
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMeasurement uncertainty associated with shipboard sample collection and filtration for the determination of the concentration of iron in seawater†
Robert
Clough
a,
Geerke H.
Floor‡
b,
Christophe R.
Quétel
b,
Angela
Milne
a,
Maeve C.
Lohan§
a and
Paul J.
Worsfold
 *a
*a
aBiogeochemistry Research Centre, School of Geography, Earth and Environmental Sciences, Plymouth University, Plymouth, PL4 8AA, UK. E-mail: pworsfold@plymouth.ac.uk
bInstitute for Reference Materials and Measurements, Joint Research Centre – European Commission, 111 Retieseweg, B-2440 Geel, Belgium
First published on 18th August 2016
Abstract
A flow injection with chemiluminescence detection (FI-CL) method was used to determine the concentration of dissolved iron in seawater samples collected in the South Atlantic during the GEOTRACES GA10 cruise that took place from 24th December 2011–27th January 2012 on board the R.R.S. James Cook (cruise JC068). Six different sample collection and filtration strategies were used. Open ocean (shallow and deep) and coastal (shallow and deep) samples were collected and five sub-samples from each collection were filtered through a cartridge filter. For the deep open ocean sample, separate sub-samples were also filtered through a membrane disc filter. In addition, deep open ocean sub-samples were also taken from five separate sampling bottles. Each sub-sample (29 in total) was analysed six times (giving 174 discrete measurements in total) and the within sub-sample precision was in the range 1.4–12.2%. There was no statistically significant difference for the deep, open ocean sample between the mean results obtained with the two different filter types or the single sample bottle versus separate sample bottle sub-samples. Application of classical ANOVA showed that the relative combined standard uncertainty for each of the six sampling strategies ranged from 2.3–3.8%. This approach did not include an estimation of sampling bias. Application of robust ANOVA to the deep open ocean samples showed that contributions to the total variance were 0% from the different sample collection and filtration strategies, 42% from the sub-sample precision and 58% from between sub-sample measurements.
1. Introduction
Reliable determinations of dissolved iron concentrations in marine waters are needed to enhance our understanding of the impact of iron on ocean productivity and processes (e.g. ocean acidification). Iron typically exists at sub-nmol L−1 concentrations in the ocean and in different labile physico-chemical forms, which makes it analytically challenging to measure. Moreover, seawater composition, including iron concentrations and speciation, varies over spatial and temporal scales, meaning that measurement uncertainty needs to be quantified in order to determine if there is any ‘real’ environmental variability or seasonality in a dataset.According to the Guide for Uncertainty in Measurements,1 a measurement begins with an appropriate specification of the measurand, i.e. the particular quantity (defined by the analyte/technique, unit of measurement, matrix and sampling location) intended to be measured. From the above it is apparent that the specification of the measurand for ‘iron in seawater’ measurements could easily vary depending on the Fe species targeted and how representative the collected seawater samples are. In addition, any sample treatment, such as filtration, will lead to an operationally defined measurand, with implications for traceability. Therefore the sample collection and treatment strategy is a fundamental part of the measurement procedure and as such must be taken into account for the estimation of the combined uncertainty associated with the measurement results, as well as for establishing the traceability of these results.2 The sampling target should also be defined and for the shipboard collection of seawater this is typically a body of water at a specified depth (±5 m).
Recent efforts in chemical oceanography to compare datasets on a global scale and over seasonal/annual timescales has been demonstrated in the GEOTRACES Intermediate Data Product 2014 (see http://www.bodc.ac.uk/geotraces/data/idp2014/), which includes the analysis of samples collected at “cross-over” stations, i.e. common sampling locations for different cruises.3 Comparability is dependent on the measurand(s), and thus the objectives chosen for the sample collection and filtration strategies and the associated measurement procedures. For example, one objective of a sample collection strategy could be to establish the dissolved Fe concentration in a given seawater sample (from one or more measurement results from the same sample). Another objective could be to establish (from measurement results from several samples) the average concentration of a specific range of Fe species at a given location in the global ocean.
To improve the reliability of dissolved iron data, whatever the defined measurand, all sources of uncertainty should be identified and quantified and then a combined uncertainty estimated.1 Common practice is to use precision on replicate measurement results for a single sample, but this can underestimate the total combined uncertainty. However, even with more robust uncertainty estimations, often only the analytical process is described and the sample collection uncertainty is not taken into account.
