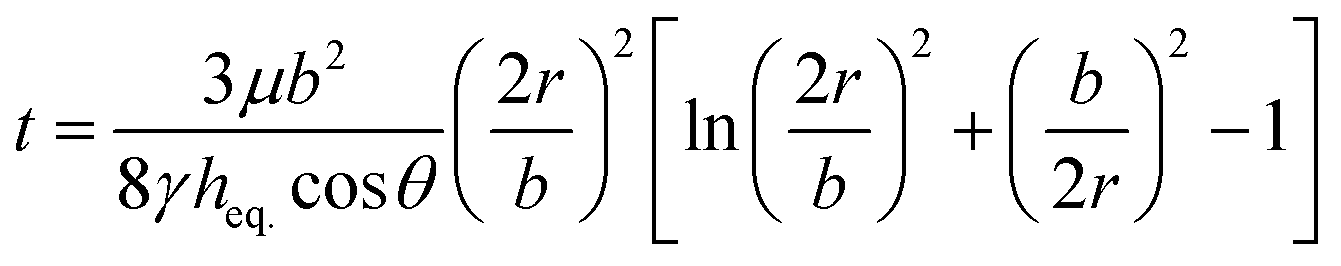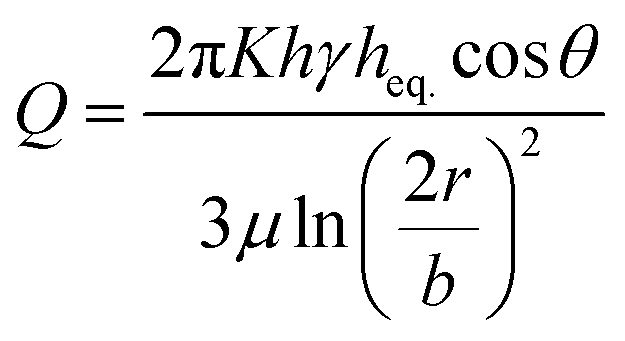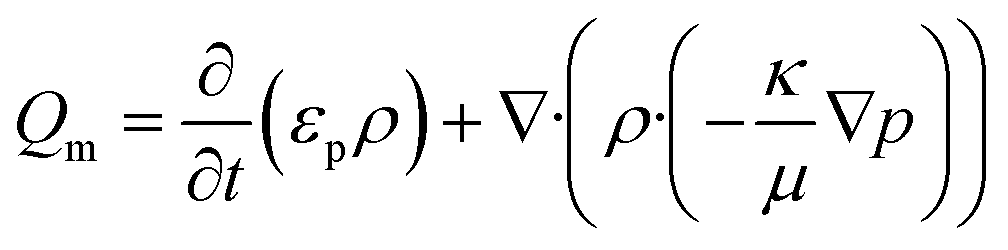DOI:
10.1039/C6AY01926A
(Paper)
Anal. Methods, 2016,
8, 6965-6973
Development of a paper-based analytical device for colorimetric detection of uric acid using gold nanoparticles–graphene oxide (AuNPs–GO) conjugates†
Received
6th July 2016
, Accepted 10th August 2016
First published on 9th September 2016
Abstract
A novel paper-based device for colorimetric detection of uric acid was developed based on the peroxidase-like activity of gold nanoparticles–graphene conjugates (AuNPs–GO). Theoretical models were utilized for designing a device that was further verified by simulation and experimentation. Detection of uric acid was then carried out by initially soaking the paper in an AuNPs–GO solution and subsequently pipetting TMB–H2O2 solution on it. Detection was achieved by measuring the color change when uric acid interacted with AuNPs–GO. A significant reduction in the blue coloration of the paper to white color was observed in a brief time of 5 minutes. Moreover, a very low detection limit of 4 ppm for uric acid was obtained on paper by visualization with the naked eye. The assembly of AuNPs within the GO matrix resulted in many active sites, which were the reason for the low detection limit. The practicality of the device was also examined by testing it on several blood serum samples. The results were in agreement with clinical results and also showed a high selectivity towards uric acid. The device provides a real-time, rapid and easy-to-use platform for the detection of uric acid for clinical purposes.
Introduction
Uric acid (UA), which is an important biochemical, is a derivatized product from purines (nitrogen-containing compound). Purines are either made inside the cell of a human body (using the endogenous purine metabolism route), by xanthine dehydrogenase (xanthine oxidase) primarily in the liver and the intestine,1 or they are generated through external food that contain exogenous purines. A uric acid concentration in the blood serum beyond a normal level (both above and below), i.e., 20.17–75.64 ppm,2 possesses a risk factor in humans. High levels of uric acid, called hyperuricemia (serum UA level > 65–70 ppm in men and >60 ppm in women), can cause diseases such as gout, hypertension, high cholesterol, high blood pressure, kidney diseases and heart diseases,1–4 while low levels, i.e. hypouricemia (serum UA levels <20.17 ppm), may cause neurodegenerative diseases like Hodgkin's disease, sarcoma, glioblastoma3,5etc. Thus, rapid clinical analysis of UA is essential for a proper health assessment.
Several sensitive detection techniques have been proposed for the detection of serum UA, such as photometric,6 electrochemical,7 capillary electrophoresis (CE),8 chemiluminescence (CL),9 and colorimetric10 techniques. The primary limitations observed in the former four methods include higher detection time, higher cost and the requirement of laboratory-based testing. On the other hand, the provision of a visual observation by the naked eye (based on color change) in the latter technique allows for simple operation by the patient himself. Generally, metals11,12 and paper10,13 are used as substrates for promoting colorimetric detection schemes. In contrast to metals, paper devices based on lateral flow assaying techniques14 are economical, easily operable, compatible with biological samples, can be modified chemically and are easy to dispose through incineration to prevent the spread of contamination. Lateral flow technology is well suited for point-of-care disease diagnostics as it allows complex bio/chemical processing to be performed without any need for external instrumentation. It fulfills the ASSURED (affordable, sensitive, specific, user-friendly, rapid and robust, equipment-free, and deliverable to users) criteria as given by WHO. Paper-based devices can be utilized for inexpensive analytical assays in areas with insufficient laboratory facilities, playing a complementary role. Several paper-based devices for detection of uric acid have been developed. The devices use a visible colorimetric change as the detection mechanism. The reported LODs by visualization through the naked eye for these devices were 33.2 ppm,10 7.26 ppm13 and 16.85 ppm.15
In the past decade, many researchers have focused on individual nanoparticles (of gold, platinum, silver etc.) because of their special physicochemical characteristics that can be utilized for the development of highly sensitive chemical and biological sensors. Gold nanoparticles (AuNPs) have especially been utilized in this domain owing to their distinguished characteristics such as biocompatibility, excellent optical properties, high surface-to-volume ratio, suitable platform for selective binding and detection of specific molecules. The target molecule triggers the aggregation of AuNPs, inducing inter-particle surface plasmon coupling and a visible color change.16 It has been reported that the catalytic properties of AuNPs decrease with increasing particle size. This is attributed to the decreased surface-to-volume ratio, resulting in fewer active binding sites.17 The catalytic activity of AuNPs can be enhanced by their immobilization on graphene oxide nanosheets, which prevents the aggregation of AuNPs within themselves18 and maintains an array of nanoparticles without clustering effects. Graphene oxide is a one-atom-thick, two-dimensional carbon lattice that is light-weight and has a large surface area.19 The AuNPs–GO conjugates show excellent synergistic peroxidase-like activity by providing a larger surface area for the adsorption of biomolecules, resulting in a substantial improvement in the limit of detection (LOD).17,20
In this study, we describe the fabrication of a portable paper-based microfluidic device for colorimetric detection of uric acid. Hybrid gold nanoparticles–graphene oxide conjugates were immobilized on a paper device for sensitive and rapid detection. The developed device was successfully tested for the detection of uric acid in human blood serum samples, demonstrating future potential for clinical applications.
Experimental
Reagents and materials
Graphite flakes (acid treated 99%), sodium nitrate (98%), potassium permanganate (99%), hydrogen peroxide (40% wt), sulphuric acid (98%), sodium hydroxide (NaOH), nitric acid, hydrochloric acid (35%) and Whatman qualitative filter paper grade 1 were purchased from Sigma Aldrich, India. Gold(III) chloride trihydrate (HAuCl4–3H2O), uric acid (2,6,8-trioxypurine, 99% AR) and 3,3′,5,5′-tetramethylbenzidine (TMB) were obtained from Loba Chemie Pvt. Ltd., India. All chemicals used were of analytical grade. Milli-Q ultrapure water was used in the synthesis of graphene oxide, and high performance liquid chromatography (HPLC) water, purchased from Merc Life Science Pvt. Ltd., India, was used for preparation of solutions and cleaning of the filter paper throughout the experiment. Human whole blood serum samples were obtained from IIT Kanpur, India, and all analyses were performed at the health care center at GSVM medical college. All experiments were performed in compliance with the relevant laws and national guidelines (Ethical Guidelines for Biomedical Research on Human Participants, provided by Indian Council of Medical Research), and the ethical clearance for the same was provided by the GSVM Medical College, Kanpur, which also approved all of our experiments. Also, all experiments with the samples were performed with informed consent obtained from the persons who provided the samples.
