
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of an HPLC-MS method for the determination of natural pteridines in tomato samples
Elísabet
Martín-Tornero
*a,
David González
Gómez
b,
Isabel
Durán-Merás
a and
Anunciación
Espinosa-Mansilla
a
aDepartment of Analytical Chemistry and Research Institute on Water, Climate Change and Sustainability (IACYS), University of Extremadura, Badajoz, 06006, Spain. E-mail: elisabetmt@unex.es; iduran@unex.es; nuncy@unex.es; Tel: +34924289376
bDepartment of Didactic of Experimental Sciences, University of Extremadura, 10003, Cáceres, Spain. E-mail: dggomez@unex.es
First published on 1st August 2016
Abstract
In plants, reduced pteridines are folate biosynthesis intermediates, and the presence of these analytes in biofortification processes is considered crucial. A simple liquid chromatography-mass spectrometry (LC-ESI-MS) method has been optimized for the determination of natural pteridines in tomato samples. A solid phase extraction (SPE) step using ISOLUTE ENV cartridges has been employed for cleaning up the samples. Eleven pteridines have been assayed but only four of them have been detected and quantified in tomatoes. The stability of the pteridines and hydropteridines in tomato extracts has been studied. Validation parameters have been evaluated and good linearity (R2 > 0.99 in all cases) and precision (interday relative standard deviation values were lower than 10%) were obtained. The amounts (as μg per g of fresh sample) found of each pteridine were 0.019, 0.44, 0.043 and 0.087 for neopterin, 7,8-dihydroneopterin, 6-hydroxymethylpterin and pterin-6-carboxylic acid, respectively.
1. Introduction
Folates are involved in many critical pathways as a one-carbon source, including DNA, RNA, and protein methylation, as well as DNA synthesis.1,2 Folate deficiency has been associated with an increased risk of several diseases and disorders such as megaloblastic anemia, spina bifida and anencephaly,3,4 with neurodegenerative disorders such as Alzheimer's disease,5 a high risk of cardiovascular diseases6 and various types of cancer.7 Recently, a review about the effect of folates on health has been published.8Unlike plants, humans and other mammals are not able to synthesize folates, and this deficiency must be supplemented through the diet, and plant foodstuffs are the main source of folates.9,10 However, in some cases, the amount of folates in vegetables is not enough to achieve the minimum daily requirements,11 and different mechanisms have been proposed to increase the folate intake such as: adding synthetic folic acid to basic food (fortification); taking folic acid tablets (supplementation); or through a promising alternative, increasing the content of folates in plants by genetic engineering or biofortification.12–16
Several potential strategies to enhance folate synthesis and its accumulation in plants through biofortification have been described. One of these options is the over-expression of the enzymes that are limiting steps in tetrahydrofolate biosynthesis.9 Other procedures to induce genetic modifications in plants, based on increasing the pteridine synthesis, which are intermediates in biosynthesis of folate, have been recently reported.8,17–19 Pteridines are bicyclic compounds made up of a pyrimidine and a pyrazine ring, that occur in a wide range of living systems, including plants, where significant quantities of these analytes have been found. Chemically, folate molecules are composed of a pterin, a p-aminobenzoic acid (PABA) and a glutamate chain. In plants, pteridines are synthesized in the cytosol, PABA in the chloroplast and folates in the plant mitochondria, according to the pathways of Fig. 1.
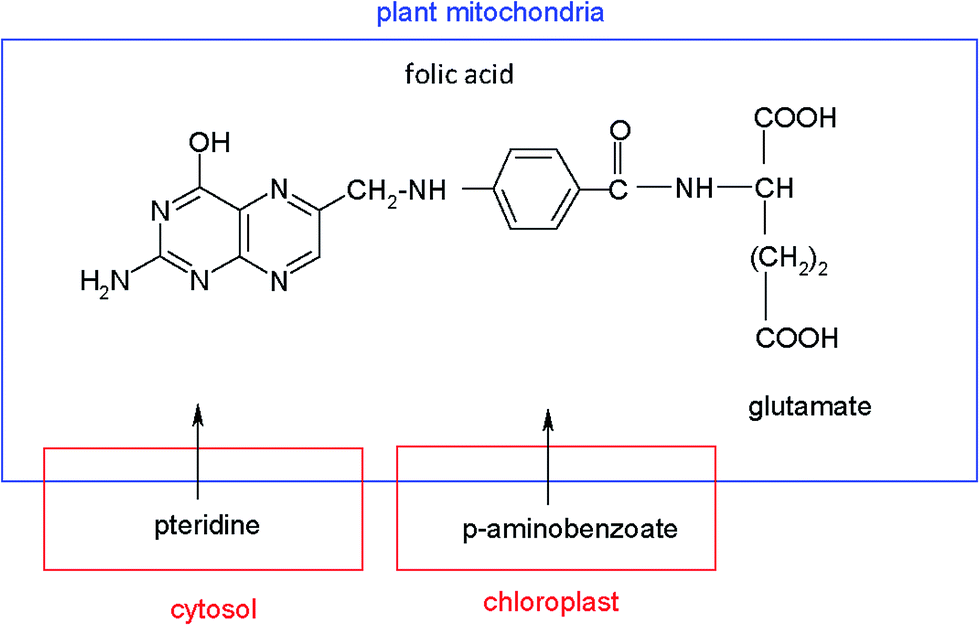 | ||
| Fig. 1 Schematic pathway of folate biosynthesis in plants. | ||
A relevant research study20 shows the possibility of increasing the amount of pteridines in tomatoes by genetic engineering. In the mentioned paper, authors indicate that pteridine synthesis capacity drops in ripe tomato fruit, and this decline can be modified by the specific overexpression of GTP cyclohydrolase I, the first enzyme of pteridine synthesis. Although the levels of folate were significantly increased by the above modifications, the levels of PABA and pteridines are still high, which implies that other substances that inhibit the synthesis of folate exist in transgenic vegetables. Therefore, considering the future implementation of folate biofortification in plant-based foods, we should take into account the accumulation of these intermediates, and therefore it is relevant to establish methods for their analysis.11
Research studies about the content of pteridines in vegetable samples are very scarce. The lack of data of pteridine levels in plants contrasts with the abundant information about the presence of these compounds in animals and bacteria. The occurrence and quantification of unconjugated pteridines in food resources, such as beans, bananas and spinach, has been reported, and characteristic pteridine patterns were observed in each product.21 Another study has shown that plants contain small amounts of 7,8-dihydroneopterin (NH2) and 6-hydroxymethyl-7,8-dihydropterin (6HMDHPT) (detected as their oxidized forms), neopterin (NEO) and 6-hydroxymethylpterin (6HMPT).22 Crude leaves extracted from transgenic crop lines were analyzed by HPLC, and the total pteridinic compounds were expressed as NEO.14 These authors indicate that the levels of pteridines in crude extracts of non-transgenic plants are very low, but the concentration increases up to 1100-fold in transgenic plants.