The Eurachem/EUROLAB/CITAC/Nordtest/RSC Analytical Methods Committee Guide “Measurement uncertainty arising from sampling: a guide to methods and approaches”4 includes sampling and sample preparation, in addition to the analytical process, in a holistic approach to determining the uncertainty associated with the overall measurement process, i.e. “the measurand is defined in terms of the value of the analyte concentration in a sampling target, rather than in just the sample delivered to the laboratory”. It describes two main approaches to the estimation of uncertainty from sampling; an empirical approach (also called “experimental”, “retrospective”, or “top-down”) and a modelling approach (also called “theoretical”, “predictive” or “bottom-up”). Robust analysis of variance (ANOVA) is one empirical approach that can be used to estimate the overall measurement uncertainty, including sample collection.5 The ANOVA approach is straightforward to apply using widely available software packages such as Excel® and the output can be used to give an estimate of the separate contributions from replicate analysis (i.e. the short term analytical repeatability) and the between measurement variability (which, with a suitable experimental design, will include uncertainty in sample collection and filtration). However the classical ANOVA approach is not able to estimate bias in either sampling or analysis, for which more complex methods are available.5,6
The aim of this work was to use ANOVA to determine the relative uncertainty associated with the way in which seawater samples are collected and conditioned for the determination of dissolved iron concentrations. It was not an aim to investigate the relative uncertainty associated with sampling per se. Flow injection with chemiluminescence detection (FI-CL) was chosen as the method of detection for this study because it is a commonly used shipboard and laboratory based method for the determination of dissolved iron concentrations7–10 and the uncertainties associated with the analytical step have already been systematically investigated.11 The specific objectives were to collect and analyse replicate, filtered seawater samples from the Atlantic Ocean covering a range of dissolved Fe concentrations (from deep and shallow waters and from coastal and open ocean locations) and to compare the results for different ways of collecting and filtering the samples, i.e. sub-samples from the same sampling bottle versus samples from separate bottles and the use of cartridge filters versus membrane filters (which are commonly used to collect marine particulate matter).
2. Experimental
2.1 Sample collection
Seawater samples were collected from two stations during the South Atlantic GEOTRACES GA10 cruise that took place from 24th December 2011–27th January 2012 on board the R.R.S. James Cook (JC068). A schematic diagram of the sample collection and filtration strategies applied is shown in Fig. 1 and was designed to test whether the type of filtration applied to samples would influence measurement results. The possibility of a difference between results from one sampling bottle and results from several sampling bottles filled within a 10 m depth range was also tested. A titanium-frame CTD (conductivity/temperature/depth) fitted with 24 trace metal clean 10 L Ocean Test Equipment water samplers (Ocean Test Equipment Inc., Fort Lauderdale, Florida, USA; henceforth described as OTE bottles) deployed on a Plasma® rope was used for water column sampling. The 10 m depth range was a constraint of the sampling equipment which required a delay between the closing of each OTE bottle during recovery of the CTD. On recovery, the OTE bottles were transferred into a trace metal clean sampling container and were lightly pressurised (1.7 bar) with high purity compressed air filtered in-line using a 0.2 μm PTFE filter capsule (Millex-FG 50, EMD Millipore, Merck KGaA, Darmstadt, Germany). | ||
| Fig. 1 Strategies for the collection and filtration of seawater samples (as defined in Table 1). | ||
Eight discrete samples from separate OTE bottles (see unique sample numbers in Table 1) were collected and all (except those collected from separate OTE bottles, i.e. strategy S3) were sub-divided to give twenty nine sub-samples in total (see Table 1 for details). All were filtered directly into acid cleaned 250 mL low density polyethylene bottles (LDPE; Nalgene, Fisher Scientific, Loughborough, UK) under positive pressure and acidified on board with 500 μL of UpA hydrochloric acid, HCl (Romil, Cambridge, UK) to give a final acid concentration of 0.024 mol L−1 (pH 1.7). The filters used were either 0.45 μm nominal pore size, 25 mm diameter Supor® polyethersulfone (PES) membrane disc filters (Pall Life Sciences, Portsmouth, UK) or 0.8/0.2 μm nominal pore size PES cartridge filters (AcroPak500™, Pall Life Sciences, Portsmouth, UK). Table 1 also shows the sample collection locations, matrices and depths for each of the six sample collection strategies (S1–S6). All sample bottles were labelled with a unique number and also marked with either PES or ACRO to distinguish between membrane and cartridge filter types respectively. Samples from Station 18 were filtered using the same AcroPak filter but a different AcroPak filter was used for the samples collected from Station 22. All sample conditioning was carried out by the same person and samples were stored double bagged for subsequent shore based analyses.