Instrumentation
Transmission electron microscopy (TEM) was carried out on a FEI Tecnai G2 12 Twin TEM 120 kV. The Raman spectra were recorded on a confocal Raman microscope with 10× objective using an excitation laser line of 532 nm. The X-ray diffractometry (XRD) was performed on a Bruker D8 Advance X-ray diffractometer with Cu Ka (k = 0.154 nm) radiation (Bruker AXS, Germany). UV-Vis absorption spectrophotometry was done using an Evolution 300 spectrophotometer (Thermo Scientific).
Preparation of graphene oxide
Graphene oxide was synthesized by modified Hummers method through the oxidation of graphite. Graphite flakes (2 g) were mixed thoroughly with NaCl (100 g) for 10 minutes. This mixture was then dissolved in milli-Q ultrapure water and filtered using filter paper to remove the carbon. The remaining graphite flakes were mixed in 50 mL of H2SO4 (98%) in a flask, kept in an ice bath and stirred continuously for 8 hours. Potassium permanganate (KMnO4) was added to the suspension very slowly while keeping the temperature less than 20 °C. The mixture was continuously stirred in a reflux system at 98 °C for 30 minutes. After 35 minutes, the temperature was changed to 40 °C, triggering a change of the solution to a brown color. Further, the mixture was diluted with the slow addition of 200 mL of ultrapure water and 10 mL of an H2O2 (30%) solution. The solution was stirred for 2 hours until its color changed to bright yellow. It was then kept stationary for 8 hours to allow a settlement of the mixture after which it was filtered. The filtered mixture was washed through repeated centrifugation alternatively with 10% HCl and milli-Q ultrapure water. After centrifugation the remaining gel-like substance was vacuum dried at 60 °C for 8 hours. Raman and UV-Vis spectra were measured for characterization of GO (ESI).
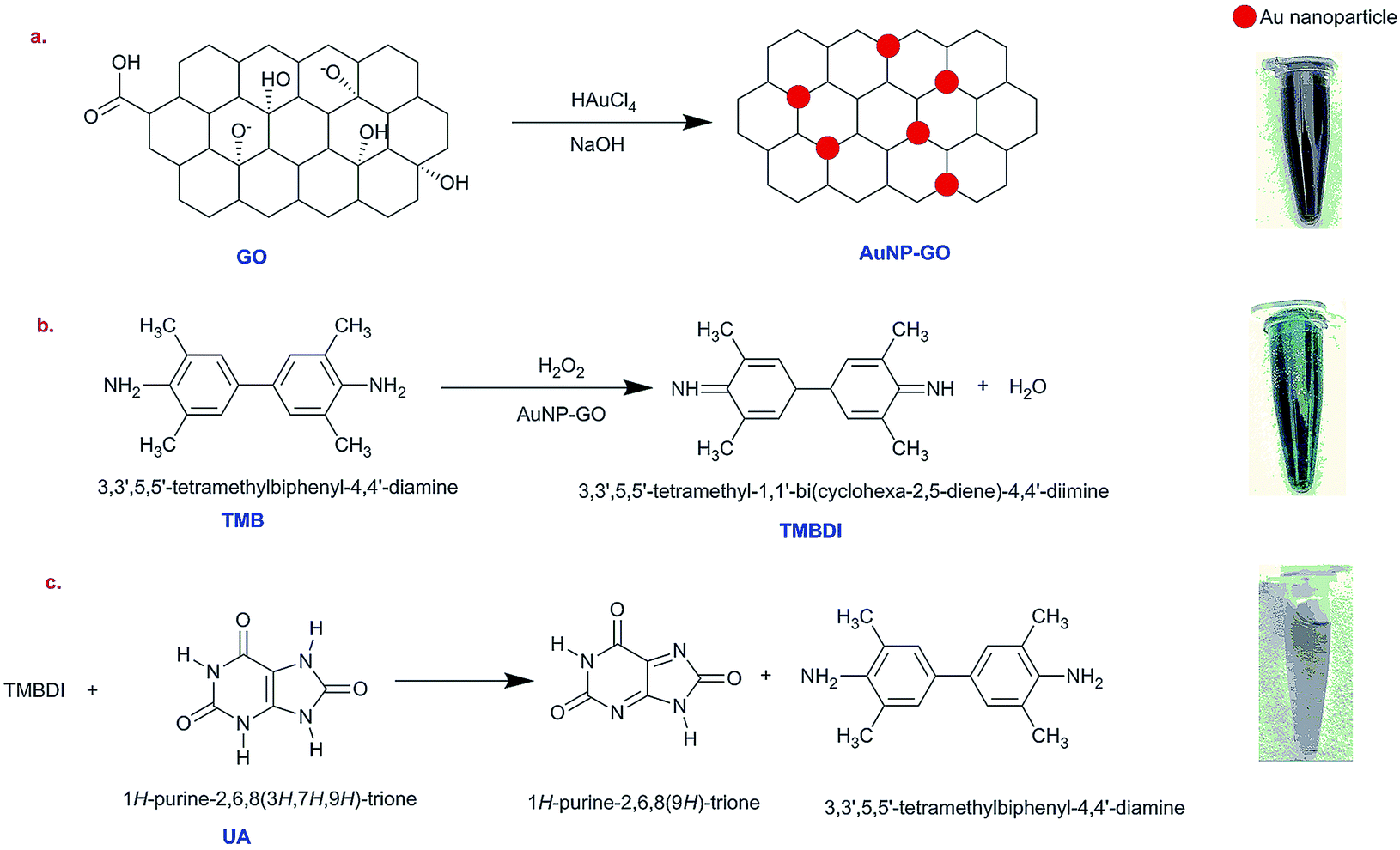
Preparation of gold nanoparticles–graphene oxide conjugates
The modified hydrothermal method was used for synthesis of the colloidal AuNPs–GO solution. A typical mixture of 10 mg GO powder, 70 mg HAuCl4·3H2O and 16 g NaOH were mixed and diluted in 40 mL of HPLC water. The mixture was then sonicated for 8 hours at room temperature. For a hydrothermal reaction to take place, the mixture was put in an autoclave with a Teflon liner at 180 °C for 12 hours. The prepared conjugates were cooled off at room temperature. The optical properties were characterized using Raman spectroscopy, ultraviolet-visible (UV-Vis) absorbance spectroscopy, Fourier transform infrared spectroscopy (FTIR) and X-ray powder diffraction (XRD). The transmission electron microscopy (TEM) was used to ensure the presence of AuNPs–GO conjugates.
Catalytic activity of the AuNPs–GO
The catalysis of graphene oxide is strongly dependent on pH, temperature, and H2O2 concentration, similar to HRP.21 The colorless 3,5,3′,5′-tetramethyl benzidine (TMB) shows peroxidase-like catalytic activity by getting oxidized in the presence of hydrogen peroxide (H2O2) and AuNPs–GO conjugates. The solution shows a yellow color22 that subsequently turns into a blue colored solution upon addition of AuNPs–GO. This bluish colored solution fades away on coming in contact with uric acid using the following mechanism.
First, TMB gets oxidized in the presence of H2O2 with the AuNPs–GO conjugates acting as a catalyst to the reaction, resulting in TMBDI (ox-TMB) and water (H2O) as the products. This oxidized product gives a blue color appearance. Second, addition of uric acid to this solution results in reduction of TMBDI back to TMB, yielding a colorless solution. The reaction mechanism is illustrated in Scheme 1.
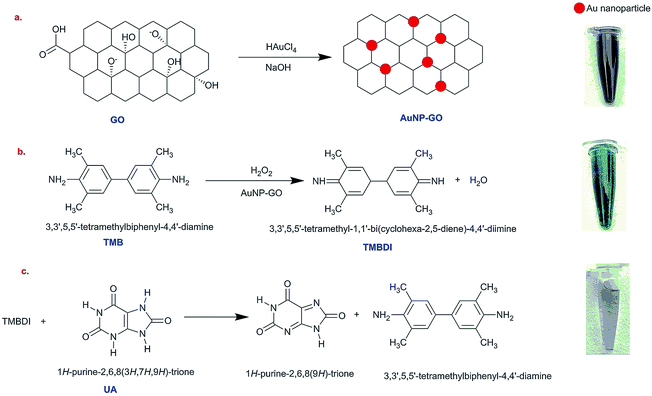 |
| | Scheme 1 Schematic illustration of the colorimetric sensor for the detection of uric acid using AuNPs–GO. | |
A diluted solution of AuNPs–GO was prepared by mixing 1 mL of stock solution with 9 mL of HPLC grade water and ultrasonicating for 6 hours to obtain a dispersed solution. A freshly prepared mixture of 0.02 (M) TMB (in methanol) and 8.82 (M) H2O2 was added to the diluted AuNPs–GO solution. Initially, the peroxidase-like activity was observed by adding 0.05 mL TMB–H2O2 to 3 mL of an AuNPs–GO solution. The effect of varying the concentration of TMB and AuNPs–GO was investigated by performing UV-Vis spectrophotometry on various solutions.