It has been described that the pteridines, NEO, monapterin (MON) and 6HMPT, as well as their potentially reduced forms and unknown pteridine glycosides, are accumulated in tomatoes.8 Díaz de la Garza et al.20 proposed a fluorimetric HPLC method for the analysis of pteridines and PABA in biofortified tomatoes. This method is based on taking representative segments of tomatoes and performing a pretreatment of the samples, using liquid–liquid extraction followed by acid I2/I− pre-oxidation, in order to transform the reduced forms into the fluorescent oxidized pteridines. A scatter plot of the total pteridine level versus folate content shows that the maximal folate concentration in plants was found at pteridine levels of about 25 nmol g−1 of fresh weight. Higher pteridine concentrations do not increase folate levels. A total pteridine content up to about 60 nmol g−1 was reported in transgenic tomatoes. Later, and using the mentioned pre-oxidation step, pteridines were analyzed in biofortified tomatoes, and the total pteridine amount was expressed as 6HMPT, but the amounts of each individual pteridine were not reported.19 Also, in this paper the authors indicated that the pericarp of velvet bean (a medicinal legume) contains 470 nmol g−1 fresh weight of total pteridine, which is 25 times higher than the pteridine content of transgenic tomatoes. Rodrigues et al. analyzed NEO, MON, 6HMPT and pterin-6-carboxylic acid (PT6C) in spinach, beets and tomatoes. The pteridine content in wild tomato cultivars, raw spinach and raw red beets was on the order of 1 nmol g−1.23
In all the above-proposed methods, fluorimetric detection has been used, and therefore, a pre-oxidation step of the sample to generate the fluorescent forms from non-fluorescent hydropteridines was necessary. MS detection allows the analysis of the pteridines in their oxidation state and the pre-oxidation step is avoided. This methodology has been explored in biological samples, such as urine or serum.24
HPLC hyphenated with mass spectrometry is the most widely applied methodology in the analysis of folates, because it allows qualitative and quantitative information of folate derivatives in a variety of foods and, recently, in tomatoes.25 However, the determination of pteridinic precursors in vegetables using LC-MS methods has been sparingly carried out. LC-MS/MS has been applied to determine pteridines in potatoes and in Arabidopsis thaliana.26 Recently, a relevant paper about the degradation of pteridines in plants during sample preparation using UHPLC-MS/MS has been published.27 In the above mentioned paper the authors report that dihydropterins are subjected to interconversion, on column, in source and auto oxidation, and they are degraded into non-pterin products during boiling. Later, Burton et al.28 established the pterinomic workflow for 15 pteridin derivatives in urine using HPLC-MS/MS revealing that previous oxidative steps were inefficient. The elimination of the preoxidation step in the analysis of pteridinic derivatives was previously recommended by Cañada et al.29 It is remarkable that Burton et al. showed that 7,8-dihydroxanpthopterin exhibited negligible in-source oxidation to xanthopterin. However data about the potential oxidation of dihydroneopterin (NH2) are not reported in this paper.
Therefore, the aim of this work was to develop a simple liquid chromatography-mass spectrometry (LC-MS) method, potentially useful for the determination of natural pteridines present in different types of samples and, particularly, detecting and determining those that exist in tomato samples. A non-oxidation step is applied and we are able to determine each pteridinic compound in its natural oxidation state. Due to the controversy about the hydropteridine stability using MS detection, a study about the stability of the pteridinic reduced forms in tomato samples has also been developed in this study. Research has been focused on those pteridinic derivatives present in tomatoes such as dihydroneopterin and a soft SPE treatment has been carried out to prevent the natural oxidation state of pteridinic compounds.
2. Experimental
2.1. Chemical reagents and solutions
NH2, NEO, 6HMPT, MON, PT6C, 6HMDHPT, biopterin (BIO), pterin (PT), isoxanthopterin (ISO), xanthopterin (XAN), and 7,8-dihydrobiopterin (BH2) were obtained from Schircks Laboratory (Jona, Switzerland). Dithiothreitol (DTT) and formic acid were from Sigma (Sigma-Aldrich S.A., Madrid, Spain). HPLC-grade acetonitrile (ACN) was purchased from Merck (Madrid, Spain). Methanol and hydrochloric acid were from Scharlau (Scharlau, Barcelona). Ultrapure water was obtained from a Milli-Q system (Waters Millipore, Milford, MA, USA).Stock standard solutions of pteridines (15–30 μg mL−1) were prepared by exact weighing of each solid pteridine, dissolution in ultrapure water by adding of 0.010 M sodium hydroxide up to pH near 10.5, and neutralization with 0.010 M hydrochloric acid. BH2 and NH2 standard solutions were prepared daily in the same way as standard solutions of pteridines, but containing 0.1% DTT to minimize the spontaneous oxidation due to environmental oxygen.29 Exposure to direct sunlight was avoided. Pteridine standard solutions were stored at −18 °C and they were stable for at least 3 weeks.
A standard working mixture solution containing 1.5 μg mL−1 of each pteridine and hydropteridine was prepared by dilution of the stock standard solutions with ultrapure water. Other solutions were prepared via serial dilutions and they were used in the generation of the calibration curves.
2.2. Instrumentation
The chromatographic studies were performed on an Agilent 1100 LC High Performance Liquid Chromatograph (Agilent Technologies, Palo Alto, CA, USA), equipped with an online degasser, quaternary pump, autosampler Agilent 1290 infinity thermostated at 5 °C, and an oven column compartment. The ChemStation software (Agilent ChemStation for LC-MS system, Rev.B.04.01) was used for controlling the instrument, data acquisition and data analysis. Chromatographic separation was achieved on an analytical column Zorbax Eclipse XDB-C18, 250–4.6 mm and 5 μm particle size (Agilent Technologies). The column temperature was set at 22 °C. Gradient mode was applied. The flow rate was set at 0.6 mL min−1 and the injection volume was 20 μL.Detection was performed with an Agilent Technologies single quadrupole mass spectrometer, model 6120, equipped with an electro-spray interface (ESI) operated in the positive ionization mode. Nitrogen was used as the nebulizer gas. Mass spectrometer values of capillary voltage, nebulizer pressure, nitrogen flow rate and temperature were adjusted to 4000 V, 40 psi, 10 mL min−1 and 300 °C, respectively. A fragmentor voltage of 100 V was selected, since it provided the best sensitivity with reference compounds. Single ion monitoring (SIM) was selected as operation mode using the target ion [M + H]+ for all the studied compounds.
