasma gas (Ar) | L min−1 | 12 | 12 |
| Nebulizer gas (Ar) | L min−1 | — | 0.9 |
| Auxiliary gas (Ar) | L min−1 | 0.8 | 0.8 |
| Freon R12 modifier | mL min−1 | 10 | — |
| Bypass gas (Ar) | L min−1 | 0.38 | — |
| Carrier gas (Ar) | L min−1 | 0.14 | — |
| Element | λ (nm) | Element | λ (nm) |
|---|---|---|---|
| Ce | 415.197 | Nd | 430.358 |
| Dy | 364.540 | Pr | 390.844 |
| Er | 369.265 | Sc | 361.384 |
| Eu | 381.967 | Sm | 360.949 |
| Gd | 364.619 | Tb | 350.917 |
| Ho | 339.898 | Tm | 342.508 |
| La | 333.749 | Y | 371.030 |
| Lu | 261.542 | Yb | 369.419 |
| In* | 325.609 | * Indium was used as the internal standard | |
3. Results and discussion
3.1 Optimization of the extraction process
To optimize the DPE procedure for REE retention on [P66614]+[BEHPA]−@Lycopodium clavatum, several experiments were performed to find optimal conditions in terms of the sample pH, amount of sorbent material, and reaction time. These preliminary experiments were done with solutions containing 100 μg L−1 of only three elements (Eu, La and Nd). Measurement of derived sample slurries was performed using conventional liquid ICP-OES analysis.As in most cation-exchange procedures, sample pH is one of the most important factors. In preliminary experiments, the pH range 2–6 was tested by adjusting the pH with different concentrations of ammonium acetate buffer, and the maximum retention for REEs was obtained for the sample solution of pH = 5.
In contrast to column-based SPE approaches, the sorbent is directly added to the analyte solution in the DPE procedure applied here. Therefore, optimization should be done to ensure that enough sorbent material is available for retaining the target elements – a prerequisite for reproducible analysis. On the other hand, an excess amount of particles requires a slower heating ramp during ETV analysis, in order to avoid extinguishing the ICP due to the sudden formation of huge amounts of CO2. Therefore, the addition of 1, 2.5, 5 and 10 mg of the particles to 10 mL sample solution was examined and the results showed that 5 mg adsorbent material was sufficient to obtain around 80% ± 3% recovery for Eu, La and Nd.
Another parameter affecting the extraction process is the reaction time. To find the optimum extraction time, 10 mL samples of the 100 μg L−1 analyte with 5 mg particles and pH 5 were shaken smoothly by hand for 2, 5, and 10 min. Accordingly, 80% recovery for Eu, La and Nd can be obtained after 5 min reaction time and a longer time does not improve the analyte retention. It should be mentioned that ultrasonic agitation has a negative effect on the recovery because it probably leads to the release of the ionic liquid from the surface of the Lycopodium particles.
Summing up, the ideal conditions for REE extraction (investigated for Eu, La and Nd) are: pH = 5, amount of adsorbent particles = 5 mg, and extraction time = 5 min.
3.2 ETV-ICP-OES measurement
First, the ETV program was optimized for best performance in terms of matrix pyrolysis and analyte-release. To avoid extinguishing the plasma by the rapid formation of CO2 (decomposition of the sorbent particles), a heating ramp from room temperature to 400 °C during 40 s, followed by a plateau of 30 s at this temperature was found to ensure stable plasma conditions. Lower pyrolysis temperatures resulted sometimes in incomplete sample pyrolysis, which resulted in extinguishing the plasma during the following vaporization step, whereas higher pyrolysis temperatures can result in analyte losses. The vaporization temperature was optimized in terms of quantitative analyte release, as well as in terms of graphite-tube life-time (too high temperatures result in a reduced life-time of the tube). In order to vaporize refractory elements such as Y, a gaseous modifier (Freon R12) was added to the carrier gas. Under optimized conditions, a second measurement of the same boat resulted in signals equal to the blank of an empty boat.ETV analysis of samples was carried out as follows: in the first step, the loaded boat was heated up to 400 °C in 40 s, then was held for 30 s at that temperature. This step allows the slow pyrolysis of the organic material (Lycopodium particles and ionic liquid) present in the investigated sample slurry, thereby a separation from the less volatile REEs could be accomplished. Then, by increasing the temperature to 2150 °C with the maximum heating rate, only the remaining inorganic sample constituents including the target elements are introduced into the plasma. The whole heating program requires 90 s and the analyte signals are continuously recorded with the software of the ICP-OES spectrometer. The amount of sample volume added into the graphite boats was first optimized (10–40 μL), and was found to be optimal at 30 μL. Higher sample volumes resulted in detector saturation for some elements (i.e., the highest spiked standard-addition samples). The temperature program and typical time-resolved analyte signals are illustrated in Fig. 3. The analyzed samples were a digested tea sample (Fig. 3a), as well as a method blank (all reagents, including microwave digestion, see Fig. 3b). Both samples were pre-treated with the above-mentioned DPE procedure and analyzed by means of ETV as described.