Colorimetric detection of uric acid
A stock solution of 16.2 mg of uric acid in 0.01 (N) NaOH was prepared, and different UA concentration solutions (1, 5, 10, 20, 30, 40, 50, 60, 70, 80, 90 and 100 ppm) were obtained by dilution of the stock solution in HPLC water. The prepared solutions were mixed with 4 mL of dilute AuNPs–GO and 0.05 mL of the TMB–H2O2 solution. The solutions were then used for UV-Vis spectra measurements.
Paper-based microfluidic device
Device design.
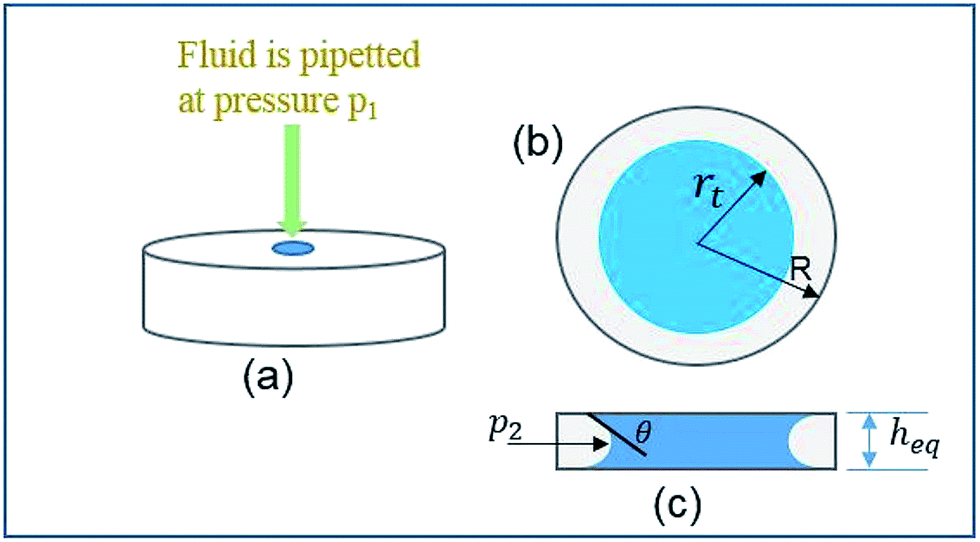
To make the analysis portable and user-friendly, a paper-based microfluidic device was fabricated. Prior to the fabrication of the device, an analytical approach was utilized to calculate an optimum diameter for the device's circular paper strip. In order to design the optimal parameters, the change in the wicking radius and mass flow rate with respect to time were theoretically modeled and further simulated. The overall scheme of the wetted paper strip is schematically represented in Fig. 1.
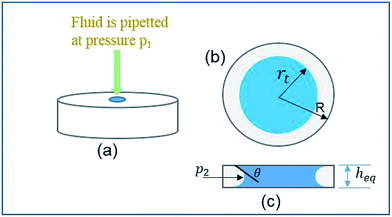 |
| | Fig. 1 Schematic of the principle of fluid flow through a circular paper strip. | |
To predict the motion of the liquid through the paper strip, the wicking radius (r) and mass flow rate (Q) were calculated using eqn (1) and (2), respectively.23 The values of the parameters used for the calculations are listed in Table 1.
| | 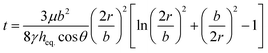 | (1) |
| | 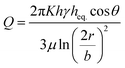 | (2) |
where
h is the thickness of the paper,
r is the wicking radius,
b is the initial diameter of the fluid droplet,
heq. is the equivalent thickness of the radial capillary,
θ is the wetting angle between the liquid sample and the cellulose fibers and
K is the parameter for moisture content in the paper.
Table 1 Parameters used in theoretical calculation
| Parameter |
Numerical value |
| Thickness of paper strip (h) |
180 μm |
| Droplet diameter (b) |
2.57 mm (0.1 mL volume) |
| Moisture constant (K) |
0.47 |
| Surface tension (γ)@25 °C |
71.97 mN m−1 |
| Viscosity (μ)@25 °C |
0.89 mPa s |
Subsequently, a finite element analysis was carried out using the porous media and subsurface flow module of COMSOL multiphysics 4.3b. The model solves for the flow of water through a porous filter paper. The flow fields were obtained by solving Darcy's law for an incompressible fluid in 2D.
| | 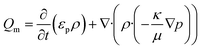 | (3) |
where
εp and
κ are the porosity and permeability of the porous media,
ρ and
μ are the density and viscosity of the fluid, and ∇
p is the pressure gradient. A zero pressure constraint was set as the boundary condition at the outlet. A transient analysis was performed for a range of 0 to 60 seconds.
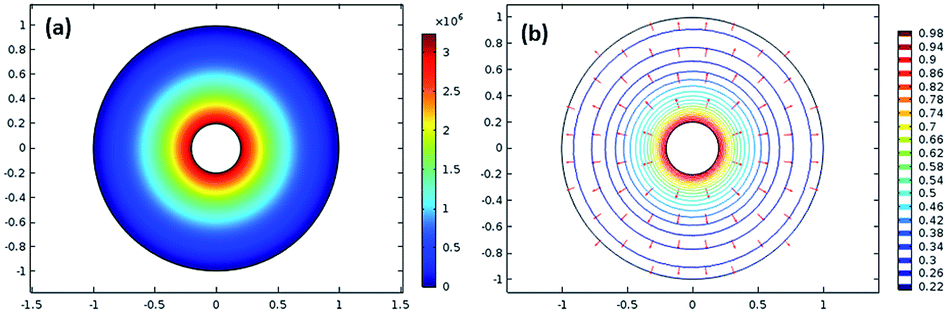
Fig. 2(a) shows the surface plot of pressure, and
Fig. 2(b) depicts the velocity contour along with the arrow surface to visualize the velocity vector.
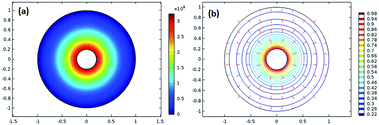 |
| | Fig. 2 (a) Surface plot of the pressure and (b) velocity contour along with the arrow surface. | |
The mass flow rate measurements were then performed experimentally as well. A paper strip (Whatman filter paper grade 1) of 110 mm diameter was placed in a Petri dish and kept on a side stand. A camera (Nikon DSLR D5200) was placed underneath this setup. All the images were captured at 10× zoom with a time interval of 1 s. A small amount of diluted uric acid solution (0.1 mL) was dispensed at the center of the paper strip. A total of 129 images were captured. Image J software was used for the measurement of the wetting area (considering the area to be circular), through which the radius for each second was calculated.
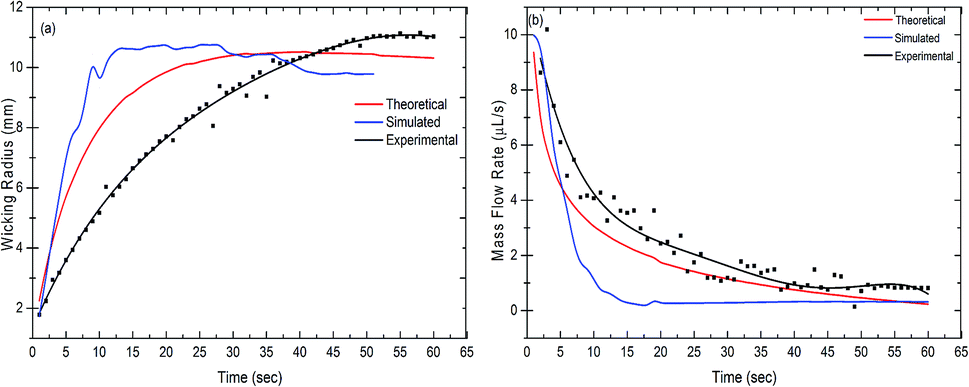
The wicking radius and mass flow rate were plotted with respect to time for the theoretical, simulated and experimental values (Fig. 3(a) and (b)). The mass flow rate of the fluid decreased with time whereas the wicking radius increased. As the fluid comes in contact with the paper, it starts wicking the fluid as a result of the capillary force between the liquid and the fibers in the paper. The fluid front moves forward by filling the subsequent pores in the paper. With the increase in the wicking radius, the number of pores increases, hence increasing the total volume to be filled by the fluid. This results in a reduction in the mass flow rate with time. It was observed that the trend shown in the theoretical results was closely followed by the simulation and experimental results. It was evident from the graph that approximately after 45 seconds, the mass flow rate became steady in all the cases. Also, the wicking radius after 45 seconds stayed constant at a value of 10.69 mm, 9.51 mm and 11 mm for the theoretical, simulated and experimental results, respectively.