Calibration curves and analytical figures of merit were performed by means of the ACOC program, developed by our research group, in MatLab code.30
2.3. Extraction of tomato samples
Red ripe stage tomatoes were bought in local supermarkets. On the day of purchase and after washing, tomatoes were finely triturated and frozen in liquid nitrogen prior to lyophilization. The lyophilized tomatoes were stored at −18 °C until needed. About one gram of lyophilized tomatoes was exactly weighed, and pteridines were extracted two times, with 10 mL each time, of the methanolic/water pH 12 (1/1, v/v) mixture alkalized with sodium hydroxide. In each extraction, the sample was sonicated for 15 minutes, and centrifuged at 3000 r.p.m. (10 minutes). The supernatants were recombined, neutralized, filtered, and diluted to 25 mL with ultrapure water. The tomato extract was stable for at least 10 days.2.4. Purification of tomato extracts
Purification of samples was carried out by solid phase extraction (SPE). Aliquots of 3.0 mL of the tomato extracts were passed through an ISOLUTEENV+ (hydroxylated polystyrene–divinylbenzene copolymer) cartridge, previously conditioned with 5 mL of methanol and 5 mL of ultrapure water. The elution of the retained pteridines was carried out with 3.0 mL of acetonitrile![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :water, 80:20 (v/v). Finally, 1.0 mL of the eluate was evaporated to dryness by a nitrogen stream, and the residue was re-dissolved with 1.0 mL of ultrapure water and filtered through a 0.2 μm PTFE filter for LC-MS analysis.
:water, 80:20 (v/v). Finally, 1.0 mL of the eluate was evaporated to dryness by a nitrogen stream, and the residue was re-dissolved with 1.0 mL of ultrapure water and filtered through a 0.2 μm PTFE filter for LC-MS analysis.
2.5. Quantification of pteridines present in tomato samples
The standard addition method was used to calculate the concentrations of each pteridine in tomato samples. Spiked lyophilized tomato samples, at five different concentration levels for each pteridine in the range between 30 and 200 ng mL−1, were prepared. Each sample was prepared by adding 800 μL of the tomato extract (or 200 μL in the case of the determination of NH2), into a vial containing variable volumes of pteridine and hydropteridine standard working mixtures, and diluting to 1 mL with ultrapure water. Peak areas of the Extracted Ion Chromatogram (EIC) were used for the quantification of all the assayed species.2.6. Sensitivity, precision and accuracy
Limits of detection (LODs) were calculated according to the Long and Winefordner criterion31 (k = 3), and limits of quantification (LOQs) as (LOD/3) × 10, using spiked tomato samples at five concentration levels (by triplicate). The accuracy of the proposed method was calculated for tomato samples spiked at five concentration levels between 30 and 200 ng mL−1 in triplicate. Intraday (n = 6) and interday (n = 5) precision assays were carried out in samples containing NH2, NEO, 6HMPT and PT6C at the LOQ amount, and expressed as the relative standard deviation (RSD).3. Results and discussion
3.1. Optimization of MS variables