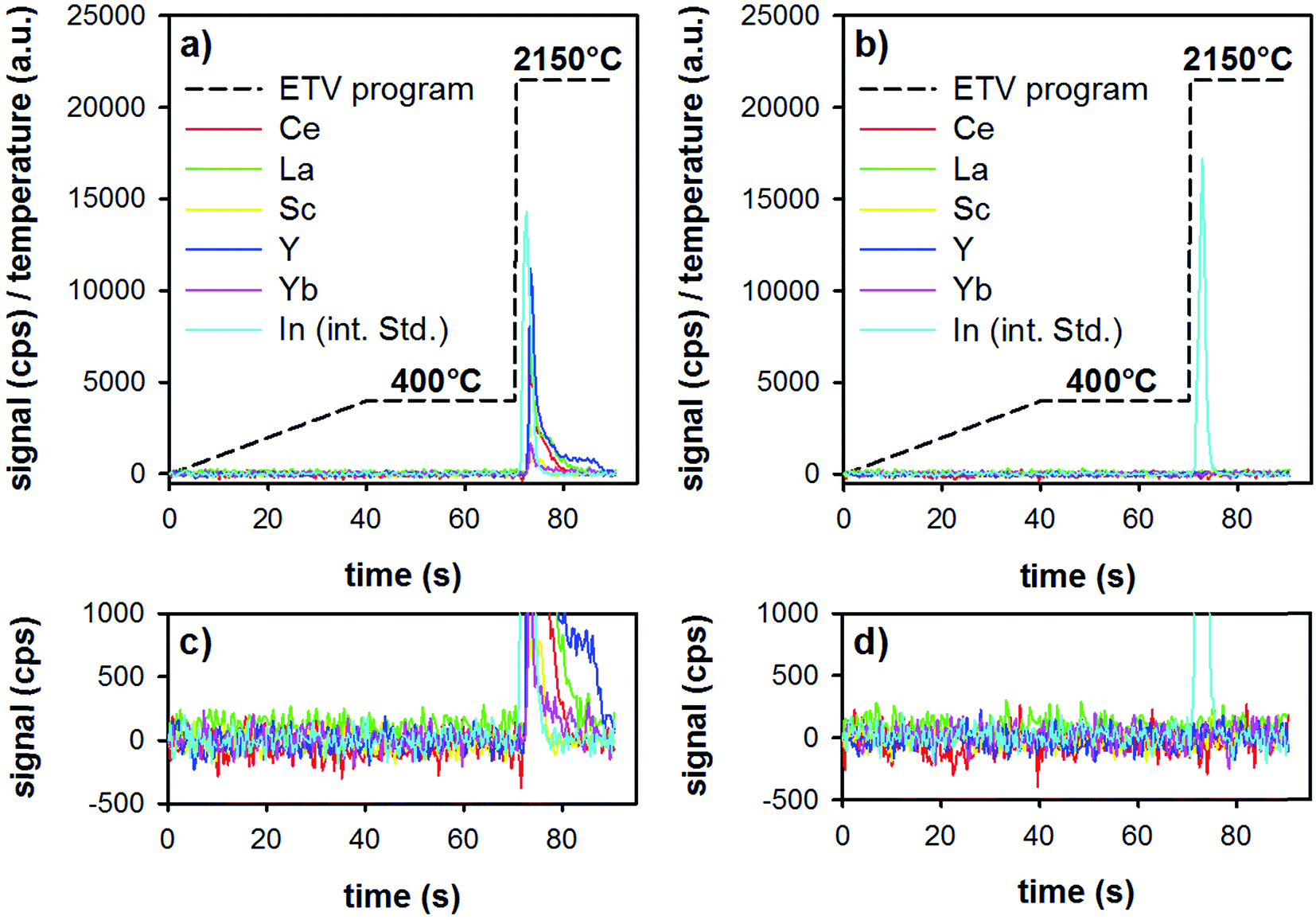 | ||
| Fig. 3 Heating program (dashed black line) used for ETV-ICP-OES analysis and the recorded signals for Ce, La, Sc, Y, Yb, and In (as the internal standard) in a tea sample pre-treated by the method presented here (a and c), and in a method blank (b and d). ((c and d) are enlarged versions of (a and b), to see the background situation more clearly). | ||
It is clear from Fig. 3 that during the first 70 s of the ETV program, there is no signal of the REEs or of the internal standard. The two insets (Fig. 3c and d) show an enlarged version of the background. This shows that the method blank is negligible, and that matrix pyrolysis does not lead to earlier release of analytes. Upon elevating the temperature to 2150 °C, analytes and the internal standard are evaporated smoothly, resulting in sharp transient signals. Repeated analysis of one boat showed that the analytes are released quantitatively already during the first firing of the ETV program.
3.3 Figures of merit
By applying the optimized parameters obtained for REE retention on [P66614]+[BEHPA]−@Lycopodium clavatum, the enrichment factor, reproducibility and the limit of detection were calculated for all investigated elements. For this purpose, an aqueous calibration row was prepared and these solutions were then treated with the discussed DPE approach. The reproducibility of measurement was determined by replicate pre-concentration and measurement (n = 4) of a sample solution containing 20 ng mL−1 of the investigated REEs, leading to relative standard deviations ranging from 4% (Nd) to 18% (Dy). Based on the conditions used for sample pre-concentration – 10 mL applied sample volume and 500 μL final volume – it is possible to achieve a theoretical enrichment factor of 20. By comparing the slopes of the calibration curves before and after pre-concentration, the actual enrichment factor was determined to be 17.8.The LOD was calculated from the standard deviation of 12 blank measurements, according to the 3 s criterion. To obtain the LOD in the non-digested tea sample, a sample intake of 0.5 g dry tea, digested and diluted to a final volume of 35 mL was considered. As can be seen in Table 3, typical detection limits are around 50 ng g−1 in the dry tea samples.
| Element | Ce | Dy | Er | Eu | Gd | Ho | La | Lu |
|---|---|---|---|---|---|---|---|---|
| LOD | 30 | 8 | 20 | 9 | 60 | 50 | 40 | 2 |
| Element | Nd | Pr | Sc | Sm | Tb | Tm | Y | Yb |