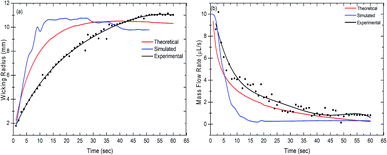 |
| | Fig. 3 Measurement of (a) the wicking radius and (b) mass flow rate as a function of time. The measured values are theoretical, simulated and experimental, respectively. | |
Prototype fabrication.
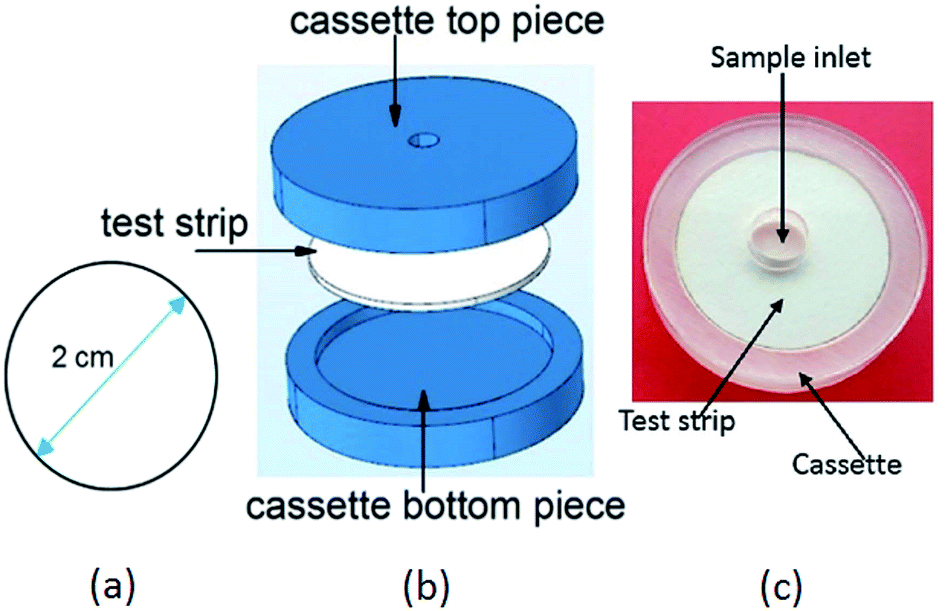
By means of a CO2 pulsed laser beam machine, small circular strips were cut out of Whatman filter paper (grade 1). Based on the above calculations, the optimal diameter for the paper device was found to be 2 cm. Fig. 4(a) shows the paper strip with the optimal diameter, (b) gives an exploded view of the device and (c) shows the fabricated device prototype.
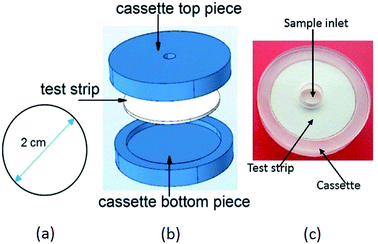 |
| | Fig. 4 Device for uric acid detection: (a) optimized dimension of the paper device (b) exploded view of the device. The paper device was housed in a cassette. (c) Photograph of the device prototype. | |
Test strip preparation.
First, the circular paper strips were washed in hot HPLC water (90 °C) and dried to remove unwanted impurities. In the second step, they were soaked in a 0.5 mL diluted solution of AuNPs–GO. In the next step, 0.05 mL of the prepared TMB–H2O2 solution was pipetted onto them. A typical blue color was observed on the paper upon the addition of the TMB–H2O2 solution. Subsequently, 0.1 mL of the uric acid solution (freshly prepared in HPLC water) with different concentrations ranging from 4, 10, 15, 20, 30, 40, 50, 65 to 95 ppm was pipetted onto these paper strips.
Preparation of human blood serum samples
In order to examine the practicability of the device, a prototype was manufactured and tested with several blood serum samples. The collected samples were prepared in accordance with the accepted procedures. Blood serum was extracted from the collected human blood samples by centrifuging them at 2000 rpm for 10 minutes. The extracted human serum samples were used without any pretreatment. They were then analyzed for the presence of uric acid using the paper strips. To further check the accuracy of the device, the results were validated by comparing them with the conventional clinical test results.
The human blood serum contains competitive entities that can potentially reduce ox-TMB, e.g. glucose, cysteine, alanine, glutathione, serum albumin etc. Hence, an investigation of the human blood serum samples was also carried out in order to ensure the selectivity for uric acid of the catalytic reduction of ox-TMB. First, the serum samples were spiked with known concentrations of uric acid. Further, UV-Vis spectrophotometry was performed to calculate its recovery.
Results and discussion
Characterization of the AuNPs–GO
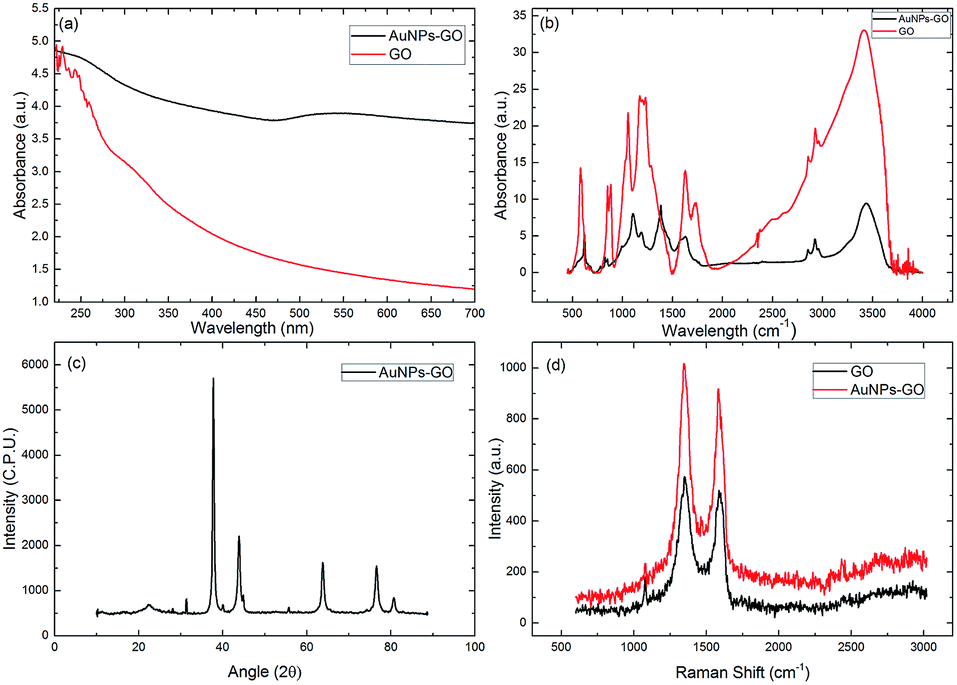
Fig. 5(a) shows the UV-Vis absorption spectra of graphene oxide and AuNPs–GO conjugate solution. In the GO spectrum the peak is at 225 nm, owing to the π–π* transition of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, and the shoulder peak is at 297 nm, corresponding to the n–π* transition of the CO bond.17 The first absorption peak of the AuNPs–GO at 225 nm indicates the presence of graphene oxide, and the second peak at 538 nm is due to the surface plasmon resonance (SPR), substantiating the successful loading of gold nanoparticles on the graphene oxide sheet.
C bond, and the shoulder peak is at 297 nm, corresponding to the n–π* transition of the CO bond.17 The first absorption peak of the AuNPs–GO at 225 nm indicates the presence of graphene oxide, and the second peak at 538 nm is due to the surface plasmon resonance (SPR), substantiating the successful loading of gold nanoparticles on the graphene oxide sheet.