The optimization of MS parameters and LC separation conditions was based on previous experiments developed by our research group for the determination of pteridines in biological fluids.24Flow injection analysis (FIA) of each pteridine standard solution was performed with the aim to optimize fragmentor voltage (FV), capillary voltage (CV), nebulizer pressure (NP), nitrogen flow rate and temperature, in ESI positive and negative modes, in order to obtain the highest sensitivity. Three FV values of 50, 75 and 100 V, in negative mode, and two FV values of 75 and 100 V, in positive mode, were assayed. Two CV values of 4000 and 4500 V were tested in both modes. The best results were obtained with 100 and 4500 V for FV and CV respectively in negative mode, and with 100 V for FV and 4000 or 4500 V for CV, in positive mode. The results show that the protonated molecular ion [M + H]+ can be selected as the target ion of the analytes due to the presence of easily protonated amino groups in the molecules. Also, the electrospray in positive mode is more stable than in negative mode. The instrumental variables were optimized to obtain the highest sensitivity of the [M + H]+ ion, using the SIM mode with FV and CV at 100 and 4500 V, respectively.
NP was varied between 10 and 55 psi and the abundance remains constant for psi values higher than 30. The best signal/noise ratio was obtained for a gain value of 15. The nitrogen flow rate and temperature do not significantly affect the abundance and 10 mL min−1 and 300 °C were selected for later studies.
3.2. Optimization of the LC separation
Once MS parameters were established, chromatographic conditions were optimized in order to attain an adequate elution of the target compounds and a short analysis time. The mobile phase composition was evaluated considering the different polarities of the pteridines analyzed, and the identical molecular-ions for NEO and MON, and for XAN and ISO. The presence of very low amounts of organic solvents such as methanol or ACN, in the mobile phase, generates a drastic diminishing of the capacity factor of the more polar compounds. However, a notable amount of organic solvent is necessary to elute the less polar compounds. On the other hand, a slightly acidic medium is necessary to avoid the formation of charged pteridines that are poorly retained. These facts indicate the necessity of applying a gradient mode for the elution of the pteridinic compounds and, in this sense, various analytical gradients were evaluated with different formic acid solutions, and ACN or methanol as organic phases. The better resolution and the higher ion abundance were obtained with mixtures of 0.1% formic acid containing 2% of ACN (solution A), and pure ACN containing 2% of formic acid (solution B). The optimized gradient was: 100% of eluent A during the first 8 min, increasing the percentage of eluent B up to 20% (in 8 min, 2.5% min−1). These conditions were maintained for 4 min and, finally, the eluent B content was decreased to the initial conditions (0% B), and the column was re-equilibrated for 10 min. The flow rate was set constant at 0.6 mL min−1, the injection volume was 20 μL, and the column temperature was kept at 22 °C. Fig. 2 shows the EICs obtained from a stock standard mixture of the 11 pteridines under these optimized conditions. Moreover in Table 1, the retention times for each pteridine and the m/z relation for the [M + H]+ ions, in a standard solution containing the eleven pteridinic and hydropteridinic derivatives, are summarized. In the mentioned table, resolution values in the EIC chromatogram obtained for those compounds with identical molecular-ions, such as NEO-MON and XAN-ISO, are shown. | ||