|---|---|---|---|---|---|---|---|---|
| LOD | 90 | 40 | 5 | 80 | 40 | 30 | 4 | 5 |
3.4 Influence of the sample matrix on DPE
In contrast to the synthetic aqueous standard solutions used for method development, real samples contain also the sample matrix, which might have an impact on the enrichment procedure. To study the effect of the digested tea matrix on the recovery of REEs, the slope obtained by standard addition onto digested tea (tea sample 4, see Table 1) was compared with the slope of aqueous standards. Both sets of samples were pre-concentrated with the described DPE approach and analyzed by means of ETV-ICP-OES. For each calibration curve, the standard uncertainty of the slope was also calculated. The ratio of standard addition slope to aqueous calibration slope for all the investigated elements is shown in Fig. 4, together with the corresponding expanded combined uncertainty (k = 2).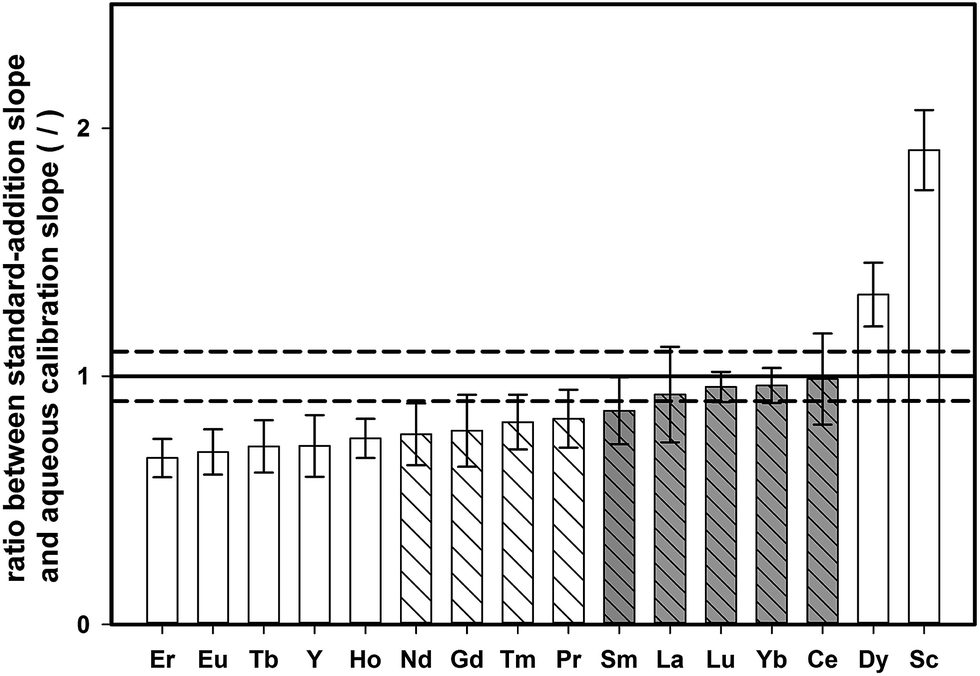 | ||
| Fig. 4 Ratio of the slopes obtained from standard-addition calibration and aqueous calibration (both in the range 0–10 μg L−1, the error corresponds to the expanded combined uncertainty of the ratio, k = 2, and the dashed lines correspond to ±10% deviation from a nominal ratio of 1). | ||
If there is no matrix effect, the ratio of both slopes is expected to be 1, which applies to Sm, La, Lu, Yb, and Ce. However, in the case of Er, Eu, Tb, Y, Ho, Nd, Gd, Tm, and Pr, the ratio is below 1, indicating that the slope of the standard addition curve was flatter than the slope of an aqueous calibration. This outcome indicates that the recovery of these elements is decreased in the presence of the tea matrix. On the other hand, Dy and Sc showed the opposite trend revealing higher signals for aqueous standards than for matrix-containing tea digests. However, when accepting a deviation within 10% of the nominal ratio of 1, most elements could be quantified with external aqueous calibration (Nd, Gd, Tm, Pr, Sm, La, Lu, Yb, and Ce).
For some elements, external calibration with aqueous standards might lead to under/overestimation of true contents. Matrix-adjusted calibration (i.e., a blank-corrected standard addition curve) was investigated as an alternative quantification approach to provide correct results for all elements investigated. Although this approach showed good results when performing spike-recovery experiments on one single tea sample (tea sample 4), with recoveries ranging from 90% to 124%, the analysis of different types of green tea showed that analyte-recovery also slightly changes due to variations in the sample matrix. Therefore, the concept of two-point standard-addition was relied upon for correct quantification in all of the following experiments, since it is less work-intensive as matrix-matched calibration for every tea sample.