| Station and location | Sample matrix | Depth (m) | Salinity | Filter type | OTE bottle no. | Unique sample number | Number of sub-samples | Sample collection and filtration strategy |
|---|---|---|---|---|---|---|---|---|
| Station 18 (40°S 42°25′W) | Open ocean (deep) | 4736 | 34.67233 | Cartridge (AcroPak) | 5 | 1955 | 5 | S1/S3 |
| Membrane disc (PES) | 5 | S2 | ||||||
| Open ocean (deep) | 4734 | 34.67229 | Cartridge (AcroPak) | 6 | 1956 | 1 | S3 | |
| Open ocean (deep) | 4730 | 34.67237 | Cartridge (AcroPak) | 7 | 1957 | 1 | S3 | |
| Open ocean (deep) | 4729 | 34.67208 | Cartridge (AcroPak) | 8 | 1958 | 1 | S3 | |
| Open ocean (deep) | 4726 | 34.67242 | Cartridge (AcroPak) | 9 | 1959 | 1 | S3 | |
| Open ocean (shallow) | 22 | Cartridge (AcroPak) | 24 | 2022 | 5 | S4 | ||
| Station 22 (36°32′S 53°06′W) | Coastal (shallow) | 23 | Cartridge (AcroPak) | 24 | 2376 | 5 | S5 | |
| Coastal (deep) | 1295 | Cartridge (AcroPak) | 9 | 2361 | 5 | S6 |
2.2 FI-CL reagents and on-shore sample handling
Concentrated HCl, ammonia (NH3, 20–22%) and glacial acetic acid (CH3CO2H), all SpA grade, were obtained from Romil. Hydrogen peroxide, Merck Suprapur grade, was obtained from VWR (Lutterworth, UK). Luminol (5-amino-2,3-dihydro-1,4-phthalazinedione), sodium carbonate and triethylenetetramine (TETA) were obtained from Sigma Aldrich (Gillingham, UK). All high purity water (HPW), 18.2 MΩ cm, was drawn from an ElgaStat Maxima system (Marlow, UK). All weighing was performed using an analytical balance (OH1602/C, Ohaus, Thetford, UK). The accuracy of the balance was checked daily before use with F1 Class certified weights (KERN, Albstadt, Germany). All facilities were managed under ISO 9001:2008 certification.All shore-based sample and reagent handling was undertaken in an ISO 14644-1 Class 5 laminar flow hood (Bassaire, Southampton, UK) situated within an ISO 14644-1 Class 6 clean room. Reagent and sample containers were cleaned using established cleaning protocols for trace metals.12,13
2.3 FI-CL measurement method
The FI-CL method, based on the method of Obata et al.,14 is the same as that used by Floor et al.11 and the manifold used is shown in Fig. 2. In summary, there were three peristaltic pumps (Minipuls 3, Gilson, Luton, UK), one PTFE manually operated three port valve (Valve 1; Omnifit), one three port solenoid valve (Valve 2) and one two-way six port electronically actuated valve (Valve 3; VICI, Valco Instruments, Schenkon, Switzerland). The detector was a photomultiplier tube (PMT; Hamamatsu H 6240-01, Hamamatsu Photonics, Welwyn Garden City, UK) containing a coiled, transparent PVC flow cell (volume 40 μL). Manifold tubing was 0.8 mm i.d. PTFE. The preconcentration column, loaded with Toyopearl AF Chelate 650M resin (Tosoh Bioscience, Stuttgart, Germany), was made of polyethylene with LDPE frits with an internal volume of 200 μL (Global FIA, Fox Island, USA). Two poly(methyl methacrylate) columns (1 cm long, 1.5 mm i.d., volume 70 μL), also loaded with Toyopearl AF Chelate 650M resin retained with HDPE frits (BioVion F, 0.75 mm thick, 22–57 μm pore size), were used to clean up the buffer and column rinse solutions.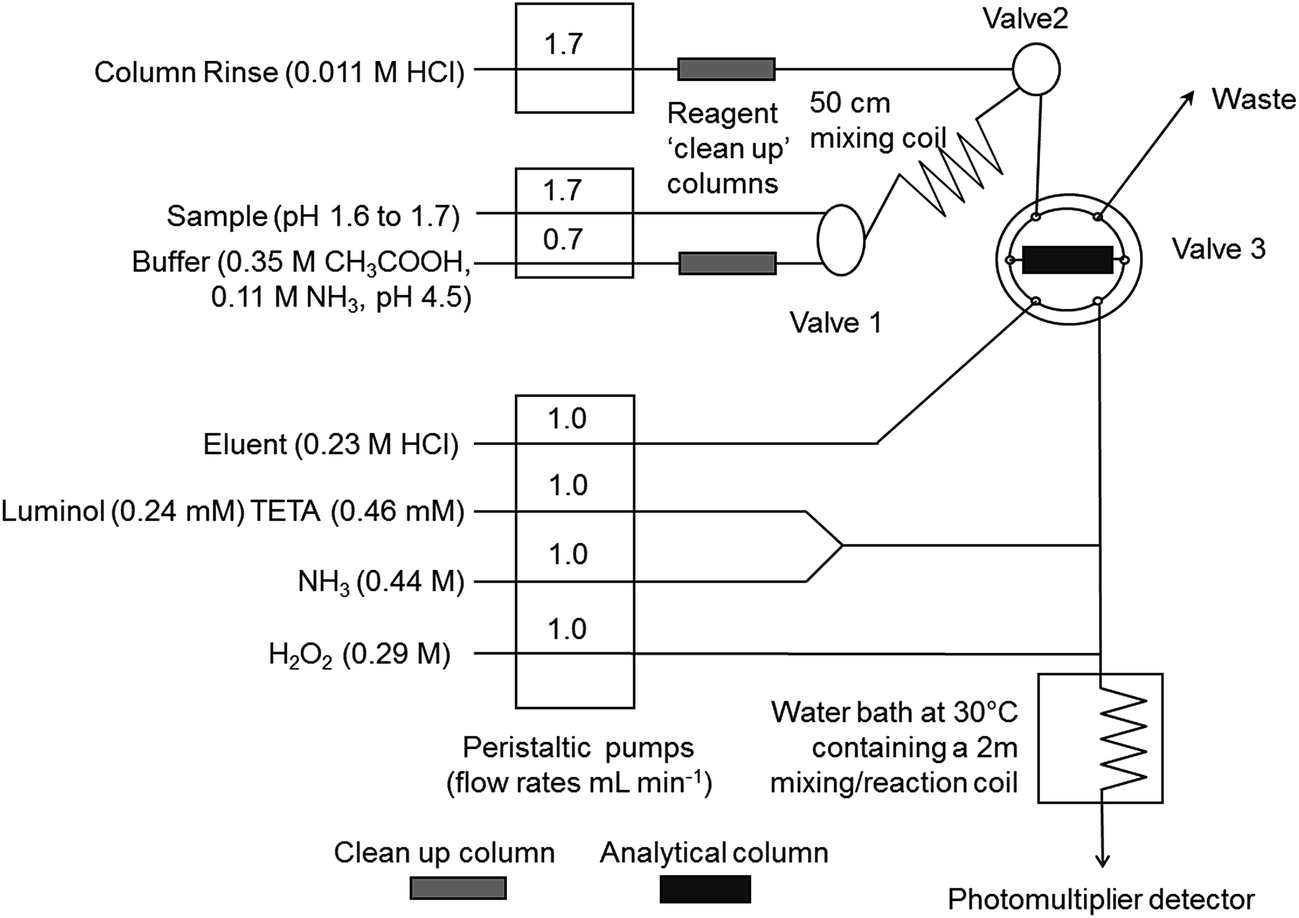 | ||
| Fig. 2 FI-CL manifold for the determination of dissolved iron concentrations in seawater. Reproduced, with permission, from reference 11 (G. H. Floor, R. Clough, M. C. Lohan, S. J. Ussher, P. J. Worsfold and C. R. Quétel, Limnology and Oceanography Methods, 2015, 13, 673-686). | ||