| Fig. 2 EICs obtained from the analysis of a standard pteridine mixture with the optimized separation method. | ||
| Pteridinic derivative | Chemical structure | t R (min) | m/z [M + H]+ | Resolution EICs mode |
|---|---|---|---|---|
| a Fragmentor voltage: 100 V, capillary voltage: 4500 V, nebulizer pressure: 30 psi, temperature: 300 °C. | ||||
| NEO |
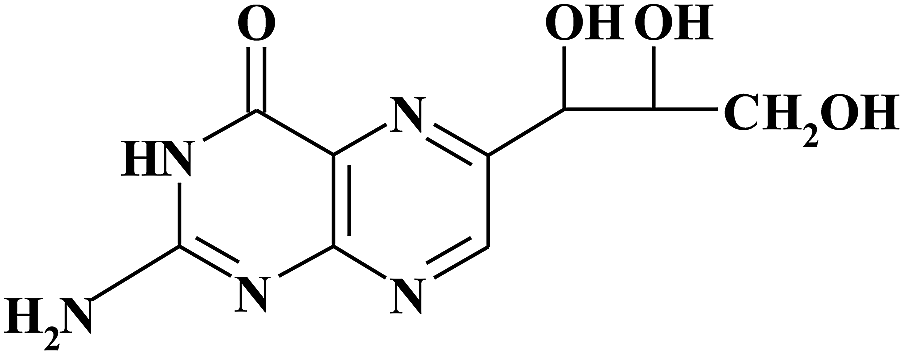
|
5.90 | 254 | R MON/NEO = 2.98 |
| NH2 |
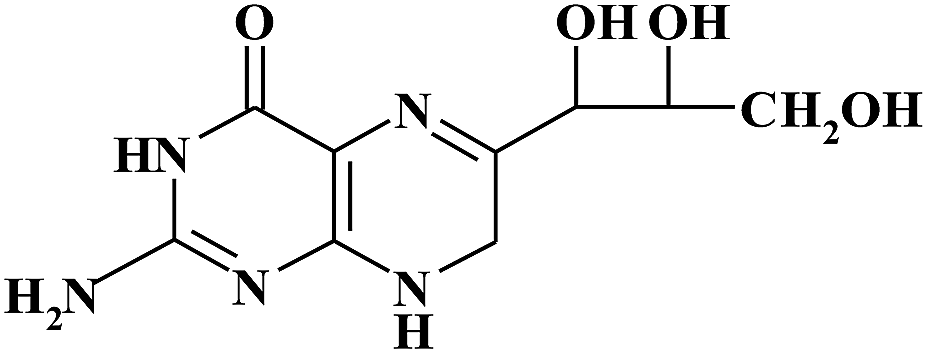
|
6.00 | 256 | |
| MON |
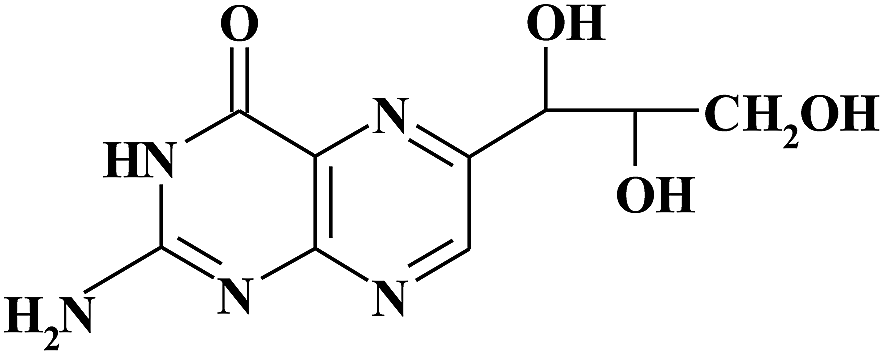
|
6.86 | 254 | |
| 6HMDHPT |
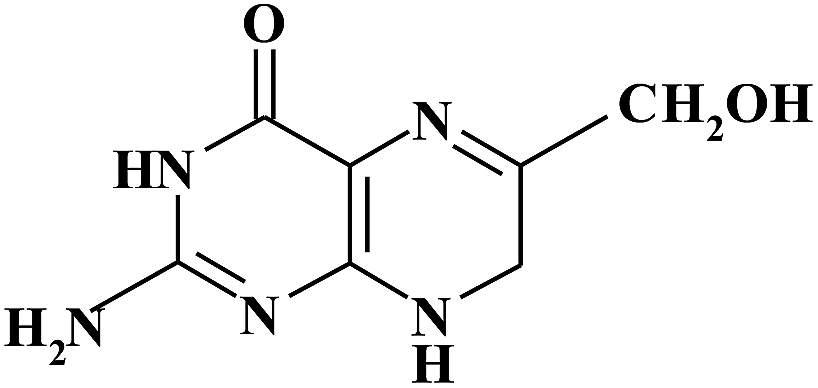
|
8.00 | 196 | |
| XAN |
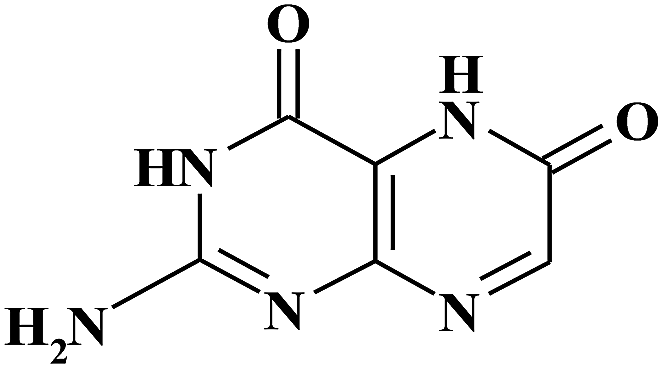
|
9.40 | 180 | |
| BH2 |
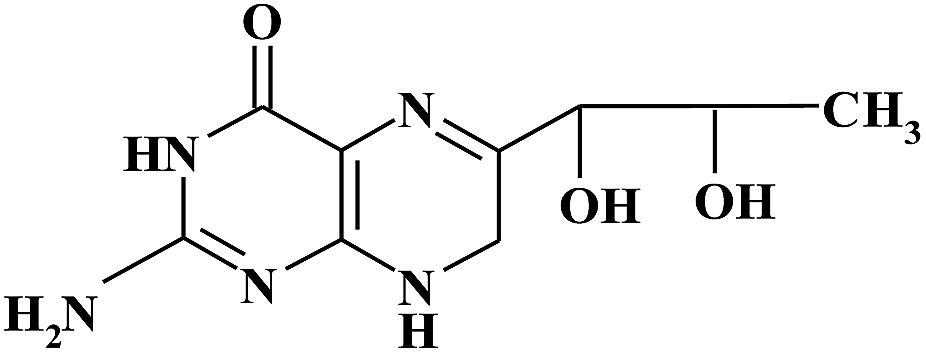
|
9.90 | 240 | |
| BIO |
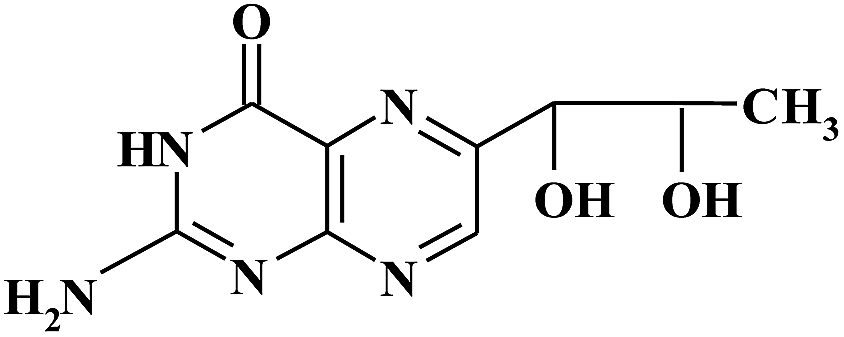
|
10.72 | 238 | |
| PT |
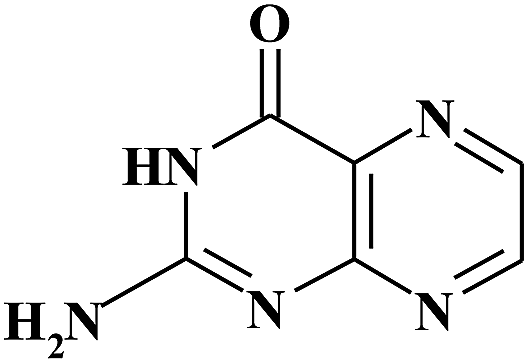
|
11.73 | 164 | R ISO/XAN = 10.48 |
| 6HMPT |
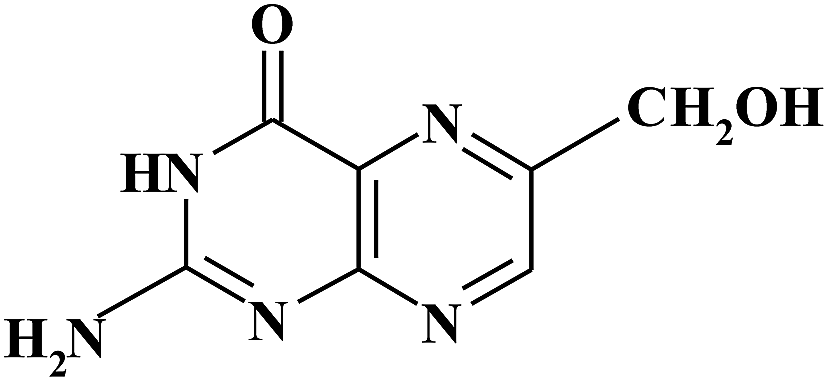
|
12.28 | 194 | |
| ISO |
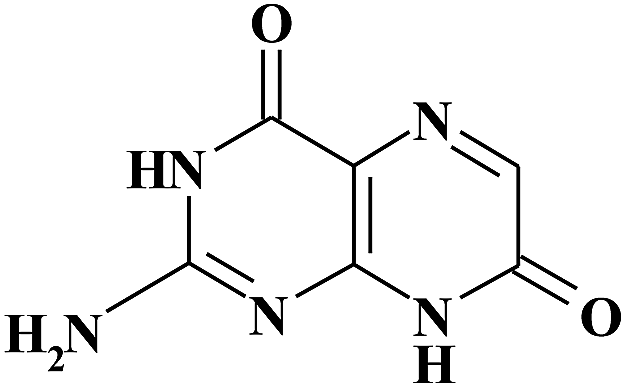
|
13.76 | 180 | |
| PT6C |
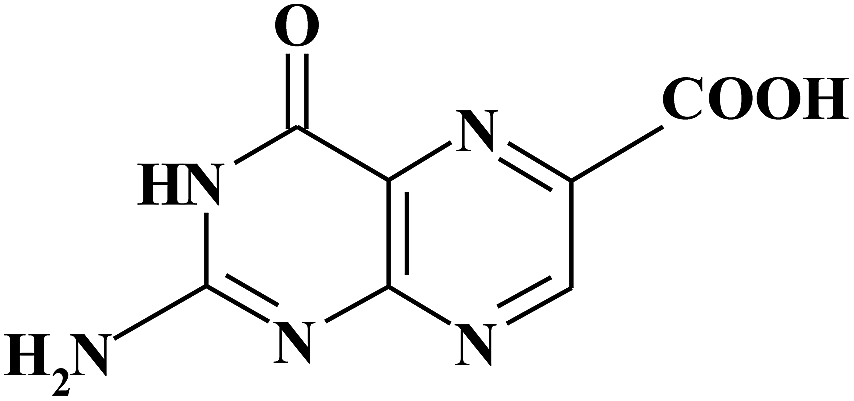
|
18.12 | 208 | |
3.3. SPE cleanup process in tomato samples
When MS detection is used in the analysis of complex matrix samples, such as vegetables, a cleanup step prior to chromatographic separation is usually recommended. The cleanup step is intended to protect the chromatographic column, and minimize the matrix effect. SPE is the selected methodology in most of the food bibliographic data due to its easy use, speed and variety of adsorbents. In our case, tomato samples were cleaned by SPE, using the ISOLUTEENV (200 mg) cartridge with the hydroxylated polystyrene–divinylbenzene copolymer as the sorbent. In the first place, the cartridge was conditioned with 5 mL of 100% methanol and then flushed with 5 mL of ultrapure water. The extraction procedure was assayed with 3.0 mL of the extracts of tomatoes spiked with pteridines between 150 and 300 μg mL−1. With the object of verifying if the pteridines were retained in the cartridge, the eluate was injected into the chromatographic system. The absence of any signal at the same retention time of the selected compounds indicates that pteridines have been retained. The elution of pteridines was tested with different solvents, such as ACN, methanol, and different mixtures of the organic solvents in ultrapure water, 80/20, 50/50 and 20/80 (v/v, organic solvent:ultrapure water), in order to achieve the maximum recovery. A pneumatic manifold allowed us to elute simultaneously twenty four tomato samples using a precise and repetitive flow of eluent. The best recovery values were obtained using between 1.5 and 3 mL of acetonitrile–water 80:20, v/v. The repeatability of the SPE extraction procedure was checked with nine independent extracts, and the average recovery values ranged between 80% for XAN and ISO and 118% for BH2. Similar recovery values were obtained when the elution was performed with 1.5 mL or 3 mL of the elution mixture.