3.5 Analysis of tea samples
For method validation, one tea-sample (tea-sample 11) was digested, and analyzed also using quadrupole ICP-MS in the kinetic energy discrimination mode (KED-mode). Details regarding the ICP-MS method can be found in the ESI.† For this tea-sample, most elements were below the respective detection limits, but for three elements, it was possible to obtain quantitative data by both methods. For those values, good agreement between ICP-MS results (La: 720 ± 40 ng g−1, Ce: 410 ± 20 ng g−1, Y: 240 ± 20 ng g−1) and the proposed ETV-ICP-OES procedure (La: 700 ± 80 ng g−1, Ce: 440 ± 20 ng g−1, Y: 180 ± 20 ng g−1) was obtained.Dispersed particle extraction was applied for enrichment of rare earth elements in ten different green tea samples (see Table 1), followed by measuring the pre-concentrates by means of ETV-ICP-OES. Quantification was carried out by means of standard-addition. To this end, one aliquot of the digested tea sample was pre-treated directly, whereas another aliquot was spiked with REEs and only then was subjected to the procedure. Table 4 summarizes the results of the investigated samples. For Ce, La, and Y all samples provided signals distinctly different from those of procedural blanks. The Gd and Tb contents were below the respective detection limits in all tea samples. For some elements (Dy and Eu), there was no significant variation observed in between the samples, whereas other elements (Ce and La) showed concentrations differing by up to a factor of five. The obtained concentrations are generally in good agreement with typical REE-concentrations in tea samples. According to the results of green tea reference material GBW07605 analysis by ICP-OES21, Ce, La, and Y have concentrations of 0.92, 0.57 and 0.37 μg g−1 in tea leaves. In another work regarding the determination of REEs in four different tea samples by using ICP-MS8 the range of concentration for Ce, La, and Y are respectively 0.62–0.99, 0.82–1.84 and 0.13–0.20 μg g−1. These values are in similar order to the concentrations obtained in our work for Ce, La and Y including 0.340–1.800, 0.390–2.000, 0.090–0.540 μg g−1 respectively.