All samples were oxidised by adding 1 μL of 10 mM hydrogen peroxide per 1 mL of sample at least 30 min before analysis to ensure that all Fe was present as Fe(III). All reagent solutions were prepared at least 12 h before use and the concentrations used are shown in Fig. 1. The analytical blank was measured using the “closed sample valve” method, i.e. the wash solution and ammonium acetate buffer were pumped through the preconcentration column with the same load time as was used for the subsequent samples in that run. One measurement consisted of six replicates run contiguously, which is typical of FI-CL measurements, and the variability of these analytical replicates is therefore referred to throughout as the short term analytical repeatability. Each replicate measurement consisted of the following analytical cycle. The column was conditioned for 10 s with 0.011 M HCl. Sample and buffer were then loaded simultaneously for 60–180 s (see Table 2), depending on the magnitude of the CL signal. The amount of sample loaded onto the column was determined gravimetrically. The column was washed with 0.011 M HCl for 20 s. The Fe on the column was then eluted with 0.23 M HCl for 120 s. Between each sample, the sample line was washed with HPW for 30 s followed by uptake of the fresh sample for 180 s. After each analytical session all lines were flushed with 0.01 M HCl for 10 min and then with HPW for 15 min and HPW was left in the lines. All signals were normalised to the loaded calibration standard/sample mass and are reported as V g−1 (volts per gram). An aliquot of Atlantic seawater from 40°S containing 0.45 nmol L−1 Fe was used to measure instrumental drift at the start and end of each analytical run.
| Sample/sampling strategy | Source of variation | Sum of the squares (SS) | Degrees of freedom (df) | Mean square (MS) | F | P-value | F crit |
|---|---|---|---|---|---|---|---|
| Control material | Between groups | 0.05071 | 2 | 0.02536 | 3.754 | 0.04766 | 3.682 |
| Within groups | 0.1013 | 15 | 0.006755 | ||||
| Total | 0.1520 | 17 | |||||
| S1 | Between groups | 0.09410 | 4 | 0.02352 | 12.58 | <0.00001 | 2.758 |
| Within groups | 0.04672 | 25 | 0.001868 | ||||
| Total | 0.1408 | 29 | |||||
| S2 | Between groups | 0.03594 | 3 | 0.01198 | 4.792 | 0.01126 | 3.098 |
| Within groups | 0.04999 | 20 | 0.0025 | ||||
| Total | 0.08593 | 23 | |||||
| S3 | Between groups | 0.03838 | 3 | 0.01279 | 3.115 | 0.04918 | 3.098 |
| Within groups | 0.08213 | 20 | 0.004106 | ||||
| Total | 0.1205 | 23 | |||||
| S4 | Between groups | 0.0002 | 3 | 6.663 × 10−5 | 0.9378 | 0.44090 | 3.0984 |
| Within groups | 0.001421 | 20 | 7.105 × 10−5 | ||||
| Total | 0.001621 | 23 | |||||
| S5 | Between groups | 0.008945 | 3 | 0.002982 | 5.020 | 0.00935 | 3.098 |
| Within groups | 0.01187 | 20 | 0.0005939 | ||||
| Total | 0.02082 | 23 | |||||
| S6 | Between groups | 0.1631 | 4 | 0.04079 | 3.635 | 0.01817 | 2.758 |
| Within groups | 0.2805 | 25 | 0.01122 | ||||
| Total | 0.4436 | 29 |
2.4 Data treatment
The FI output data were collected and reported as peak heights as this is the commonly used parameter for FI-CL measurements in both the oceanographic and flow injection communities. Peak area measurements were also made (data not shown) and followed the same trends as the peak height data. All sample loadings were mass normalised. Further data treatment was carried out in Excel®. Data were subject to a Grubbs test for outliers prior to processing.For each set of n replicate measurements of a sub-sample;
| Within sub-sample precision (%, n = 6) = (σwithin/meanwithin) × 100 | (1) |
Analysis of variance (ANOVA) was applied to each of the S1–S6 datasets and the output from Excel® gave the mean square “between groups” and “within groups”, designated as MSbetween and MSwithin respectively, for each dataset. See ESI Table 2† for the equations used to calculate MSbetween and MSwithin. Equations (2)–(4), derived from Linsinger et al.15 and van der Veen et al.16 and already applied to uncertainty estimations for the determination of dissolved iron amount content using FI-CL,11 were then used to calculate relative standard uncertainties as shown below. All standard uncertainties are reported as relative values and are denoted by “urel”.