3.4. Application to the analysis of pteridines in tomato samples
In the first place and with the object of focusing the research on compounds that are present in tomatoes, an unspiked aliquot of the tomato extract and a spiked sample with a standard mixture of pteridines was injected into the chromatographic system. The extracted-ion chromatograms (EICs) obtained in SIM mode are shown in Fig. 3. Only four pteridines were detected in the tomato samples: NEO, NH2, 6HMPT and PT6C but, due to bibliographic data that indicate the presence of MON in tomatoes,19 this pteridine was also investigated in later studies. From this point, the studies were centered in these five pteridines and hydropteridines. | ||
| Fig. 3 EICs of an unspiked tomato sample and a spiked tomato sample. | ||
3.5. Stability of the pteridines in the tomato extract
Due to the known instability of the aqueous solutions of hydropteridines, aliquots of the same tomato extract, spiked with NEO, NH2, 6HMPT and PT6C, were analyzed for 20 days, to establish the stability of the extracted tomato solution. The average value of the relative abundance of three injections for the selected pteridines during 20 days is represented in Fig. 4a, which shows that for NEO, 6HMPT and PT6C, it remains constant for the first ten days, and decreases after that. NH2 is the pteridine presenting minor stability, remaining unchanged only for 7 days, and their relative abundance decreases a third of its initial value after 20 days. A week was fixed as the maximum time for analysis after preparation of the tomato sample.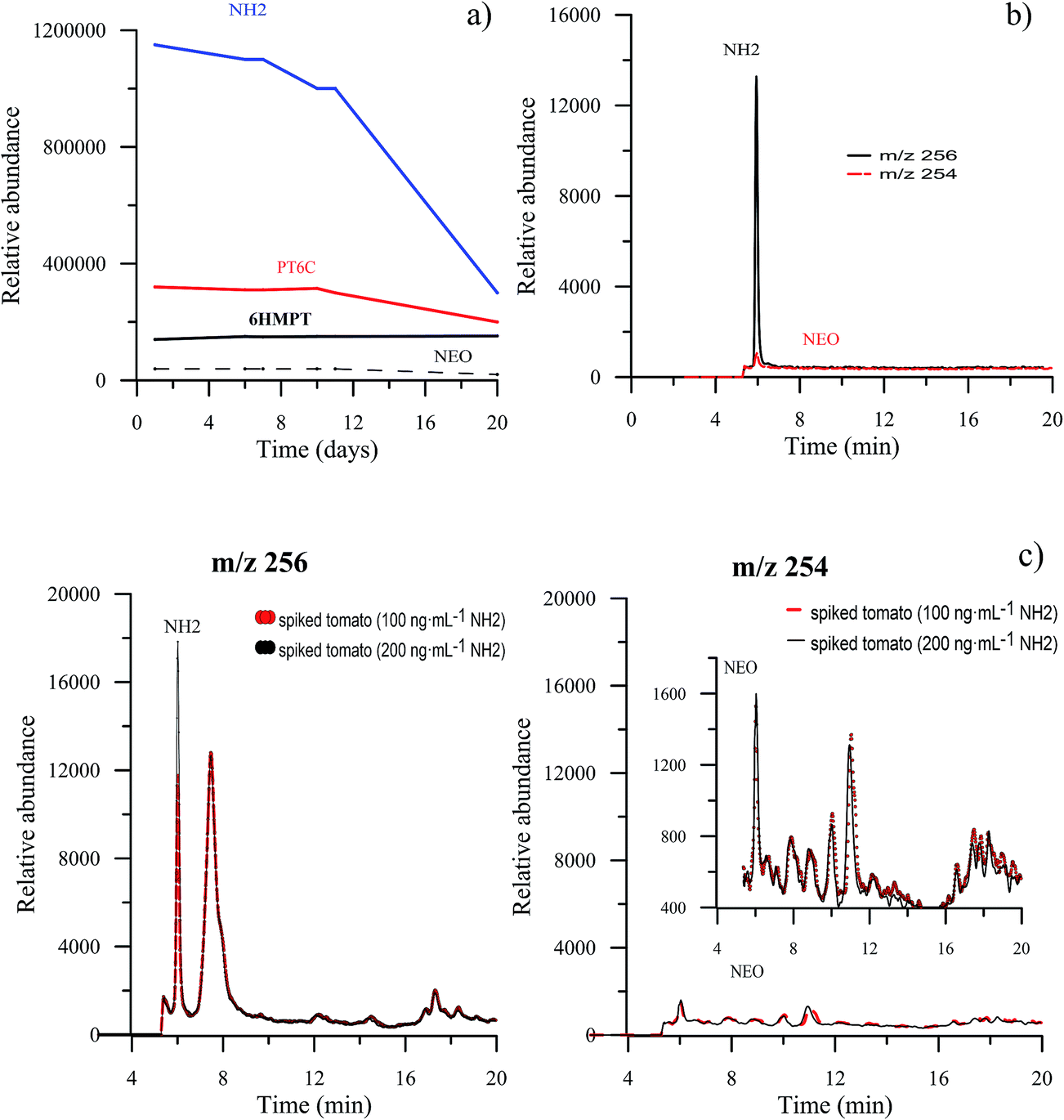 | ||
| Fig. 4 (a) Stability of the pteridines detected in tomatoes. (b) Extracted ion chromatograms of a standard solution of NH2 at 256 and 254 m/z values. (c) Extracted ion chromatograms of tomato extracts spiked with NH2 at 100 and 200 ng mL−1. | ||
Furthermore, and in order to test if the target compounds undergo degradation and/or interconversion processes, tomato extracts were individually spiked with each pteridine and subjected to the SPE clean-up step. For each solution, the signals in the MS detector were simultaneously monitored for the m/z of all ions. This study has also been done with standard solutions. We have seen no evidence of degradation of the pteridines when they are treated with this procedure. As hydropterines are more unstable and easily oxidizable, an exhaustive study was carried out with NH2. In the first place, and with a standard solution of NH2, subjected to the SPE clean step, EICs at m/z values of 256 and 254, corresponding to NH2 and NEO (oxidized pteridine) respectively, were obtained. Fig. 4b shows both chromatograms, where it can be appreciated a very small signal corresponding to NEO, practically negligible compared with the signal from NH2 indicating that the oxidation is minimal. In the second place, we have studied the tomato sample. For this, we have compared the extracted ion chromatograms, for m/z values of 256 and 254, obtained from spiked tomato samples at two different levels, 100 ng mL−1 and 200 ng mL−1 of NH2. In Fig. 4c, it can be observed that the NH2 signal (at m/z 256) increases when the amount of NH2 increases, as expected. However, the signal corresponding to NEO (m/z 254) remains constant. This allows us to confirm that in this matrix, and with the sample treatment proposed, in the presence of organic solvent and at room temperature, NH2 is stable. These data are contradictory with the results reported by Van Daele and co-workers.27 However, results similar to ours have been recently described by Burton et al.,28 who found negligible in-source oxidation of 7,8-dihydroxanthopterin to xanthopterin.
3.6. Study of the matrix effect
Before performing the calibration and quantification of the pteridines, the influence of the matrix over the HPLC-ESI-MS method was evaluated. For this, a comparison of the slopes between external calibration curves in ultrapure water, and standard addition curves with tomato extracts spiked with NH2, NEO, MON, 6HMPT and PT6C, at five different concentrations in the range 0–200 ng mL−1, in triplicate, was carried out.The matrix effect was studied with extracts of tomatoes before and after the SPE cleanup. In both cases, for each pteridine, the regression plot was obtained and the comparison between the slopes of external calibration and standard addition was accomplished applying the F and t statistical tests at the 95% confidence level.32 The matrix effect, expressed as percentage, was calculated as: % matrix effect = 100 × (tomato/water slope ratio) − 100.
When the non-SPE clean up extracts were analyzed, statistical differences are observed between both external standard and standard addition calibration slopes. This fact indicates a matrix effect for the analysis of all pteridines exhibiting an absolute value in the range 29.5–69.2%. For most of the pteridines, a matrix suppression effect was observed, although for 6HMPT an ion enhancement effect was observed. When SPE clean up treatment was used, a softer matrix effect was observed with absolute values between 6.3 and 50.4%, particularly for NH2, NEO and PT6C, however for MON the results were not improved. Then, the standard addition methodology, previous SPE cleanup, was recommended for analyzing pteridines in tomato samples.
3.7. Validation of the method
Linearity has been established in cleaned SPE lyophilized tomato extracts, in the range LOQ–300 ng mL−1. Different linearity ranges were obtained depending on the compound and statistical parameters were calculated. Good linearity was observed for the five pteridines, and determination coefficients (R2) were higher than 0.99 for all analytes. The results of the least squares regression analysis for the standard calibrations and detection and quantification limits are summarized in Table 2. LODs ranged between 8 and 12 ng mL−1. The lowest limit of detection was found for NEO (8 ng mL−1) and very similar LOD values were obtained for MON, NH2 and PT6C. LOQs ranged from 27 to 41 ng mL−1. To evaluate the repeatability (intraday precision, n = 6) of the method, RSDs were evaluated with solutions of SPE clean tomato extracts containing 27 ng mL−1 of NEO, 40 ng mL−1 of MON, 41 ng mL−1 of NH2, 35.0 ng mL−1 of 6HMPT, and 40.7 ng mL−1 of PT6C. Interday precision (reproducibility) was analyzed over 5 consecutive days at the same concentration levels. Intraday precision ranged from 1.1% for NEO and 6HMPT up to 4.1% for NH2, and interday precision values are lower than 9.6% for all analytes, which indicates the good repeatability of the proposed method.| Parameters | NEO | MON | NH2 | 6HMPT | PT6C |
|---|---|---|---|---|---|
| a Sb: slope standard deviation. b Sa: intercept standard deviation (n = 15). c α = β = 0.05. d LOD, limit of detection according to the Long–Winefordner criterium (k = 3). e LOQ, limit of quantification: LD × 10/3. | |||||
| Slope ± Sba | 690 ± 17 | 1341 ± 44 | 617 ± 2 | 2708 ± 83 | 520 ± 18 |
| Intercept ± Sab | 6331 ± 1856 | 1182 ± 535 | 33990 ± 223 |
56853 ± 9260 |
22113 ± 1971 |
| R 2 | 0.9948 | 0.9935 | 0.9903 | 0.9906 | 0.9886 |
| Linearityc (%) | 98 | 97 | 98 | 97 | 98 |
| LODd (ng mL−1) | 8.1 | 12.0 | 12.3 | 10.5 | 12.2 |
| LOQe (ng mL−1) | 27.0 | 40.0 | 41.0 | 35.0 | 40.7 |
3.8. Analysis of tomato samples
Finally, the proposed LC-ESI-MS method was applied to the analysis of tomato samples. The standard addition methodology was employed and the tomato extracts were spiked at five concentration levels, for each pteridine, in the range between 30 and 200 ng mL−1. Samples were prepared in triplicate, and the results are shown in Table 3 as mean values and recoveries. Accuracy was evaluated through the calculation of percent recoveries for each pteridine and, in all cases, satisfactory values between 83 and 117% were obtained. The amounts of pteridines obtained from lyophilized tomatoes are also shown in Table 3. For the oxidized pteridines, the concentrations found are 0.075 nmol g−1 of NEO, 0.22 nmol g−1 of 6HMPT, and 0.42 nmol g−1 of PT6C. And for the hydropteridine NH2, 1.72 nmol g−1 was found. These results are in accordance with those previously reported by Rodrigues et al. These authors, in a previous paper, have quantified several pteridines in wild-type tomatoes using HPLC-fluorimetric detection.23 The reported pteridine content was on the order of 1 nmol g−1, but in this procedure NH2 was not detected due to the previous oxidation step, necessary in the HPLC-fluorimetric method.| Analytes | Added (ng mL−1) | Founda (ng mL−1) | Recoverya (%) | Amountb (μg pteridine per g of lyophilized tomatoes ± confidence interval) | Amountb (μg pteridine per g of fresh tomatoes ± confidence interval) |
|---|---|---|---|---|---|
| a Mean value for three individual replicates for each added amount. b α = 0.05. | |||||
| NEO | 0 | <LOQ | — | 0.29 ± 0.10 | 0.019 ± 0.006 |
| 29.92 | 37.98 | 83 | |||
| 49.37 | 57.07 | 89 | |||
| 89.76 | 102.87 | 100 | |||
| 149.60 | 157.59 | 97 | |||
| 198.97 | 209.19 | 99 | |||
| NH2 | 0 | 54.53 | — | 6.90 ± 1.15 | 0.44 ± 0.11 |
| 29.72 | 83.45 | 97 | |||
| 49.60 | 106.89 | 106 | |||
| 99.20 | 149.57 | 96 | |||
| 148.80 | 211.75 | 106 | |||
| 198.40 | 245.86 | 96 | |||
| MON | 0 | <LOD | — | <LOD | — |
| 30.00 | <LOQ | — | |||
| 49.50 | 90.60 | 112 | |||
| 90.00 | 137.70 | 114 | |||
| 150.00 | 191.26 | 104 | |||
| 199.50 | 242.18 | 104 | |||
| 6HMPT | 0 | <LOQ | — | 0.68 ± 0.19 | 0.043 ± 0.012 |
| 30.08 | 49.48 | 82 | |||
| 49.63 | 65.11 | 82 | |||
| 90.27 | 115.06 | 100 | |||
| 150.40 | 169.39 | 96 | |||
| 200.03 | 222.35 | 99 | |||
| PT6C | 0 | 42.02 | — | 1.33 ± 0.34 | 0.087 ± 0.011 |
| 30.16 | 77.23 | 117 | |||
| 49.76 | 96.26 | 109 | |||
| 90.48 | 144.35 | 113 | |||
| 150.08 | 192.10 | 100 | |||
Taking into account the lyophilized process applied, and the average water content (93.5 ± 2.0%) of the assayed tomato samples, the concentrations of the pteridines have also been expressed as μg pteridine per g of fresh tomatoes, and 0.019 ± 0.006; 0.44 ± 0.11; 0.043 ± 0.012 and 0.087 ± 0.011 for NEO, NH2, 6HMPT and PT6C, respectively were obtained. The obtained results indicate that NH2 is the most abundant compound and NEO is the compound in a smaller concentration. MON was not detected under the assayed conditions.
4. Conclusions
Therefore, a new analytical HPLC-ESI-MS method has been developed to determine precursors of folates in tomatoes as an alternative to fluorimetric detection that does not allow the determination of hydropteridine compounds or HPLC-MS/MS because not everyone has this technology available.The proposed method allows a simple determination of the natural forms of pteridines in tomatoes by LC-ESI-MS. On the other hand, the use of a simple quadrupole analyzer eases its use as an easy and robust detector in routine analysis. With the object to keep the natural composition of the reduced pteridines in the tomato samples, preoxidation steps and boiling processes are avoided. Under the proposed conditions, the oxidation of the dihydroneopterin in tomatoes is negligible. This method could be useful for monitoring pteridine formation in biofortification studies to provide overproduction of folates in tomatoes, and it could be easily modified with similar aims to analyze other vegetables.
Abbreviations
| ACN | Acetonitrile |
| BH2 | 7,8-Dihydrobiopterin |
| BIO | Biopterin |
| CV | Capillary voltage |
| DTT | Dithiothreitol |
| EICs | Extracted ion chromatograms |
| ESI | Electro-spray interface |
| FIA | Flow injection analysis |
| FV | Fragmentor voltage |
| ISO | Isoxanthopterin |
| 6HMDHP | 6-Hydroxymethyl-7,8-dihydropterin |
| 6HMPT | 6-Hydroxymethylpterin |
| LC-ESI-MS | Liquid chromatography-mass spectrometry |
| LC-MS | Liquid chromatography-mass spectrometry |
| LOD | Limits of detection |
| LOQ | Limits of quantification |
| MON | Monapterin |
| NEO | Neopterin |
| NH2 | 7,8-Dihydroneopterin |
| PABA | p-Aminobenzoic acid |
| PT | Pterin |
| PT6C | Pterin-6-carboxylic acid |
| RSD | Relative standard deviation |
| SIM | Single ion monitoring |
| SPE | Solid phase extraction step |
| TIC | Total ion chromatogram |
| XAN | Xanthopterin |
Acknowledgements
The authors are grateful to the Ministerio de Economía y Competitividad of Spain (Project CTQ2014-52309-P) and the Junta de Extremadura (GR15090-Research Group FQM003), both co-financed by the European FEDER funds, for financially supporting this work. E. Martín-Tornero thanks Ministerio de Economía y Competitividad of Spain and European social founds for a FPI grant with reference BES-2015-075407 (Order ECC/1402/2013, de 22 of July, modified by the order ECC/1820/2014, of 26 of September, and by the order ECC/2483/2014, of 23 of December; BOE no. 140 of 12/06/15).References
- R. J. Cook, Arch. Biochem. Biophys., 2001, 392, 226–232 CrossRef CAS PubMed.
- P. J. Stover, in Folate in Health and Disease, ed. L. B. Bailey, CRC Press, Taylor & Francis Group, Gainesville, FL, USA, 2nd edn, 2009, pp. 49–74 Search PubMed.
- J. I. Rader and B. O. Schneemann, Pediatrics, 2006, 117, 1394–1399 CrossRef PubMed.
- P. De Wals, F. Tairou, M. I. Van Allen, S. Uh, R. B. Lowry, B. Sibbald, J. A. Evans, M. C. Van den Hof, P. Zimmer, M. Crowley, B. Fernandez, N. S. Lee and T. Niyonsenga, N. Engl. J. Med., 2007, 357, 135–142 CrossRef CAS PubMed.
- S. Seshadri, A. Beiser, J. Selhub, P. F. Jacques, I. H. Rosenberg, R. B. P. D'Agostino, W. F. Wilson and P. A. Wolf, N. Engl. J. Med., 2002, 346, 476–483 CrossRef CAS PubMed.
- O. Stanger, Cell. Mol. Biol., 2004, 50, 953–988 CAS.
- S. W. Choi and S. Friso, Clin. Chem. Lab. Med., 2005, 43, 1151–1157 CrossRef CAS PubMed.
- D. Blancquaert, S. Storozhenko, K. Loizeau, H. De Steur, V. De Brouwer, J. Viaene, S. Ravanel, F. Rebeille, W. Lambert and D. Van Der Straeten, Crit. Rev. Plant Sci., 2010, 29, 14–35 CrossRef CAS.
- J. Scott, F. Rébeillé and J. Fletcher, J. Sci. Food Agric., 2000, 80, 795–824 CrossRef CAS.
- E. J. Konings, H. H. Roomans, E. Dorant, R. A. Goldbohm, W. H. Saris and P. A. Van den Brandt, Am. J. Clin. Nutr., 2001, 73, 765–776 CAS.
- D. DellaPenna, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 3675–3676 CrossRef CAS PubMed.
- D. DellaPenna, Science, 1999, 285, 375–379 CrossRef CAS PubMed.
- E. Bouis, J. Nutr., 2002, 132, 491–494 Search PubMed.
- T. Hossain, I. Rosenberg, J. Selhub, G. Kishore, R. Beachy and K. Schubert, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5158–5163 CrossRef CAS PubMed.
- A. D. Hanson and J. F. Gregory III, Annu. Rev. Plant Biol., 2011, 62, 105–125 CrossRef CAS PubMed.
- D. Blancquaert, H. De Steur, X. Gellynck and D. Van Der Straeten, J. Exp. Bot., 2014, 65, 895–906 CrossRef CAS PubMed.
- G. J. Basset, E. P. Quinlivan, S. Ravanel, F. Rébeillé, B. P. Nichols, K. Shinozaki, M. Seki, L. C. Adams-Phillips, J. J. Giovannoni, J. F. Greegory III and A. D. Hanson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 1496–1501 CrossRef CAS PubMed.
- G. J. Basset, R. Ravanel, E. P. Quinlivan, R. White, J. J. Giovannoni, F. Rébeillé, B. P. Nichols, K. Shinozaki, M. Seki, J. F. Gregory III and A. D. Hanson, Plant J., 2004, 40, 453–461 CrossRef CAS PubMed.
- R. I. Díaz de la Garza, J. F. Gregory III and A. D. Hanson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4218–4222 CrossRef PubMed.
- R. Díaz de la Garza, E. P. Quinlivan, S. M. Klaus, G. J. Basset, J. F. Gregory III and A. D. Hanson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 13720–13725 CrossRef PubMed.
- M. Kohashi, K. Tomita and K. Iwai, Agric. Biol. Chem., 1980, 44, 2089–2094 CAS.
- A. Goyer, V. Illarionova, S. Roje, M. Fischer, A. Bacher and A. D. Hanson, Plant Physiol., 2004, 135, 103–111 CrossRef CAS PubMed.
- V. Rodrigues da Silva, E. P. Quinlivan, A. D. Hanson and J. F. Gregory, FASEB J., 2007, 21, A346 Search PubMed.
- A. Jiménez, E. Martín-Tornero, M. C. Hurtado Sánchez, I. Durán Merás and A. Espinosa-Mansilla, Talanta, 2012, 101, 465–472 CrossRef PubMed.
- K. Tyagi, P. Upadhyaya, S. Sarma, V. Tamboli, Y. Sreelakshmi and R. Sharma, Food Chem., 2015, 179, 76–84 CrossRef CAS PubMed.
- D. Blancquaert, S. Storozhenko, J. Van Daele, C. P. Stove, R. G. Visser, W. E. Lambert and D. Van der Straeten, J. Exp. Bot., 2013, 64, 3899–3909 CrossRef CAS PubMed.
- J. Van Daele, D. Blancquaert, F. Kiekens, D. Van der Straeten, W. E. Lambert and C. P. Stove, Food Chem., 2016, 194, 1189–1198 CrossRef CAS PubMed.
- C. Burton, H. Shi and Y. Ma, Anal. Chim. Acta, 2016, 927, 72–81 CrossRef CAS PubMed.
- F. Cañada-Cañada, A. Espinosa-Mansilla, A. Muñoz de la Peña and A. Mancha de Llanos, Anal. Chim. Acta, 2009, 648, 113–122 CrossRef PubMed.
- A. Espinosa-Mansilla, A. Muñoz de la Peña and D. González Gómez, Chem. Educ., 2005, 10, 337–346 CAS.
- G. L. Long and J. D. Winefordner, Anal. Chem., 1983, 55, 712A–724A CrossRef CAS.
- D. L. Massart, B. G. M. Vandeginste, L. M. C. Buydens, S. De Jong, P. J. Lewi and J. Smeyers-Verbeke, in Handbook of Chemometrics and Qualimetrics: Part A, Elsevier, Amsterdam, 1997, vol. 20 Search PubMed.
| This journal is © The Royal Society of Chemistry 2016 |