| Ce | Dy | Er | Eu | |
|---|---|---|---|---|
| Tea 1 | 340 ± 5 | <8 | <20 | <9 |
| Tea 2 | 850 ± 360 | 270 ± 10 | 1200 ± 60 | 40 ± 10 |
| Tea 3 | 1500 ± 330 | 280 ± 30 | 800 ± 120 | 20 ± 5 |
| Tea 4 | 1700 ± 180 | 290 ± 10 | <20 | <9 |
| Tea 5 | 600 ± 130 | <8 | <20 | <9 |
| Tea 6 | 1800 ± 90 | 290 ± 10 | 880 ± 30 | 30 ± 5 |
| Tea 7 | 480 ± 160 | 240 ± 15 | 840 ± 30 | 20 ± 10 |
| Tea 8 | 180 ± 70 | <8 | <20 | 20 ± 5 |
| Tea 9 | 1000 ± 280 | 230 ± 5 | 1300 ± 10 | 20 ± 5 |
| Tea 10 | 850 ± 130 | 240 ± 5 | <20 | 20 ± 5 |
| Gd | Ho | La | Lu | |
|---|---|---|---|---|
| Tea 1 | <60 | <50 | 390 ± 10 | 10 ± 5 |
| Tea 2 | 1300 ± 170 | 870 ± 440 | 20 ± 5 | |
| Tea 3 | <50 | 1400 ± 400 | 20 ± 5 | |
| Tea 4 | <50 | 2000 ± 100 | 10 ± 5 | |
| Tea 5 | <50 | 780 ± 120 | 10 ± 5 | |
| Tea 6 | 830 ± 20 | 2000 ± 160 | 20 ± 5 | |
| Tea 7 | 870 ± 120 | 540 ± 80 | 20 ± 10 | |
| Tea 8 | 730 ± 100 | 370 ± 110 | <2 | |
| Tea 9 | 1100 ± 70 | 750 ± 210 | 30 ± 20 | |
| Tea 10 | <50 | 790 ± 110 | 20 ± 5 |
| Nd | Pr | Sc | Sm | |
|---|---|---|---|---|
| Tea 1 | <90 | <40 | <5 | <80 |
| Tea 2 | 7300 ± 480 | <40 | 70 ± 5 | 1000 ± 120 |
| Tea 3 | 5200 ± 400 | 260 ± 80 | 70 ± 5 | <80 |
| Tea 4 | <90 | <40 | <5 | <80 |
| Tea 5 | 3700 ± 400 | <40 | 30 ± 5 | <80 |
| Tea 6 | 5400 ± 250 | 180 ± 30 | 40 ± 5 | <80 |
| Tea 7 | 5000 ± 430 | 120 ± 10 | 50 ± 5 | <80 |
| Tea 8 | 4600 ± 330 | <40 | <5 | <80 |
| Tea 9 | 7800 ± 860 | <40 | 50 ± 5 | 1000 ± 190 |
| Tea 10 | 3700 ± 410 | 160 ± 5 | 40 ± 10 | <80 |
| Tb | Tm | Y | Yb | |
|---|---|---|---|---|
| Tea 1 | <40 | <30 | 140 ± 10 | 20 ± 5 |
| Tea 2 | 810 ± 150 | 460 ± 130 | 60 ± 10 | |
| Tea 3 | 640 ± 100 | 240 ± 40 | 40 ± 10 | |
| Tea 4 | <30 | 180 ± 5 | 30 ± 5 | |
| Tea 5 | <30 | 160 ± 20 | 20 ± 5 | |
| Tea 6 | 670 ± 50 | 540 ± 10 | 50 ± 10 | |
| Tea 7 | 570 ± 20 | 400 ± 110 | 50 ± 10 | |
| Tea 8 | 440 ± 10 | 90 ± 10 | <5 | |
| Tea 9 | 880 ± 20 | 460 ± 10 | 50 ± 5 | |
| Tea 10 | 490 ± 80 | 450 ± 80 | 60 ± 5 |
4. Summary and outlook
In this work, the concept of dispersed particle extraction has been further explored by ETV-ICP-OES for direct measurement of the analyte-containing sorbent particles. Compared to conventional slurry analysis, distinct improvements in sensitivity could be achieved; furthermore sample introduction by ETV allows the separation of the analyte and matrix in time. In order to achieve this goal, the use of an organic sorbent material instead of commonly applied silica based materials was necessary. For this purpose, Lycopodium clavatum spores coated with an ionic liquid acting as the cation exchanger were used, which could be completely volatilized during the matrix-pyrolysis step in the ETV program.The method was applied to the analysis of REEs in digested tea samples, with typical detection limits around 50 ng g−1 in the dry tea. The measurement procedure was shown to produce good results in terms of analyte recovery when performing spike-recovery experiments in combination with matrix-adjusted calibration. However, since slight differences in the matrix between different tea-samples persisted, two-point standard addition was relied upon for quantification. The results obtained for ten tea samples are in the same concentration range as reported by other authors.8,21