The relative standard uncertainty (coverage factor k = 1) corresponding to the short-term analytical repeatability;
| urel_repeatability = ((√(MSwithin) × 100/meanbetween)/√n | (2) |
The relative standard uncertainty (coverage factor k = 1) corresponding to the between measurement uncertainty (which will encompass uncertainties from a range of effects);
| urel_between measurement = (((√((MSbetween − MSwithin)/n)) × 100/meanbetween)/√m) | (3) |
The relative combined standard uncertainty, which assumes that both components are random and uncorrelated;
| urel_combined = √((urel_repeatability)2 + (urel_between measurement)2) | (4) |
3. Results and discussion
3.1 Sample collection and filtration
Section VI of the GEOTRACES ‘cookbook’ for micronutrient sample collection and sample-handling17 is recommended as a general guide for practical aspects of seawater sample collection and handling strategies for the determination of trace elements. For the determination of dissolved iron (and other trace metals) concentrations the use of appropriate filter types and filtration protocols is also critical10 and may impact on the relative overall measurement uncertainty. Therefore in order to decide if results for dissolved iron in deep, open ocean water from different sampling campaigns can be compared it is important to know what influence the sample collection strategy, i.e. single versus multiple sample bottles (S1 and S3) and filtration strategy, i.e. the type of filter used (S1 and S2), may have on the quantity measured, and what contribution this can make to the relative overall uncertainty of the result. In addition, it is important to consider the influence of the level of dissolved Fe concentration on the relative overall uncertainty. Hence samples were collected and filtered in the same way from four locations (S1/S2/S3, S4–S6), with contrasting dissolved Fe concentrations (S1S2/S3 = 0.39 nM, S4 = 0.27 nM, S5 = 1.27 nM and S6 = 1.04 nM). The sample collection and filtration strategies used in this work shown in Fig. 1 and Table 1 were therefore designed to investigate these influences.3.2 Data acquisition and preliminary evaluation
There were three analytical runs; sub-samples from strategies S4 and S5 were analysed on day 1 (8 h run time), sub-samples from strategies S1, S2 and S3 were analysed on day 2 (10 h run time) and sub-samples from strategy S6 were analysed on day 5 (5 h run time). The stability of the measurement method was monitored by replicate analysis (n = 6) of a control material (Atlantic seawater sample from 40°S) at the beginning and end of each analytical run (and also after 4.5 h of the analytical run on day 2). The means of these replicate analyses for the start and end of each analytical run showed no significant difference (P = 0.05) for any of the analytical runs based on a two way t-test (tcalc all <1.5; ttab = 2.23).The control material data for the analytical run on day 2 (m = 3) were further explored using one way ANOVA to investigate signal stability during the analysis of sub-samples from S1, S2 and S3. The ANOVA output (Table 2) showed that there was a significant difference between the control material results over the 10 h analysis period (P = 0.048). Therefore the short-term analytical repeatability (eqn (2)) and the signal stability for the control material over the 10 h analysis period, hereafter called the within-sequence-stability (eqn (3)), were calculated using the approach outlined in the literature to determine “between-bottle” homogeneity.15,16 Using this approach it is only possible to calculate two separate uncertainty components, the second of which (calculated using eqn (3)) may actually cover a range of effects and the assumption made here is that the within-sequence-stability is the dominant effect for the control material. For the seawater samples (S1–S6) the uncertainty linked to the collection and filtration strategy applied will also be included in this component. The relative standard uncertainty corresponding to the short-term analytical repeatability for the control material was 2.4% and the within-sequence-stability over 10 h was 2.3%. These data show the importance of regularly analysing a control material during a FI-CL run in order to minimise the uncertainty contribution from any signal instability and supports the practical recommendation of Floor et al.11 that a control material should be run every 2 h and the system recalibrated if the value is outside of a specified range.
The objective of this work was to compare results for different modes of sample collection and filtration and the strategy was based on empirical method #2 in the “Measurement uncertainty arising from sampling” Guide.4 Hence analytical bias was not determined but Floor et al.11 concluded that results for three seawater reference materials, using the same FI-CL method as was used in this work, were in agreement with consensus values within their reported uncertainty statements.
The dataset for n replicate measurements of each set of m sub-samples for the six sample collection strategies (S1–S6) is shown in ESI Table 1.† Prior to comparing the results for the different sample collection strategies the overall dataset was assessed for outliers. Twenty nine sub-samples were analysed and results were rejected from sub-samples PES 1955-5, 2022-5 and 2376-4 (from strategies S2, S4 and S5 respectively). These three results had a positive bias of 46%, 150% and 37% respectively, with RSDs for six replicate FI-CL measurements of 5.4%, 3.0% and 0.9% respectively. This suggests contamination at some point in the overall process for these three sub-samples and highlights the importance of rigorous cleaning and careful sample handling. It also shows the benefit of collecting and analysing several sub-samples (including the use of separate sample bottles) as a means of identifying random sources of contamination.