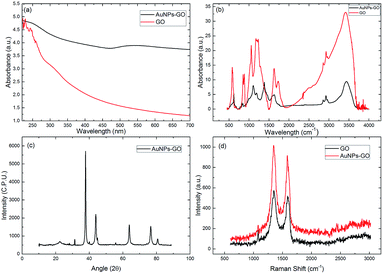 |
| | Fig. 5 (a) UV-Vis absorption spectra of the GO and AuNPs–GO, (b) FTIR spectra of the GO and AuNPs–GO, (c) XRD spectra of the AuNPs–GO and (d) Raman spectra of the GO and AuNPs–GO. | |
Additionally, to study the functionalization of AuNPs–GO conjugates, Fourier transform infrared (FTIR) spectroscopy was carried out as shown in Fig. 5(b). The graphene oxide spectrum has strong absorption bands at 3412, 1736, 1626, 1172, and 1054 cm−1, corresponding to stretching vibrations from O–H, CO, CC, C–O and C–O alkoxy, respectively. The absorption bands at 2926 and 2850 cm−1 ensure the presence of the CH2 bond. The spectrum of AuNPs–GO shows similar peaks as GO, except for the decreased absorption at 1627 and 1104 cm−1, corresponding to the COOH and C–OH groups.24 This decrease can be attributed to the gold nanoparticles using the oxygen functional groups present on the graphene oxide sheets as nucleation sites.
Also, the crystal structure of the AuNPs–GO was examined by XRD analysis (Fig. 5(c)). The peaks at angles 38.33°, 44.56°, 64.64°, 77.45° and 82.62° have been assigned to the face-centered cubic bulk gold (111), (200), (220), (311) and (222) phases, respectively, which are in agreement with the standard values of gold (JCPDS 04-0784). Further, the peak at 11.3° confirms the existence of GO.
The Raman spectra of the GO and AuNPs–GO are shown in Fig. 5(d). The two prominent peaks at 1353 cm−1 and 1606 cm−1 in the GO are assigned to the D and G peaks, respectively. The D band is attributed to the structural imperfections due to the attachment of hydroxyl and epoxide groups on the carbon basal plane. The G band is attributed to the tangential stretching mode of the E2g phonons of the sp2 bonded carbon. In the case of the AuNPs–GO conjugates, similar peaks at 1348 cm−1 and 1591 cm−1 with higher intensities were observed. The relative intensity ratios of the D and G bands (ID/IG) for the GO and AuNPs–GO were 0.86 and 0.97, respectively. It may be due to the fact that sp2 cannot be regenerated through chemical reduction.24
The morphology of the AuNPs–GO conjugates was demonstrated through transmission electron microscopy (TEM). Fig. 6 shows the aggregation of the Au nanoparticles (appearing as dark dots) on the surface of the GO. Analysis of the TEM images using Image J revealed an average particle size of 6 nm for the AuNPs–GO.
 |
| | Fig. 6 TEM images of the AuNPs–GO conjugates: (a) at a lower resolution, (b) at a higher resolution. (c) Graphene oxide sheet and (d) SAED pattern. | |
Catalytic activity of AuNPs–GO
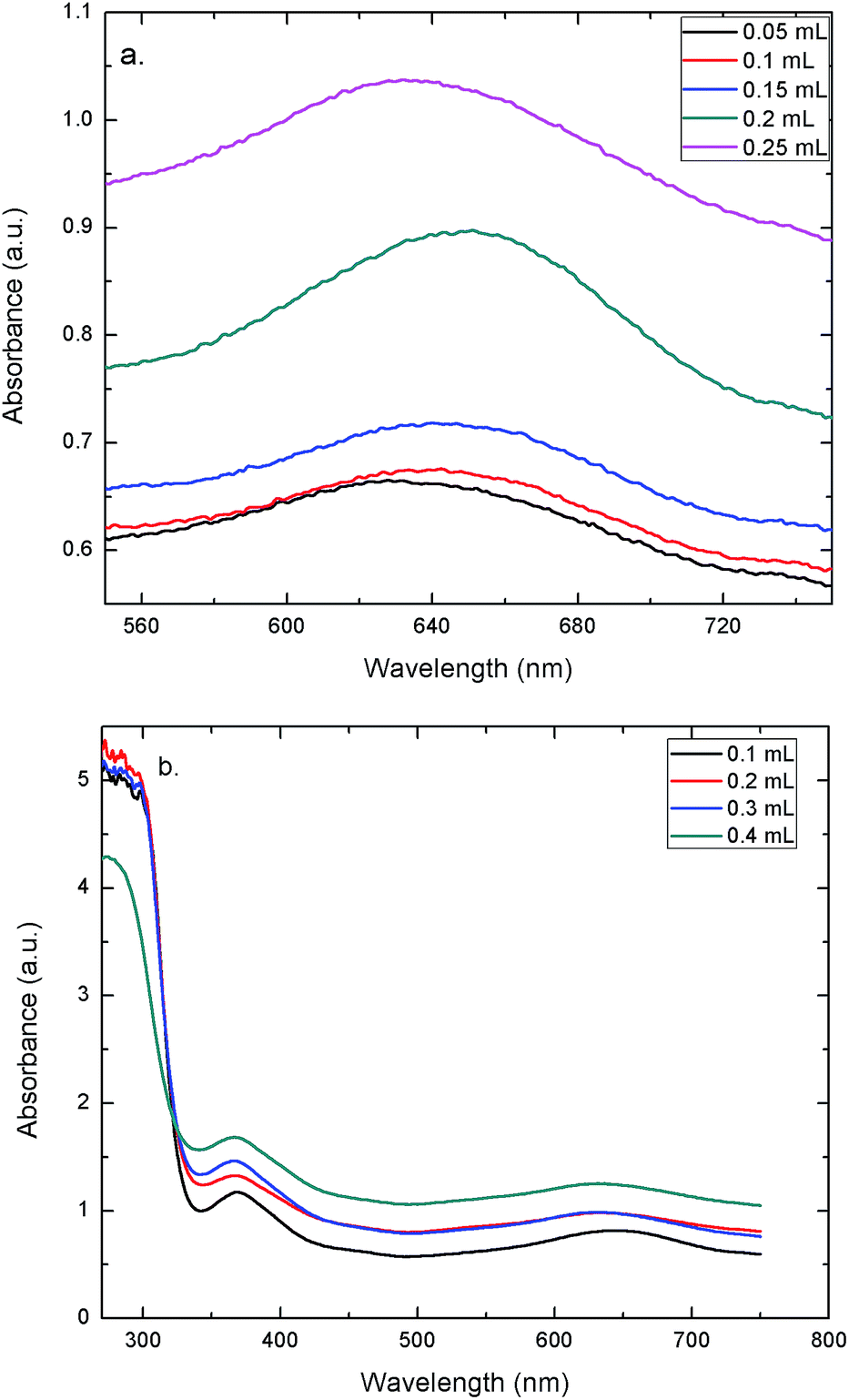
To evaluate the peroxidase-like activity of the AuNPs–GO, the catalytic oxidation of TMB in solution with H2O2 was tested. The concentration of TMB was varied in order to study its effect on the change in the color of the solution. Fig. 7(a) shows the UV-Vis spectra of solutions at varying concentrations of TMB. Clearly, the peak at 635 nm validates the blue color of the solution. The graph demonstrates that increasing the concentration led to an increase in the absorbance. Hence, the solution shows a deeper blue color with an increase in the TMB concentration. UV-Vis spectra of solutions with varying concentrations of the AuNPs–GO revealed the same trend (Fig. 7(b)). Increasing the concentration of the AuNPs–GO would increase the number of binding sites for the target molecule (TMB), implying an increase in the amount of bound TMB molecules in the solution. Hence, the solution demonstrates an increase in the absorbance with increasing AuNPs–GO concentration, which is an expected behavior. Based on this data, the minimum values for TMB and AuNPs–GO required to visualize the blue color by the naked eye were found. The values were 0.05 mL for TMB (last peaking line in Fig. 7(a)) and 0.1 mL for the AuNPs–GO solution (last peaking line at 635 nm in Fig. 7(b)). These amounts were used for the experiments carried out on Whatman filter papers.