The method presented here is a first proof of principle to show the applicability of natural bio-particles coated with ionic liquid for extracting and pre-concentrating REE elements. The method will be further optimized in terms of the pre-concentration factor as well as the digestion procedure, in order to mineralize the organic sample-matrix of the tea entirely. Then, it should be possible to rely upon aqueous external calibration, which would enhance the sample throughput.
Furthermore, the procedure could be easily adapted to other tasks by simply changing the ionic liquid used for analyte extraction. With the use of task-specific ionic liquids, improvements not only in sensitivity but also selectivity are expected, compared to conventional SPE methods.
Acknowledgements
The research presented in this article was financed by the Austrian Science Fund under project number P25030-N28.References
- R. C. Borges, D. I. T. Favaro, V. G. Caldas, D. da Costa Lauria and A. V. B. Bernedo, Instrumental neutron activation analysis, gamma spectrometry and geographic information system techniques in the determination and mapping of rare earth element in phosphogypsum stacks, Environ. Earth Sci., 2016, 75, 705 CrossRef.
- M. Nishiura, F. Guo and Z. Hou, Half-Sandwich Rare-Earth-Catalyzed Olefin Polymerization, Carbometalation, and Hydroarylation, Acc. Chem. Res., 2015, 48, 2209–2220 CrossRef CAS PubMed.
- T. E. Lister, P. Wang and A. Anderko, Recovery of critical and value metals from mobile electronics enabled by electrochemical processing, Hydrometallurgy, 2014, 149, 228–237 CrossRef CAS.
- S. Kulaksız and M. Bau, Anthropogenic dissolved and colloid/nanoparticle-bound samarium, lanthanum and gadolinium in the Rhine River and the impending destruction of the natural rare earth element distribution in rivers, Earth Planet. Sci. Lett., 2013, 362, 43–50 CrossRef.
- G. Pagano, F. Aliberti, M. Guida, R. Oral, A. Siciliano, M. Trifuoggi and F. Tommasi, Rare earth elements in human and animal health: state of art and research priorities, Environ. Res., 2015, 142, 215–220 CrossRef CAS PubMed.
- C. Vidaud, D. Bourgeois and D. Meyer, Bone as Target Organ for Metals: The Case of f-Elements, Chem. Res. Toxicol., 2012, 25, 1161–1175 CrossRef CAS PubMed.
- X. Wang, J. Su, L. Zhu, N. Guan, X. Sang and Y. Ze, Hippocampal Damage and Alterations of Inflammatory Cytokine Expression in Mice Caused by Exposure to Cerium Chloride, Arch. Environ. Contam. Toxicol., 2013, 64, 545–553 CrossRef CAS PubMed.