3.3 Comparison of the results for the six different sample collection strategies (S1–S6)
The within sub-sample precision and the mean sub-sample peak height for the six sample collection strategies are presented in Table 3. Of most interest are the three deep, open ocean collection strategies (strategies S1, S2 and S3 in Table 1) as they all sampled the same water mass (within a 10 m depth range) and had the same salinity (34.672) and would therefore be expected to give the same mean peak heights unless the collection and/or filtration strategy used (and/or the within-sequence-stability) impacted on the measurement result. A two way t-test showed that there was no significant difference between the mean sub-sample peak heights for cartridge and membrane disc filtered sub-samples from the same OTE bottle (S1 and S2; tcalc = 0.269, ttab = 2.36) or for sub-samples from the same OTE bottle compared with sub-samples from separate OTE bottles (strategies S1 and S3; tcalc = 0.418, ttab = 2.36). Two way ANOVA confirmed that there were no significant differences resulting from the type of filter used (sample collection strategies S1 and S2; P = 0.999) or from collecting samples from a single OTE bottle versus multiple OTE bottles (sample collection strategies S1 and S3; P = 0.102).| Sample collection and filtration strategy | FI-CL load time (s) | Within sub-sample precision (n = 6) (%) | Mean sub-sample peak height (V g−1) |
|---|---|---|---|
| S1 | 90 | 3.4–6.1 | 0.855 |
| S2 | 90 | 3.8–7.6 | 0.845 |
| S3 | 90 | 3.3–12.2 | 0.872 |
| S4 | 180 | 4.1–9.4 | 0.122 |
| S5 | 120 | 1.4–6.3 | 0.579 |
| S6 | 60 | 3.9–4.7 | 2.38 |
Cartridge filters are typically used to collect samples for dissolved Fe concentration measurements and can be used at a higher flow rate than membrane filters.18 However membrane filters must be used to collect samples for particulate Fe measurements. In addition, when at sea, there are often competing demands on the water budget and constraints on ship time, which would make it advantageous to use membrane filters to simultaneously collect both particulate and dissolved samples providing that there was no significant difference between the two filtration methods for dissolved iron concentration measurements. Data comparing filter types for the determination of dissolved iron are sparse19,20 but a GEOTRACES intercalibration showed no difference (within analytical uncertainty) in neodymium concentrations measured in Atlantic seawaters using five different filter types and nominal pore sizes (0.2–1.0 μm).21 The results from the current study show that membrane filters could be used instead of cartridge filters for the determination of dissolved iron and that, in spite of the differences in filter design and nominal size cut-off, applying ANOVA to the data from this experimental design showed that there was no significant difference in the dissolved Fe species collected (at least for this particular water mass).
The demands on the water budget may also require collection of the same water mass from more than one OTE bottle and the results show that this would also be a reasonable strategy as there was no significant difference between the single bottle and multiple bottle dissolved Fe concentration measurements.
3.4 Contribution of sample collection and filtration to the relative overall uncertainty for each sampling strategy
The discussion in Section 3.3 focused on the variability of mean FI-CL results for 4–5 replicate sub-samples for each of the separate sample collection and filtration strategies. It showed that there were no significant differences between the two filtration methods or the use of single versus multiple OTE bottles for sample collection. The application of ANOVA to each dataset (S1–S6) of n replicate measurements for each of m replicate sub-samples generates mean square outputs that can be used (see eqn (2) and (3) in Section 2.4) to determine the relative contributions of uncertainty in the FI-CL measurement step (i.e. the short-term analytical repeatability, urel_repeatability) and the uncertainty between measurements (urel_between measurement) to the overall uncertainty. In this work, urel_between measurement will include both the uncertainty linked to the collection and filtration strategy applied, possibly influenced by the water mass characteristics, and the within-sequence-stability over the analytical session arising from any variability in response over the length of time required (10 h) to measure all of the different replicate sub-samples and control material. It may also include contributions from other, unspecified factors. The relative combined standard uncertainty (urel_combined) can be obtained by the quadratic combination of urel_repeatability and urel_between measurement (eqn (4)).The output data from the application of these equations (Table 4) show that urel_repeatability (ranging from 1.7–3.0%) was of a similar magnitude to urel_between measurement (ranging from <0.1–3.1%). ESI Table 2† shows the calculations used to obtain these values. This range for urel_repeatability is in agreement with the urel_repeatability of 1.7% (n = 6) for FI-CL reported by Floor et al.11 for the same determination. The relative combined standard uncertainty (urel_combined) ranged from 2.3–3.8%. The ANOVA outputs in Table 2 however show that there were significant differences (P < 0.05) between sub-sample measurements for all sample collection and filtration strategies except for S4. Nonetheless, the control material had a similar urel_between measurement (2.3%) to the value reported by Floor et al.11 of 2.8% for within-sequence stability. This suggests that the within-sequence-stability was the dominant contributor to urel_between measurement and highlights again the importance of analysing a control material frequently during a FI-CL run. It also supports the conclusion that for this particular set of samples, using FI-CL detection, there was no significant difference between the different sample collection and filtration strategies used.
| Sample collection and filtration strategy | Short-term analytical repeatability k = 1 (%) | Between-measurement variability k = 1 (%) | Relative combined standard uncertainty k = 1 (%) |
|---|---|---|---|
| Control material | 2.4 | 2.3 | 3.3 |
| S1 | 2.1 | 3.1 | 3.8 |
| S2 | 2.4 | 2.4 | 3.4 |
| S3 | 3.0 | 2.2 | 3.7 |
| S4 | 2.8 | <0.1 | 2.8 |
| S5 | 1.7 | 1.7 | 2.4 |
| S6 | 1.8 | 1.3 | 2.3 |
Further assessment of the combined data for S1, S2 and S3 using robust ANOVA,22i.e. with three sampling targets, the first four sub-samples for each target (to provide a balanced design) and six replicate analyses for each sub-sample, provided further evidence that there was no significant difference between the different sample collection and filtration strategies used (i.e. the three targets) and that urel_repeatability and urel_between measurement were of a similar magnitude. The relative contributions to the total variance using robust ANOVA were 0% from the different sample collection and filtration strategies used, 42% from the short term analytical repeatability and 58% from between sub-sample measurements.
In other situations the within-sequence-stability may not be the dominant contributor to urel_between measurement. Firstly, if an experimental design with additional statistical power could be implemented, e.g. one based on the recommendations given in ref. 4, the application of ANOVA may lead to a different conclusion. The implementation of such a strategy would, however, require dedicated and expensive ship time. Secondly, for methods with a lower analytical uncertainty or a more constant short-term analytical repeatability compared with FI-CL, there may be a greater contribution from sample collection and filtration. For example, the use of FI with collision/reaction cell – quadrupole ICP-MS detection for the determination of dissolved iron concentration in seawater reference materials gave a relative combined standard uncertainty of 4% for GEOTRACES GD (1.035 nmol L−1)23 compared with the 5–6% reported for FI-CL.11 For further comparison, Milne et al. reported a short-term analytical repeatability for iron in NASS-5 (3.29 nmol L−1) using isotope dilution ICP-MS of 2.3% (n = 3).24 In these examples the method would potentially be more sensitive to variations caused by any uncertainty in sample collection and filtration. However, the salinity data for the deep open ocean samples (S1, S2 and S3) shows that there was a negligible difference (RSD = 0.0004%, Table 1) in this physical property between these samples, which implies that sample collection did not contribute significantly to the overall measurement uncertainty.
Additionally, the scale of observation is critical. In this study the relative sample collection and filtration uncertainty for a specific location at a specific time was investigated. However, chemical oceanography datasets are compared on a global scale and over seasonal/annual timescales. The sample collection uncertainty will potentially be greater when taking into account variations over relatively small temporal (e.g. days per week) and/or spatial (e.g. sub-km) scales. The sample collection strategy could therefore be refined to take sample replicates at slightly different times and locations so as to include this portion of the uncertainty in that of the measurement values but this would require dedicated (and expensive) ship time. Finally, it should be noted that the urel_combined range reported here (2.3–3.8%) is lower than the 6% relative standard uncertainty for the dissolved iron amount content determined using FI-CL peak height measurements reported by Floor et al.11 but this value included the relative uncertainty on the sensitivity coefficient (i.e. calibration slope), which was not assessed in the current study.
4. Conclusions
An experimental design for the collection of seawater samples was successfully used to determine the between-measurement variability (<0.1–3.1%), which included sub-sampling and filtration, but excluded spatial and temporal variations and uncertainty on the calibration slope. Results suggested that sample collection and filtration parameters as tested in this study were not major contributors to the relative combined uncertainty estimated for these FI-CL measurement results. Firstly, variability of the combined effect of the within-sequence-stability and the uncertainty associated with the sample collection and filtration parameters tested showed no trend with the sampling strategies. Secondly, there was no significant difference in the results obtained from sub-sampling from a single OTE bottle as compared with samples taken from different OTE bottles. There was also no significant difference in from the use of 0.45 μm pore size, 25 mm diameter Supor® polyethersulfone (PES) membrane disc filters (Pall) or 0.8/0.2 μm PES cartridge filters (AcroPak500™). Membrane discs are used to sample marine waters for particulates and these results suggest that the filtrate from this process could be used for the determination of iron concentrations in the dissolved phase as an alternative to the use of cartridge filters. However, this is based on one deep, open ocean water mass and further studies would be required to confirm the generic applicability of this statement. In addition, the approach used does not include an estimation of sampling bias. Nonetheless, this study reconfirms that an uncertainty based on the standard deviation of replicate analysis of a single sample underestimates the true uncertainty of the measurement.Acknowledgements
This work was financially supported by the EMRP via JRP-ENV05 (Metrology for ocean salinity and acidity, G.F.) and JRP-ENV05-REG1 (R.C.). The EMRP is jointly funded by the EMRP participating countries within EURAMET and the European Union. This study was also supported by National Environmental Research Council (NERC) grant NE/H004475/1 (M.C.L.). The authors would like to thank the captain and crew of the R.R.S. James Cook.References
- JCGM, Evaluation of Measurement Data—Guide to the Expression of Uncertainty in Measurement (GUM), Joint Committee for Guides in Metrology, Paris, 1st edn, 2008 Search PubMed.