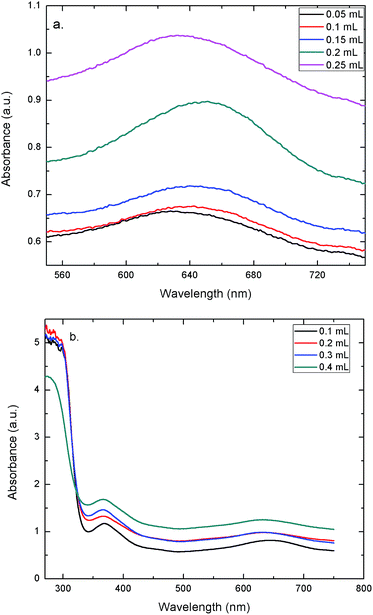 |
| | Fig. 7 UV-Vis spectra at (a) varying concentrations of TMB (b) and varying concentrations of AuNPs–GO. | |
Solution-based quantitative determination of uric acid
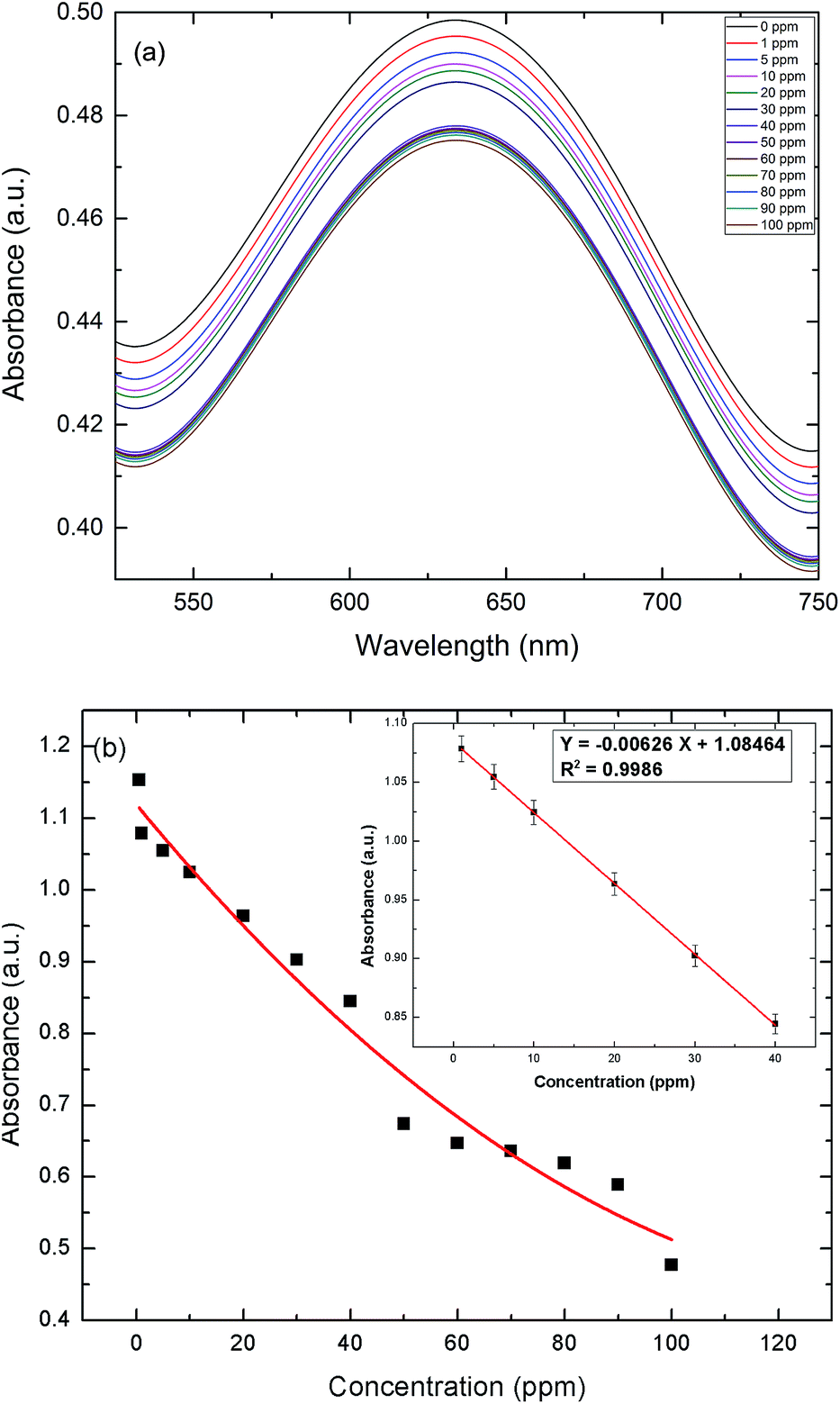
In these experiments, a colorimetric method was implemented to detect uric acid, utilizing the peroxidase-like catalytic activity of AuNPs–GO. The color variation of various solutions from blue to light black and eventually to colorless can be directly observed by the naked eye, as shown in Fig. 8. The UV-Vis spectra of the resulting solutions are shown in Fig. 9(a). The intensity of the blue color in solution decreases with an increase in the concentration of uric acid, as depicted by the decreasing absorbance. The Fig. 9(b) inset presents the linear regression curve for the varying concentrations of uric acid in solution at 635 nm. The regression equation was calculated as Y = −0.00626X + 1.08464 with 0.9986 being the correlation coefficient, where X and Y are the uric acid concentration and the absorbance, respectively. The range of uric acid was varied linearly between 1 to 40 ppm, and the LOD was calculated as 0.06 ppm. The proposed protocol was sensitive enough to detect the serum uric acid.
 |
| | Fig. 8 Photographs of solutions with varying concentrations of UA (left to right): 0 ppm, 10 ppm, 20 ppm, 30 ppm, 40 ppm, 50 ppm, 60 ppm, 70 ppm, 80 ppm and 90 ppm, respectively. | |
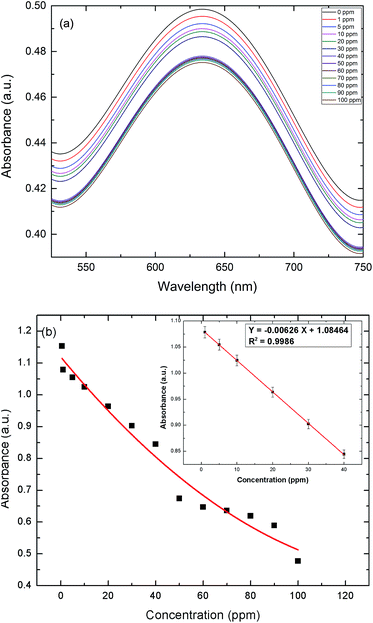 |
| | Fig. 9 (a) UV-Vis absorption spectra at varying concentrations of UA in solution. (b) Response curve for UA detection (solution) in the range of 0 to 100 ppm. Inset: the linear regression plot for UA. | |
Paper-based detection of uric acid
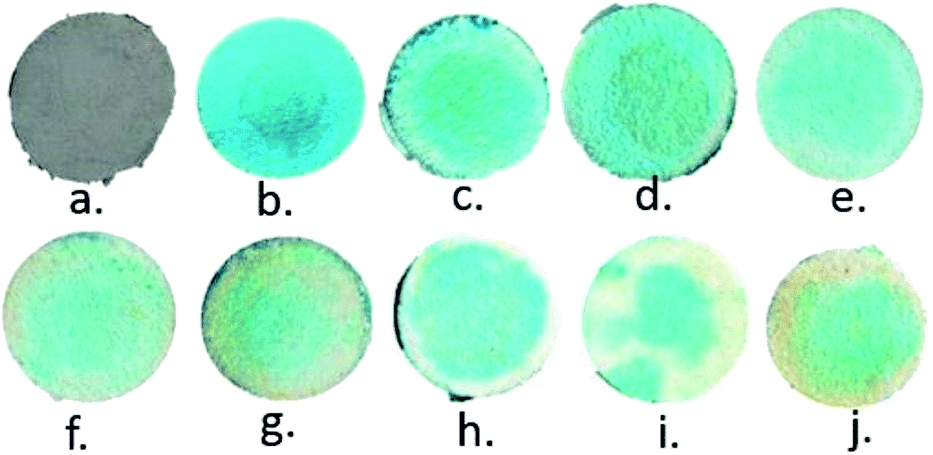
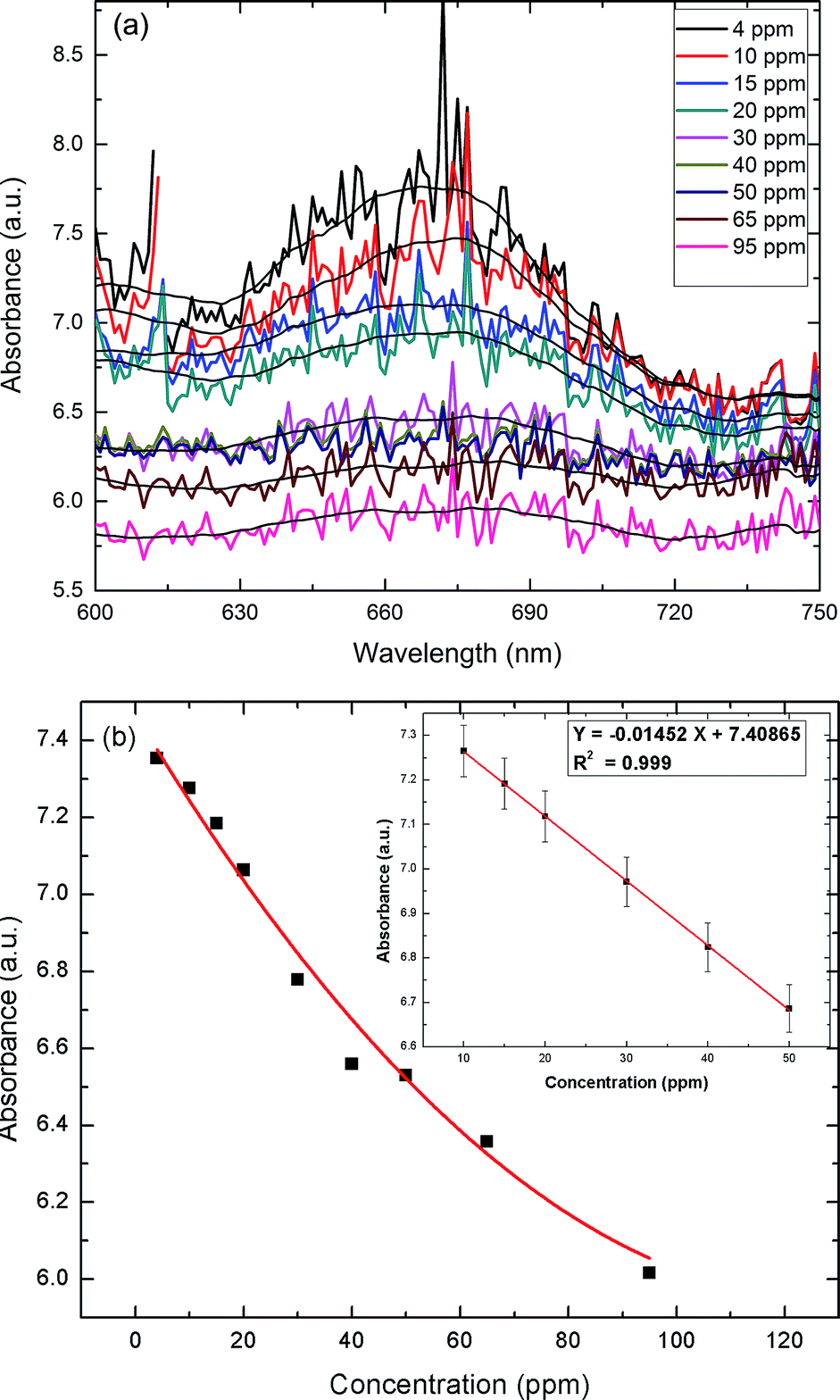
To assess the applicability of the paper strip as a detection tool, an assay was performed to determine the effect of varying the UA concentration. Addition of TMB + H2O2 to the AuNPs–GO turned the color of the circular paper strip to blue. Within five minutes, the bluish color of the paper strips faded away to different extents, depending linearly on the concentration of the uric acid (Fig. 10). The developed paper device was able to detect a color change at 4 ppm of uric acid. Solid state UV-Vis spectra of the circular strips were recorded to confirm the color change (Fig. 11(a)). The spectra were all noisy due to the paper background on which the serum was tested. If we average the data to plot the overall response with respect to the wavelength, we see one peak corresponding