- S. Su, B. Chen, M. He, B. Hu and Z. Xiao, Determination of trace/ultratrace rare earth elements in environmental samples by ICP-MS after magnetic solid phase extraction with Fe3O4@SiO2@polyaniline–graphene oxide composite, Talanta, 2014, 119, 458–466 CrossRef CAS PubMed.
- D. Wang, C. Wang, S. Ye, H. Qi and G. Zhao, Effects of Spraying Rare Earths on Contents of Rare Earth Elements and Effective Components in Tea, J. Agric. Food Chem., 2003, 51, 6731–6735 CrossRef CAS PubMed.
- A. Hokura, H. Matsuura, F. Katsuki and H. Haraguchi, Multielement Determination of Major-to-Ultratrace Elements in Plant Reference Materials by ICP-AES/ICP-MS and Evaluation of Their Enrichment Factors, Anal. Sci., 2000, 16, 1161–1168 CrossRef CAS.
- S. Chen, X. Cheng, Y. He, S. Zhu and D. Lu, Determination of the rare earth elements La, Eu, and Yb using solidified floating organic drop microextraction and electrothermal vaporization ICP-MS, Microchim. Acta, 2013, 180, 1479–1486 CrossRef CAS.
- O. T. Butler, W. R. L. Cairns, J. M. Cook and C. M. Davidson, 2014 atomic spectrometry update – a review of advances in environmental analysis, J. Anal. At. Spectrom., 2015, 30, 21–63 RSC.
- G. Alpdoğan, Solid phase extraction of Cu(II), Ni(II), Co(II), and Fe(III) ions in water samples using salicylaldehyde benzoylhydrazone on Amberlite XAD-4 and their determinations by flame atomic absorption spectrometry, Toxicol. Environ. Chem., 2015, 98, 179–188 CrossRef.
- G. Bauer, M. A. Neouze and A. Limbeck, Dispersed particle extraction—A new procedure for trace element enrichment from natural aqueous samples with subsequent ICP-OES analysis, Talanta, 2013, 103, 145–152 CrossRef CAS PubMed.
- R. Janski, M. A. Neouze and A. Limbeck, Determination of rare earth elements in saline matrices using dispersed particle extraction and inductively coupled plasma mass spectrometry, Rapid Commun. Mass Spectrom., 2014, 28, 1329–1337 CrossRef CAS PubMed.
- W. Nischkauer, M. A. Neouze, F. Vanhaecke and A. Limbeck, Extraction and pre-concentration of platinum and palladium from microwave-digested road dust via ion exchanging mesoporous silica microparticles prior to their quantification by quadrupole ICP-MS, Microchim. Acta, 2015, 182, 2369–2376 CrossRef CAS PubMed.
- A. Diego-Taboada, P. Cousson, E. Raynaud, Y. Huang, M. Lorch and B. P. Binks, Sequestration of edible oil from emulsions using new single and double layered microcapsules from plant spores, J. Mater. Chem., 2012, 22, 9767 RSC.
- J. Rowley and J. Skvarla, The elasticity of the exine, Grana, 2000, 39, 1–7 CrossRef.
- X. Cao, G. Zhao, M. Yin and J. Li, Determination of ultratrace rare earth elements in tea by inductively coupled plasma mass spectrometry with microwave digestion and AG50W-X8 cation exchange chromatography, Analyst, 1998, 123, 1115–1119 RSC.
- W. Nischkauer, E. Herincs, M. Puschenreiter, W. Wenzel and A. Limbeck, Determination of Pt, Pd and Rh in Brassica napus using solid sampling electrothermal vaporization inductively coupled plasma optical emission spectrometry, Spectrochim. Acta, Part B, 2013, 89, 60–65 CrossRef CAS.
- Y. Li and B. Hu, Cloud point extraction with/without chelating agent on-line coupled with inductively coupled plasma optical emission spectrometry for the determination of trace rare earth elements in biological samples, J. Hazard. Mater., 2010, 174, 534–540 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay02189a |
| ‡ These authors contributed equally to the work presented in this article. |
| This journal is © The Royal Society of Chemistry 2016 |