- M. H. Ramsey, Geostand. Geoanal. Res., 2016 DOI:10.1111/ggr.12121.
- R. Middag, R. Séférian, T. M. Conway, S. G. John, K. W. Bruland and H. J. W. de Baar, Mar. Chem., 2015, 177, 476–489 CrossRef CAS.
- M. H. Ramsey and S. L. R. Ellison, Eurachem/EUROLAB/CITAC/Nordtest/RSC Analytical Methods Committee Guide: measurement uncertainty arising from sampling: a guide to methods and approaches, Available from the Eurachem secretariat, Eurachem, 2007 Search PubMed.
- M. H. Ramsey, J. Anal. At. Spectrom., 1998, 13, 97–104 RSC.
- M. H. Ramsey and A. Argyraki, Sci. Total Environ., 1997, 198, 243–257 CrossRef CAS.
- E. P. Achterberg, T. W. Holland, A. R. Bowie, R. F. C. Mantoura and P. J. Worsfold, Anal. Chim. Acta, 2001, 442, 1–14 CrossRef CAS.
- A. R. Bowie, E. P. Achterberg, S. Blain, M. Boye, P. L. Croot, H. J. W. De Baar, P. Laan, G. Sarthou and P. J. Worsfold, Mar. Chem., 2003, 84, 19–34 CrossRef CAS.
- A. R. Bowie, E. P. Achterberg, P. L. Croot, H. J. W. De Baar, P. Laan, J. W. Moffett, S. Ussher and P. J. Worsfold, Mar. Chem., 2006, 98, 81–99 CrossRef CAS.
- P. J. Worsfold, M. C. Lohan, S. J. Ussher and A. R. Bowie, Mar. Chem., 2014, 166, 25–35 CrossRef CAS.
- G. H. Floor, R. Clough, M. C. Lohan, S. J. Ussher, P. J. Worsfold and C. R. Quétel, Limnol. Oceanogr.: Methods, 2015, 13, 673–686 CrossRef CAS.
- G. A. Cutter, Limnol. Oceanogr.: Methods, 2013, 11, 418–424 CrossRef CAS.
- K. S. Johnson, E. Boyle, K. Bruland, K. Coale, C. Measures, J. Moffett, A. Aguilar-Islas, K. Barbeau, B. Bergquist, A. Bowie, K. Buck, Y. Cai, Z. Chase, J. Cullen, T. Doi, V. Elrod, S. Fitzwater, M. Gordon, A. King, P. Laan, L. Laglera-Baquer, W. Landing, M. Lohan, J. Mendez, A. Milne, H. Obata, L. Ossiander, J. Plant, G. Sarthou, P. Sedwick, G. J. Smith, B. Sohst, S. Tanner, S. Van den Berg and J. Wu, Eos, 2007, 88, 131–132 CrossRef.
- H. Obata, H. Karatani and E. Nakayama, Anal. Chem., 1993, 65, 1524–1528 CrossRef CAS.
- T. P. J. Linsinger, J. Pauwels, A. M. H. Van Der Veen, H. Schimmel and A. Lamberty, Accredit. Qual. Assur., 2001, 6, 20–25 CrossRef CAS.
- A. M. H. Van Der Veen, T. Linsinger and J. Pauwels, Accredit. Qual. Assur., 2001, 6, 26–30 CrossRef CAS.
- G. A. Cutter, C. Andersson, L. Codispoti, P. Croot, R. Francois, M. Lohan, H. Obata and M. Van Der Loeff, Sampling and Sample-handling Protocols for GEOTRACES Cruises (Cookbook), 2010, http://www.geotraces.org/library/geotraces-policies/170-sampling-and-sample-handling-protocols-for-geotraces-cruises Search PubMed.
- J. T. M. De Jong, M. Boye, V. F. Schoemann, R. F. Nolting and H. J. W. De Baar, J. Environ. Monit., 2000, 2, 496–502 RSC.
- J. N. Fitzsimmons and E. A. Boyle, Limnol. Oceanogr.: Methods, 2014, 12, 246–263 CrossRef.
- C. Schlosser and P. L. Croot, Limnol. Oceanogr.: Methods, 2008, 6, 630–642 CrossRef CAS.
- K. Pahnke, T. van de Flierdt, K. M. Jones, M. Lambelet, S. R. Hemming and S. L. Goldstein, Limnol. Oceanogr.: Methods, 2012, 10, 252–269 CrossRef.
- Analytical Methods Committee, AMCTB No 64, Analytical Methods, 2014, 10.1039/c1034ay90062f.
- R. Clough, H. Sela, A. Milne, M. C. Lohan, S. Tokalioglu and P. J. Worsfold, Talanta, 2015, 133, 162–169 CrossRef CAS PubMed.
- A. Milne, W. Landing, M. Bizimis and P. Morton, Anal. Chim. Acta, 2010, 665, 200–207 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay01551d |
| ‡ Currently at GFZ German Research Centre for Geosciences, Helmholtz Centre Potsdam, Section 3.3, Earth Surface Geochemistry, 14473 Potsdam, Germany. |
| § Currently at Ocean and Earth Sciences, National Oceanography Centre Southampton, University of Southampton, SO14 3ZH Southampton, UK. |
| This journal is © The Royal Society of Chemistry 2016 |