to 674 nm. The limit of detection (LOD) of the fabricated device observed by the naked eye was around 4 ppm (i.e., 2.38 × 10−5 M), which was lower than that reported in literature (1.97 × 10−4 M10, 4.31 × 10−5 M13, 10 × 10−5 M15). The Fig. 11(b) inset presents the linear regression curve for varying concentrations of uric acid on paper at 674 nm. The equation was expressed as Y = −0.01452X + 7.40865 with 0.999 being the correlation coefficient, where X and Y are the uric acid concentration and the absorbance, respectively. The linear range of uric acid was from 10 to 50 ppm, and the LOD was calculated to be 0.56 ppm.
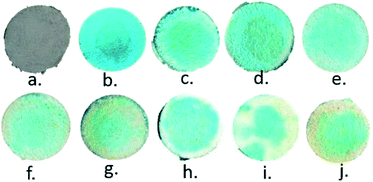 |
| | Fig. 10 Images of varying concentrations of UA on paper: (a) AuNPs–GO coated, (b) AuNPs–GO + TMB–H2O2 (c), (d), (e), (f), (g), (h), (i) and (j) after addition of UA at concentrations of 4 ppm, 10 ppm, 15 ppm, 20 ppm, 30 ppm, 40 ppm, 65 ppm and 95 ppm, respectively. | |
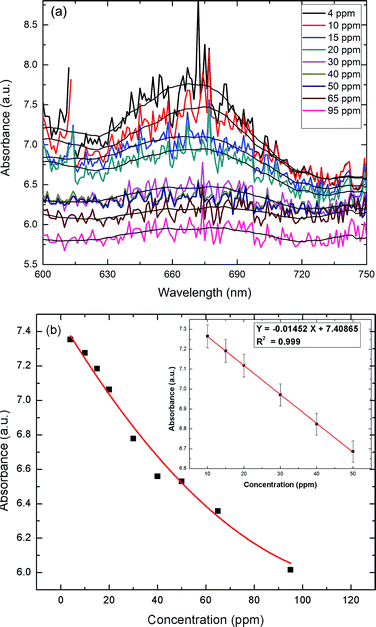 |
| | Fig. 11 (a) UV-Vis absorption spectra at varying concentrations of UA on the paper strip. (b) Response curve for UA detection (paper) in the range of 0 to 95 ppm. Inset: the linear regression plot for UA. | |
Solid state UV-Vis spectrophotometry was used for quantification since a qualitative assessment is not sufficient in such cases. However, the above method requires a laboratory, rendering the device impractical for on-site quantification. In order to address this issue, a portable colorimetric reader could be developed in the future. Several approaches have been utilized for on-site quantification of the results shown by paper-based devices.25–27 Similar approaches can be implemented in order to make the device portable and feasible for on-site detection purposes.
Paper-based analysis of human blood serum samples
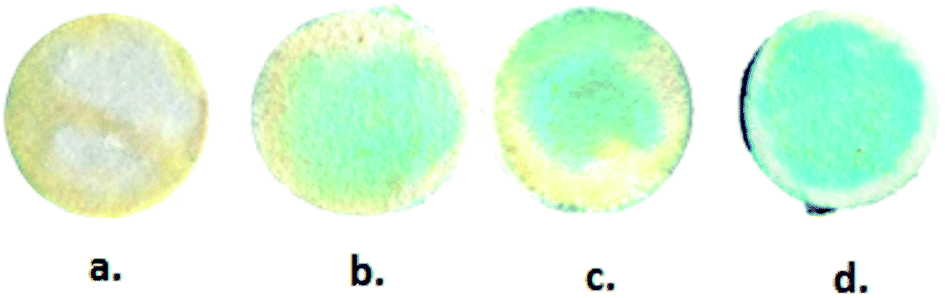
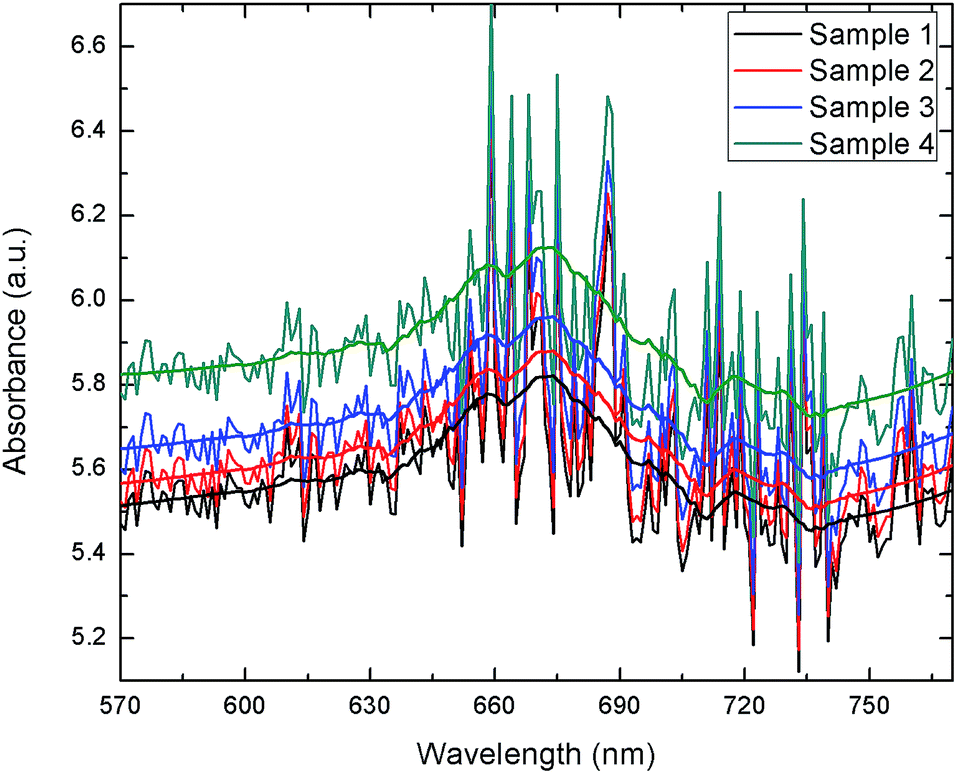
The extracted blood serum samples (0.1 mL) were pipetted onto the paper strips on which the AuNPs–GO conjugates were already deposited. The photographs shown in Fig. 12 depict the color change of the paper strips after 5 minutes of addition of the blood serum samples. UV-Vis spectra of these circular strips were recorded to calculate the UA concentration in the respective samples (Fig. 13). We did a similar averaging as before on the UV-Vis data and found a peak at 675 nm. The obtained results were validated by comparing them with the conventional clinical test results performed through the uricase-Trinder method28 given in Table 2. The results were in accordance with the clinical results.
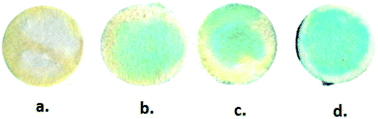 |
| | Fig. 12 Photographs of blood serum samples on the paper strips. (a) Sample 1, (b) sample 2, (c) sample 3 and (d) sample 4. | |
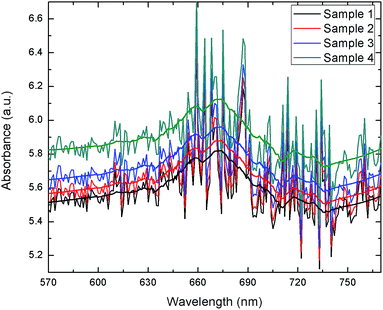 |
| | Fig. 13 UV-Vis spectra of blood serum samples on paper strips. | |
Table 2 Comparison of clinical and paper device results
| Blood serum sample |
Clinical result (ppm) |
Paper device result (ppm) |
| Sample 1 |
71 |
67.75 |
| Sample 2 |
67 |
65.67 |
| Sample 3 |
65 |
63.68 |
| Sample 4 |
52 |
57.48 |
Selectivity of the catalytic reduction of ox-TMB by uric acid
The four blood serum samples were spiked with different known concentrations of uric acid, which were 1 ppm, 1.5 ppm, 2 ppm and 3 ppm. The results shown in Table 3 show the good recovery of the added uric acid. This confirms that the other entities present in the serum had no obvious effect on the catalytic reduction of ox-TMB.
Table 3 Recovery results of spiked blood serum samples
| Blood serum sample |
Result without spiking (ppm) |
Spiking concentration of uric acid (ppm) |
Result with spiking (ppm) |
Recovery (%) |
| Sample 1 |
68.65 |
1 |
67.81 |
97.35 |
| Sample 2 |
65.97 |
1.5 |
68.55 |
101.6 |
| Sample 3 |
64.07 |
2 |
64.9 |
98.23 |
| Sample 4 |
53.02 |
3 |
57.98 |
103.5 |
Conclusions
A paper-based assay for the colorimetric detection of uric acid in blood serum using AuNPs–GO conjugates has been developed. We explored the peroxidase-like activity of AuNPs–GO by performing analyses in both the liquid (solution) and solid phase (paper). The paper device is capable of detecting uric acid at a concentration as low as 4 ppm within five minutes, which is the minimum detectable limit of detection reported for all paper based diagnostics used so far for uric acid. The results from the real blood serum samples demonstrate the device's applicability as a sensitive, easy-to-use, inexpensive and portable detection tool for point-of-care health diagnostics.
Acknowledgements
We would like to acknowledge the Center for Nanosciences and Advanced imaging at IIT Kanpur for providing the characterization facilities. We are thankful to Mr Tapan Kumar Pradhan for his assistance with flow rate experimental measurements.
Notes and references
- M. Alderman and K. J. Aiyer, Curr. Med. Res. Opin., 2004, 20, 369–379 CrossRef CAS PubMed.
- D. Lakshmi, M. J. Whitcombe, F. Davis, P. S. Sharma and B. B. Prasad, Electroanalysis, 2011, 23, 305–320 CrossRef CAS.
- M. K. Kutzing and B. L. Firestein, J. Pharmacol. Exp. Ther., 2008, 324, 1–7 CrossRef CAS PubMed.
- R. J. Johnson, D.-H. Kang, D. Feig, S. Kivlighn, J. Kanellis, S. Watanabe, K. R. Tuttle, B. Rodriguez-Iturbe, J. Herrera-Acosta and M. Mazzali, Hypertension, 2003, 41, 1183–1190 CrossRef CAS PubMed.
- V. L. Clark and J. A. Kruse, J. Am. Med. Assoc., 1990, 264, 2808–2809 CrossRef.
- J. Perelló, P. Sanchis and F. Grases, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2005, 824, 175–180 CrossRef PubMed.
- J. Du, R. Yue, F. Ren, Z. Yao, F. Jiang, P. Yang and Y. Du, Gold Bull., 2013, 46, 137–144 CrossRef.
- J. Matějčková, P. Tůma, E. Samcová and Z. Zemanová, J. Sep. Sci., 2007, 30, 1947–1952 CrossRef PubMed.
- R. D. Chaudhari, A. B. Joshi and R. Srivastava, Sens. Actuators, B, 2012, 173, 882–889 CrossRef CAS.
- A. Kumar, A. Hens, R. K. Arun, M. Chatterjee, K. Mahato, K. Layek and N. Chanda, Analyst, 2015, 140, 1817–1821 RSC.
- J. Lu, Y. Xiong, C. Liao and F. Ye, Anal. Methods, 2015, 7, 9894–9899 RSC.
- D. Wu, H.-F. Lu, H. Xie, J. Wu, C.-M. Wang and Q.-L. Zhang, Sens. Actuators, B, 2015, 221, 1433–1440 CrossRef CAS.
- X. Chen, J. Chen, F. Wang, X. Xiang, M. Luo, X. Ji and Z. He, Biosens. Bioelectron., 2012, 35, 363–368 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2009, 82, 3–10 CrossRef PubMed.
- W. Dungchai, O. Chailapakul and C. S. Henry, Anal. Chim. Acta, 2010, 674, 227–233 CrossRef CAS PubMed.
- K. Saha, S. S. Agasti, C. Kim, X. Li and V. M. Rotello, Chem. Rev., 2012, 112, 2739–2779 CrossRef CAS PubMed.
- L. Zhan, C. M. Li, W. B. Wu and C. Z. Huang, Chem. Commun., 2014, 50, 11526–11528 RSC.
- D. Carboni, B. Lasio, V. Alzari, A. Mariani, D. Loche, M. F. Casula, L. Malfatti and P. Innocenzi, Phys. Chem. Chem. Phys., 2014, 16, 25809–25818 RSC.
- N. Zhang, H. Qiu, Y. Liu, W. Wang, Y. Li, X. Wang and J. Gao, J. Mater. Chem., 2011, 21, 11080–11083 RSC.
- X. Fu, L. Chen, J. Li, M. Lin, H. You and W. Wang, Biosens. Bioelectron., 2012, 34, 227–231 CrossRef CAS PubMed.
- B. Garg, T. Bisht and Y.-C. Ling, Molecules, 2015, 20, 14155–14190 CrossRef CAS PubMed.
- P. D. Josephy, T. Eling and R. P. Mason, J. Biol. Chem., 1982, 257, 3669–3675 CAS.
- X. Wang, J. A. Hagen and I. Papautsky, Biomicrofluidics, 2013, 7, 014107 CrossRef PubMed.
- H. Zhang, D. Hines and D. L. Akins, Dalton Trans., 2014, 43, 2670–2675 RSC.
- B. Li, L. Fu, W. Zhang, W. Feng and L. Chen, Electrophoresis, 2014, 35, 1152–1159 CrossRef CAS PubMed.
- A. K. Ellerbee, S. T. Phillips, A. C. Siegel, K. A. Mirica, A. W. Martinez, P. Striehl, N. Jain, M. Prentiss and G. M. Whitesides, Anal. Chem., 2009, 81, 8447–8452 CrossRef CAS PubMed.
- W. Zhang, B. Li, L. Chen, Y. Wang, D. Gao, X. Ma and A. Wu, Anal. Methods, 2014, 6, 2066–2071 RSC.
- G. Sanders, A. Pasman and F. Hoek, Clin. Chim. Acta, 1980, 101, 299–303 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay01926a |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence