
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fingerprinting of falsified artemisinin combination therapies via direct analysis in real time coupled to a compact single quadrupole mass spectrometer†
Matthew C.
Bernier
a,
Frederick
Li
b,
Brian
Musselman
b,
Paul N.
Newton
c and
Facundo M.
Fernández
*a
aSchool of Chemistry and Biochemistry, Georgia Institute of Technology, Atlanta, GA, USA. E-mail: facundo.fernandez@chemistry.gatech.edu; Fax: +1-404-385-3399; Tel: +1-404-385-4432
bIonsense Inc., Saugus, MA, USA
cLao-Oxford-Mahosot Hospital Wellcome Trust Research Unit (LOMWRU), Laos and Worldwide Antimalarial Resistance Network, Centre for Tropical Medicine & Global Health, University of Oxford, UK
First published on 5th July 2016
Abstract
Falsified anti-malarial treatments continue to constitute a major health crisis, especially in malarious Africa. Even after detection of poor quality pharmaceuticals, it is critical that they be fully analyzed to determine their components, in order to assess their health effects and ultimately allow forensic tracing of their sources of production and distribution. Timely assessment requires robust and complete field-testing, or at the very least timely analysis after seizure or purchase. Ideally, low-cost and simple analytical equipment such as portable mass spectrometry (MS) is the best approach for achieving this quick and informative analysis. To date, Direct Analysis in Real Time (DART) MS has been successfully implemented to rapidly analyze falsified artemisinin-based combination therapies (ACTs) in laboratory settings, but this approach typically translates into high-cost and the need for high-resolution instrumentation. Here, we examine the use of DART ionization coupled with a portable low-resolution single-quadrupole instrument, and compare its success in fingerprinting anti-malarial tablets with higher resolution instrumentation. Using single quadrupole DART-MS, the same sample components were detected as with the high-resolution instrument, while needing significantly less consumables and power, and the additional advantages of increased portability and ease of use. Using Principal Component Analysis (PCA) of DART data, specific classes of falsified ACTs were identified, providing a more straightforward method for sourcing counterfeits and assessing their similarities.
1. Introduction
There are currently 3.2 billion people at risk of contracting Plasmodium falciparum malaria. This vector-borne infection does possess an efficacious treatment, the artemisinin-based combination therapies (ACTs). However, malaria remains one of the highest leading causes of death in developing countries.1–3 Despite a major reduction of malarial cases in the last 15 years (37% worldwide), and also a significant drop in the rate of malarial-related deaths (60%), there are still major challenges moving forward with the continued effort to eradicate this disease.1 One of the major hindrances to effective treatment and reduction in drug resistance is the presence of falsified drugs mimicking commercially available ACTs.4–6 This is especially a problem in developing regions, where there is a profound disease burden but little to no oversight in the monitoring of medical products.7–11 For example, many of the treatment problems are centered in sub-Saharan Africa, which in 2015 was the location of 88% of all malaria cases and 90% of malaria deaths.1 In most of Africa, the commonly distributed ACT is co-formulated oral artemether and lumefantrine, which has been the latest target of criminal counterfeiters.4,12–14 As with other types of falsified drugs, falsified ACTs frequently contain wrong active ingredients not stated on the packaging. These may be dangerous, toxic, and clinically confusing.Various field tests are used for detecting falsified anti-malarial drugs in situ.15–18 Typically, a two-step process combining testing via a handheld spectroscopic device (Raman and near infra-red)19–23 with colorimetric assays23–26 or thin-layer chromatography17,27 is used to detect and quantify the presence of one or a few active pharmaceutical ingredients (API). While this approach is relatively low-cost and rapid, it is only able to detect the presence of the expected active ingredients, and will not provide detailed compositional information regarding wrong APIs.28 To ascertain the exact chemical make-up of falsified ACTs, mass spectrometry (MS) is recognized as one of the most powerful tools as it can confirm the presence or absence of the expected APIs, the identity of wrong APIs, and provide a unique chemical fingerprint. Due to the complexity of the involved instrumentation, this type of fingerprinting by MS is typically performed in central laboratories in developed countries, which requires shipping samples outside of the regions directly affected by drug counterfeiting, and unnecessarily delays results and the corresponding interventions.29 However, the chemical information provided by MS, if acted on rapidly, can not only reveal the potential harmful health effects associated with falsified medicines, but also enable rapid sourcing of the origins of such samples.13,30 It is therefore imperative that more robust and portable mass spectrometers be developed for this purpose. The coupling of such instrumentation with sampling/ionization approaches that do not require extensive sample preparation to produce informative data, even if used by relatively unskilled personnel, can therefore be leveraged to produce a robust answer to the need for better drug chemical fingerprinting closer to the sample's origin.31
The World Health Organizations current strategy for effectively eradicating malaria by 2030 includes “universal access” to treatment along with the ability to “transform malaria surveillance into core intervention”.1 With the current lack of analytical tools in malarious areas to detect and trace falsified anti-malarial treatments, malaria surveillance will suffer and the lack of universal affordable access to efficacious treatment will result in the continued threat of vulnerable patients consuming falsified pharmaceuticals.13,32 It is possible, however, that as the costs and efficiency of portable instruments improve, they could see increased use in low-resource regions, providing rapid chemical information on falsified tablets upon discovery.33 The work described here involves a direct performance comparison between a portable single-quadrupole mass spectrometer benchmarked against a high-resolution laboratory-grade QTOF mass spectrometer for the purpose of detecting and fingerprinting falsified ACTs. Both instruments were modified to accommodate a direct analysis in real-time (DART) ambient plasma ion source, used in previous falsified analysis,28,31,34 to enable direct analysis and ionization of a large variety of molecular species found in falsified drugs.
2. Experimental
2.1 ACT tablet preparation and DART ionization/sampling
In total, 192 separate blister packs containing multiple tablets labelled as commercial brands of anti-malarial ACTs were supplied through seizure, purchase, or other third party interdiction from sources in several sub-Saharan Africa countries, by an international organization which procures and distributes lifesaving essential medicines. Samples were transported and stored at room temperature (∼23 °C) and no information as to the results of initial chemical tests were provided and hence investigators were blinded as to identity and chemical composition. Genuine WHO pre-qualified co-formulated artemether (10 mg per tablet) and lumefantrine (120 mg per tablet) blister packs were delivered together with the falsified drug shipments artemether–lumefantrine (AL) controls with which to compare chemical fingerprints.A commercial DART-SVP ion source (IonSense Inc., Saugus, MA) was used to directly sample each anti-malarial tablet. For generation of positive ions, the optimization of which has been described previously,35 a negative DC potential bias of several kV was applied to a point electrode with ultra-grade 99.999% pure helium gas (Airgas, Atlanta GA) flowing through at 2.2 L min−1 to generate a metastable helium plasma. The two grid electrodes used to filter plasma-generated ions were set to +250 V, and the source gas heater was optimized to operate at a set temperature of 500 °C. In all experiments conducted, the DART source was positioned in front of a gas-ion separator tube (GIST), which provided additional pumping of the heated helium gas preventing it from overloading the mass spectrometer vacuum system. The DART nozzle was positioned at 1 cm distance from the GIST tube.
After initial testing, it was found that inconsistent results were obtained if only sampling the surface of the tablets by DART. More consistent results were obtained by crushing the entire tablet into a fine powder which was homogenized and then sampled (0.5–1 mg) using a borosilicate glass capillary. At the DART gas temperatures used, the glass capillary generated its own set of unique ions and therefore background subtraction was performed using a blank capillary measurement. Mass calibration was performed in between each sample using a crushed acetaminophen tablet as a drift compensation standard.
Nanopure water (Barnstead Diamond) and HPLC grade methanol (99.9%) from Sigma-Aldrich (St. Louis, MO) were used for tablet extraction for comparison of DART results with electrospray ionization (ESI). Tablet extraction consisted of adding ∼1 mg of crushed tablet powder into 1 mL of water or methanol, shaking the solution, and letting the solution equilibrate for 8 hours. This extraction time was settled on only as a means of assuring a full transfer of analytes into solution and not in order to highlight any time inefficiency in the extraction process itself. After extraction, solutions were centrifuged to remove the remaining solids and diluted into 100% H2O or MeOH prior to performing MS analysis. Mass calibration for ESI-MS was performed using sodium formate in water (20 μM), which was introduced immediately following infusion of the tablet extract. Standards of mannitol, chloramphenicol, and ciprofloxacin were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification to confirm chemical formula assignments.
2.2 MS analysis
Two separate mass spectrometers were employed for DART analysis: (1) a hybrid high resolution QTOF Bruker MicrOTOF-Q1 and (2) a Waters QDa single quadrupole. In the case of the Bruker QTOF, the instrument was operated in positive mode in the range of 50 to 1000 m/z with the following settings: +3500 V capillary bias, −500 V end plate offset, 3.0 L min−1 N2 dry gas at 150 °C, 300 m/z quadrupole isolation mass, 3.0 eV ion energy offset, 2.5 mbar fore pressure, and a 4.42 × 10−7 mbar TOF pressure. For MS/MS experiments, settings were consistently set to 5 m/z isolation width with collision energies of 20 eV. The QDa was also operated in positive mode with a mass range of 30 to 800 m/z, a capillary bias of 800 V, a 33 V cone voltage, and 150 °C source temperature. For ESI experiments the QTOF instrument was used with the following settings: +4500 V capillary voltage, −500 V end plate offset, a 2.0 L min−1 and 200 °C N2 dry gas, 1.0 bar N2 nebulizing gas, and fore and TOF vacuum pressures of 2.41 mbar and 2.44 × 10−7 mbar, respectively. Cone voltages were ramped from 5 V up to 15 V, 30 V, 50 V, and 70 V at 10 Hz to monitor the in-source CID during each separate tablet experiment. The same operator performed both DART-QTOF MS and DART-QDa MS experiments.2.3 Data analysis and statistical processing
Prior to comparing the low-resolution quadrupole spectra with high resolution QTOF mass spectra, a data pre-processing script was developed in MATLAB (MathWorks, Natick, MA) using the PLS toolbox (Eigenvector Research, Manson, WA). A summary of the script used to process all spectra is shown in ESI (Fig. S1†). Briefly, the full set of mass spectra, after background subtraction, were exported from their respective software packages (MassLynx or DataAnalysis) as an (x, y) tab-delimited text file, uploaded into MATLAB, and resampled to produce data sets with a matching number of data points and identical mass range (from 50 to 1000 m/z). Following resampling, all spectra were appended into a single matrix. The most informative data window was selected, intensities were normalized, and data points binned in order to reduce the length of processing. Following these steps, the data was mean centered and PCA analysis performed within MATLAB.3. Results and discussion
3.1 Detection of falsified ACTs using high resolution DART-MS
All initial fingerprinting to determine the presence of active pharmaceutical ingredients (APIs) and identities of wrong active ingredients was performed using the Bruker QTOF instrument described above. Fig. 1 shows an overview of the most common ingredients detected, along with a breakdown of the main tablet-types or “Classes” that emerged from MS fingerprinting. For each class, a representative DART-MS spectra taken from a single tablet is shown with the proposed structures of the main components identified in each. High resolution MS analysis revealed that of all 192 samples under study, only six samples (3%) contained both artemether and lumefantrine, matching the fingerprint of genuine control tablets. The left region in Fig. 1 depicts the spectrum for a tablet taken from the set of unknown samples that was found to contain artemether/lumefantrine, matching the spectrum of a genuine reference tablet exactly, as seen in Fig. S2.† The remaining tablets fell into two main groups or classes; (1) those that contained a complex mixture of carbohydrates (CxHyOz) (Fig. 1, middle) and (2) those that were dominated by one of two common antibiotics (Fig. 1, right), ineffective in treating malaria. In total, 52% (99 tablets) were assigned to the carbohydrate-containing class II, and 45% (84 tablets) were assigned to the antibiotics-containing class III.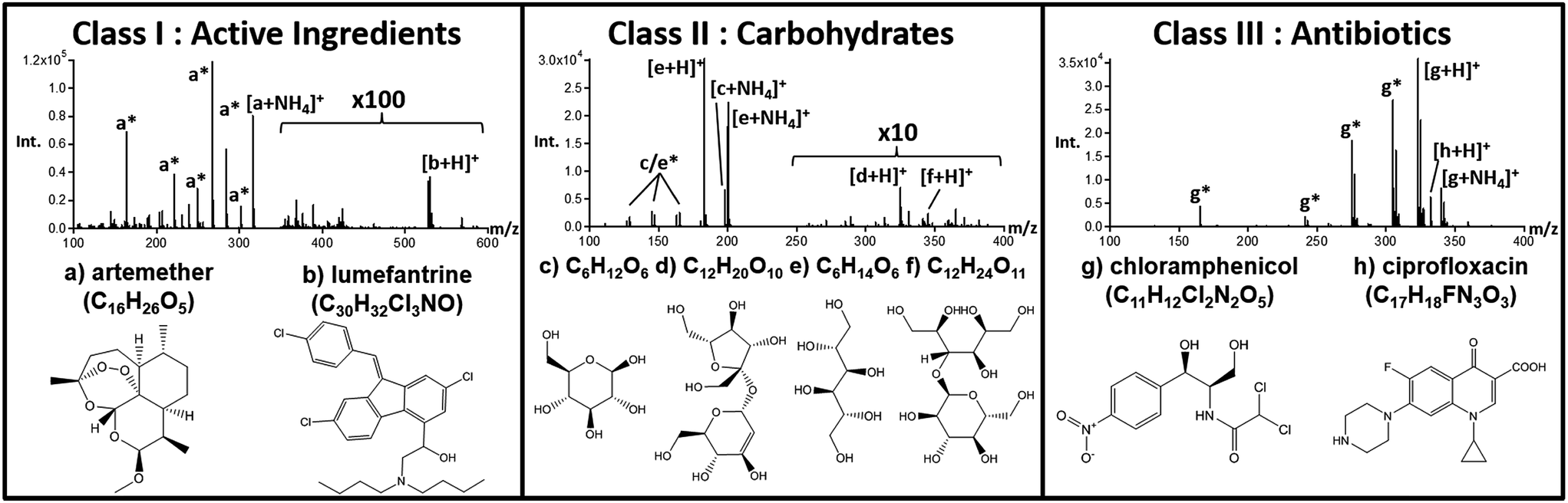 | ||
| Fig. 1 Structures of the major compounds found in the set of anti-malarial tablets analyzed using DART-MS. Class I tablets contained the expected active ingredients: (a) artemether and (b) lumefantrine. Class II tablets were those with a chemical make-up consisting mostly of carbohydrates, including those with elemental formulas (c) C6H12O6, (d) C12H20O10, (e) C6H14O6, and (f) C12H24O11. Class III tablets contained primarily the antibiotics (g) chloramphenicol and (h) ciprofloxacin. | ||
Artemether (Fig. 1a) (C16H26O5) was dominant in the DART-MS spectra of class I tablets, matching the spectra of genuine ACTs. The artemether [M + NH4]+ ion at m/z 316.21 along with several fragment ions generated directly during the plasma ionization process (all marked in Fig. 1 with a “*”) were observed.31 The artemether derived fragments included [M − CH2 + NH4]+ at m/z 302.20, [M − OHCH3 + NH4]+ at m/z 284.19, [M − OHCH3 + H]+ at m/z 267.16, [M − OHCH3 − H2O + H]+ at m/z 249.15, [M − OHCH3 − CO − H2O + H]+ at m/z 221.15, and [M − OHCH3 − C4H8O3 + H]+ at m/z 163.11. In additional to these diagnostic ions, the second API, lumefantrine (Fig. 1b, C30H32Cl3NO), was observed at m/z 528.16 as the protonated species [M + H]+ without any additional in-source fragments of significant intensity. The identity of the lumefantrine species was confirmed by the characteristic M + 2 isotope pattern matching resulting from the three chlorine atoms present on the molecule.
For class II tablets, the majority of samples produced primarily two signals: one at m/z 198.10 and one at m/z 325.11, labeled in Fig. 1c and d, respectively. These two species were assigned to [C6H12O6 + NH4]+ and [C12H20O10 + H]+, with the possible structural identities of glucose (or other hexose), and a related disaccharide. Additionally, these tablets contained signals indicative of diagnostic in-source fragments for both the monosaccharide and disaccharides, observed at m/z 289.09 ([C12H20O10 − 2(H2O) + H]+), m/z 163.06 ([C6H12O6 − H2O + H]+), m/z 145.05 ([C6H12O6 − 2(H2O) + H]+), m/z 127.04 ([C6H12O6 − 3(H2O) + H]+), m/z 97.03 ([C5H4O2 + H]+), and m/z 85.03 ([C4H4O2 + H]+). These species were the principal ones observed for 64 of the class II tablets (65%) while a separate subclass of 29 tablets (29%) also presented an additional set of signals at m/z 183.09, assigned to [C6H14O6 + H]+ (Fig. 1e), and m/z 345.14 assigned to [C12H24O11 + H]+ (Fig. 1f). Furthermore, another six samples (6%) within this class presented only a very low abundance signal for the ion at m/z 198.10 (C6O12H6), and no other distinguishable species or fragments.
The antibiotics detected in falsified ACTs of class III presented either chloramphenicol (C11H12Cl2N2O5) or ciprofloxacin (C17H18FN3O3) as the most intense species in the DART spectrum. For chloramphenicol-containing tablets (22 samples, 25%), the protonated [M + H]+ ion at m/z 323.02 and ammoniated [M + NH4]+ ion at m/z 340.05 were both present along with chloramphenicol fragments including the [M − H2O + H]+ at m/z 305.01, [M − CH4O2 + H]+ at m/z 274.00, [M − C2HOCl2 + H]+ at m/z 241.04, and [M − C2HOCl2 − CH4O2 + H]+ at m/z 165.07. Spectra for the ciprofloxacin-containing tablets, consisting of 65 samples (75%), showed primarily an intense [M + H]+ signal at m/z 332.14 with a prominent DART in-source [M − COOH + H]+ fragment at m/z 288.15. This sub-set of tablets also frequently included samples that contained both chloramphenicol and ciprofloxacin with varying observed MS intensities for each.
Despite the ability of DART-MS to generate a set of diagnostic fragments in-source, all chemical compound identity assignments were further confirmed using accurate mass measurements (2 ppm), comparisons with the NIST collision-induced dissociation (CID) database, and tandem MS/MS experiments of authentic standards. Fig. S3–S6† show these comparisons for the most abundant species (sugars, sugar-alcohols, chloramphenicol, ciprofloxacin) detected in each falsified ACT class.
3.2 Comparison of DART spectra to electrospray ionization
Although DART has become well established as a reliable tool for rapidly determining drug quality, it is also important to determine whether electrospray ionization following liquid–solid extraction would provide similar information in terms of identifying the main sample components. As with DART, no chromatography was attempted with ESI, as the goal of our study was to provide chemical fingerprint information in a short time frame. Fig. 2 shows ESI-MS spectra from the same three ACT tablets depicted in Fig. 1 extracted with two different solvents, with the DART-MS data provided for comparison.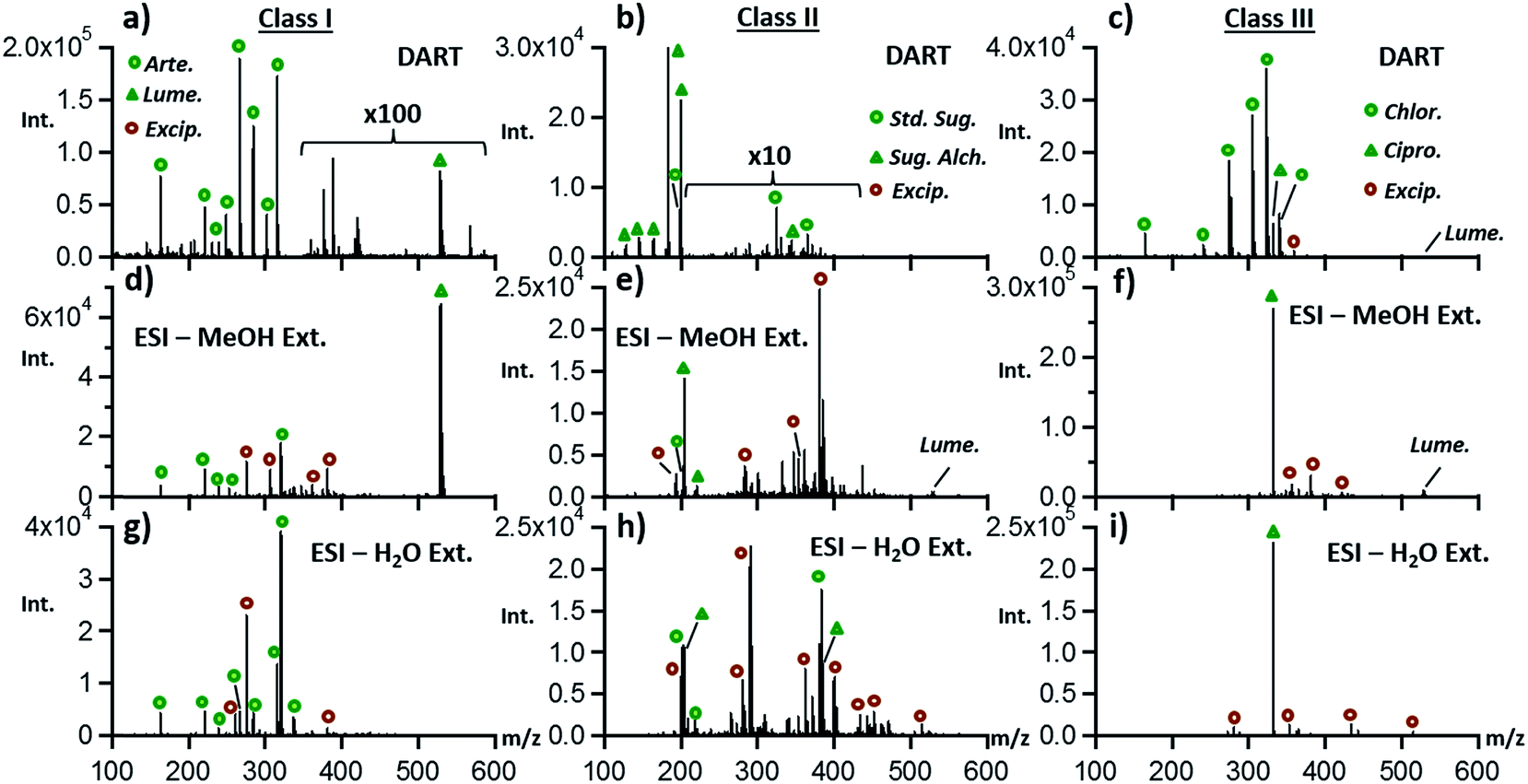 | ||
| Fig. 2 Comparison of the DART-MS (a–c) to ESI-MS (d–f) and (g–i) results for representative classes of genuine and falsified ACT tablets containing artemether/lumefantrine, sugars/sugar-alcohols, and antibiotics. Direct infusion ESI experiments were performed on either methanol or water extracts at the 1 mg mL−1 level. Samples were extracted for eight hours prior to MS analysis. | ||
Fig. 2a shows that higher sensitivity but more extensive fragmentation was obtained by DART than by ESI (Fig. 2d and g), as expected from previous internal energy deposition studies.36 Extraction of ACT tablet powder in water followed by ESI produced spectra with no detectable signal for lumefantrine, due to solubility limitations. ESI spectra also showed new species that were also detected in class II and class III samples, determined to be common tablet excipients. These were more easily detected by ESI than by DART, with DART spectra being representative mostly of the APIs, probably due to the differences in the species sampled by DART thermal desorption vs. those sampled by liquid–solid extraction. Based on a 2 ppm mass accuracy threshold, excipients detected included trimethylolpropane trimethacrylate, dihexyl maleate, and glycerol monostearate. Class II and class III samples revealed these excipients as well and additional compounds including cellulose, palmitoyl glycerol, and lactitol monohydrate.
Fig. 2b outlines the DART results for class II samples and the comparison to what was observed by ESI (Fig. 2e and h). Similar results to what was observed in the case of class I tablets were seen for these samples, with the main difference being that the abundance of sugar species was higher by ESI than by DART, as expected due to their solubility and lability to thermal degradation. Antibiotic-containing class III tablets were better fingerprinted by DART than by ESI, with chloramphenicol not being detected in ESI experiments (Fig. 2c, f and i). It is possible that the highly abundant ciprofloxacin ions observed in ESI experiments suppressed the ionization of chloramphenicol in this direct infusion experiment, thus explaining the sparse nature of the ESI data for class III samples.
3.3 PCA analysis of DART MS fingerprint data
Since DART-MS provided the clearest distinction between the three main classes of genuine and falsified ACT tablets, a multivariate approach was used to further refine this classification. The raw DART-MS data produced using the high resolution QTOF instrument was processed to enable comparisons via PCA. The full workflow of complete DART chemical fingerprinting procedure is outlined in Fig. S7.† Tablet crushing required about 30 seconds, with DART sampling of the tablet only requiring another minute, even if performed in triplicate. Fig. S7b† details the effect of the different data pre-processing steps, focusing on the artemether ion region at m/z 260–330. For this range, a mass resolution of 5500 was achieved, corresponding to a FWHM of 0.05 m/z for the m/z 316.21 peak. Resampling was carried out to produce spectral data that aligned exactly in terms of granularity, producing spectra that could be appended in a single matrix. Normalization was necessary to adjust for differences in overall signal intensity that are produced by the variability innate to DART ionization,37 where binning was used so as to avoid over sensitivity to minor spectral features by the PCA analysis step. The binning step effectively converted high-resolution data into low-resolution data, which could be viewed as undesirable from the chemical identification perspective, but greatly sped up the calculation of the PCA loadings and scores. Overall, exporting of raw data into MATLAB and subsequent data processing was the longest step in the chemical fingerprinting workflow, requiring between 1 and 2 hours.The PCA plot of the resampled and binned DART QTOF data from all 192 samples is shown in Fig. 3. The six artemether/lumefantrine genuine samples (class I) are located in the center of the plot. Their tight grouping is better observed in the zoomed-in in-set in the top right corner. The neighboring samples coded with gray circles represent falsified tablets containing sugars in low concentration. The class II tablets containing primarily monomeric sugars (C6H12O6) and low abundance of dimeric oligosaccharides (C12H22O10–H2O), are shown with yellow circles, and referred to as “carbohydrate mixture 1”. These samples form a distinct linear trend in the PCA scores plot with the samples closest to the center of the plot exhibiting progressively lower intensity of the carbohydrate species, eventually reaching the region occupied by the samples coded in gray. The pink sub-set of “carbohydrate mixture 2” samples formed a distinct sub-cluster, reflecting a fingerprint with the largest amount of alcohol–carbohydrates (C6H14O6 and C12H24O11) within the class II tablets.
 | ||
| Fig. 3 Principal component analysis of the full set of DART mass “chemical fingerprints” obtained using DART-MS on a QTOF mass spectrometer. The first three principal components are shown in the x, y, and z axes. Artemether/lumefantrine-containing tablets are shown in orange, ciprofloxacin in green, chloramphenicol in blue, sugar mix 1 (common sugars only) in yellow, sugar mix 2 (common sugars + alcohol sugars) in pink, and tablets showing only low intensity sugar patterns in gray. Higher resolution QTOF data points are marked as circles (○). | ||
Class III tablets were all clustered together, with some internal structure visible within the main cluster. The green ciprofloxacin-containing tablets progressively approached the blue-coded chloramphenicol-only samples in PCA score space. This progressive trajectory coincided with the level of mixing of the two antibiotics in any particular sample. Clearly, a subset of the class III tablets consisted of a mixture of the two compounds. It is possible, but difficult to confirm, that this trend reflected a change in the “recipe” used by counterfeiters over time to produce these “product”. Additionally, some ciprofloxacin-containing tablets had varying amounts of this wrong active ingredient, and therefore their chemical signatures progressively approached the low concentration sugar-containing fakes towards the center of the plot.
Investigation of higher principal components revealed some structure not seen in the PC1–PC2–PC3 scores plot. The PC1–PC2–PC4 plot (Fig. S8†) shows that this view produces a more defined and distinct grouping for the genuine tablets. Fig. S9† highlights the details of the four principal components and makes clear how the major artemether/lumefantrine features contribute to PC4. Additionally, it shows that PC1 and PC2 provide the largest distinctions between the ciprofloxacin peak at m/z 332, the carbohydrate peaks containing the m/z 198 and 180 peaks, and the chloramphenicol peaks at m/z 305, 323, and 340. PC3 defines the differences between spectra containing the antibiotics and glucose peaks against the sugar alcohol peaks at m/z 183 and 200 and to a lesser extent the artemether class I peaks at m/z 267 and 316. PC4 highlights the class I differences further in that it separates all of the falsified detected ingredients (both class II and III) with the artemether peaks m/z 316 and 267 along with the lumefantrine peak at m/z 528. This is significant in providing the different PCA results using PC1–PC2–PC3 compared to that of the PC1–PC2–PC4, where the AL tablet points are better isolated from the falsified tablets. Therefore, PC4 could be used for its ability to conclusively separate artemether/lumefantrine-containing samples efficiently.
3.4 DART fingerprinting with single-quadrupole instrument and comparison to QTOF results
To bring the fight against falsified drugs closer to where the problem exists, to empower local scientists & clinicians, and to deploy a more decentralized structure with faster response times, low maintenance portable/benchtop instrumentation that requires less consumables, is easily operated by personnel with minimum training, and can produce results somewhat on par with those from higher-end instruments is highly desirable.Fig. 4 shows a set of mass spectra obtained with the QDa mass spectrometer for class I, II, and III tablets, and their direct comparison with QTOF spectra. ESI Fig. S10–S14† show comparisons between spectra for additional types of samples with more detail. Essentially, spectra obtained with the QDa (Fig. 4d–f) showed the same base peaks, and similar relative abundances among species as spectra obtained with the QTOF (Fig. 4a–c). The main differences related to some variation in the degrees of fragments observed for the APIs in the various sample classes under study. For class I samples, the QDa provided significantly greater intensity of the lumefantrine peak at m/z 528.16, and detected the same artemether species at m/z 316.21, 267.16, 249.15, and 221.15. For class II, the previously identified sugar alcohol at m/z 345.14 (C12H24O11), its water loss products, the simple carbohydrate at m/z 198.01 (C6H12O6), the sugar alcohol at m/z 183.01 (C6H12O6), and the corresponding fragments were all detected. Lastly, the single-quadrupole was able to detect both antibiotics in class III tablets, as shown in Fig. 4f for a specific sample containing both ciprofloxacin and chloramphenicol. The main difference observed in this case was that the intensity of chloramphenicol was significantly lower with the low-resolution instrument. In general, it was observed that many low abundance species were not detected with the QDa, which impacted fingerprinting of tablets known to contain both ciprofloxacin and chloramphenicol, but for which only the first API was detected. This difference affected the corresponding clustering of samples in PCA, as discussed below.
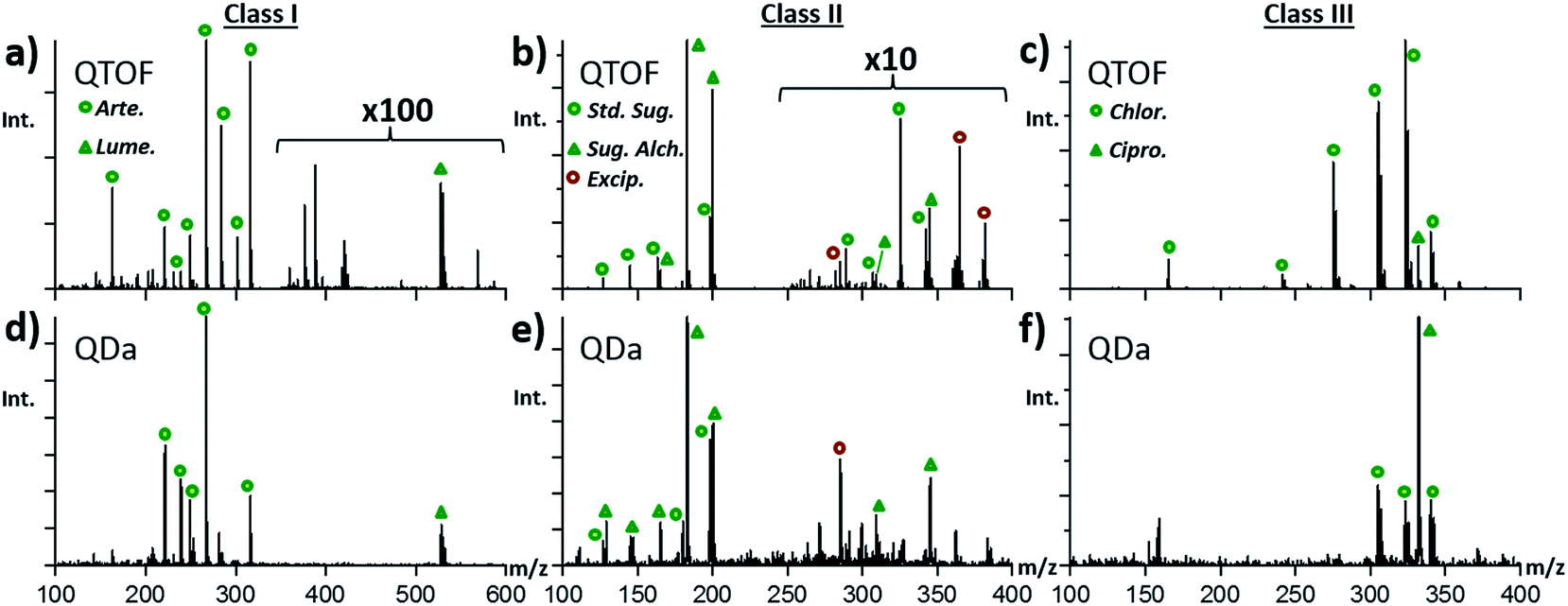 | ||
| Fig. 4 Comparison of the DART-MS spectra collected for the major types of anti-malarial treatment tablets examined using the QTOF mass spectrometer (a–c) and the QDa (d–f). | ||
A comparison of the ion intensities observed with both platforms is summarized in Fig. S15.† The relative intensity for the lumefantrine ion among all six tablets investigated where lumefantrine was a primary component was found to be 10% for the QDa instrument and only 0.04% for the QTOF (pink). This effect is due, in large part, to the mass bias associated with the time-dependent hexapole trapping enrichment technique used in this type of QTOF.38,39 As the single-quadrupole has very few settings in voltage and frequency that would bias the collection of ions in to the analyzer, it has an advantage of simplicity in its ion collection that appears to affect the overall level of each ion with respect to the QTOF where MS settings highly influence the intensities of differing mass ions. The relative intensity of the artemether species was higher for the QTOF at 20%, whereas the QDa showed an average intensity of 10%. Similar effects were observed for species in class II and class III samples, but these differences in specific ion abundances, when considered as part of a whole chemical fingerprint (i.e. the complete mass spectrum), did not influence PCA results as much as it would have been expected.
The PCA analysis conducted for the QDa dataset is shown in Fig. 5, with data being processed in an identical way as for the QTOF. The main difference observed between this plot and Fig. 3 was the reduced variability in the cluster of class III tablets. Only two tablets had PCA scores outside of their respective group, namely chloramphenicol (blue) or ciprofloxacin (green). Additionally, tablets containing the carbohydrate mix 1 (yellow) showed a tighter PCA grouping with less variability, and the lower sugar content tablets (gray) were better separated from the orange artemether/lumefantrine-containing tablets. As with the QTOF dataset, the classes were well separated using the first three principal components, but examination of PC4 (Fig. S16†) in place of PC3 exposed a grouping that better distinguished artemether/lumefantrine-containing tablets.
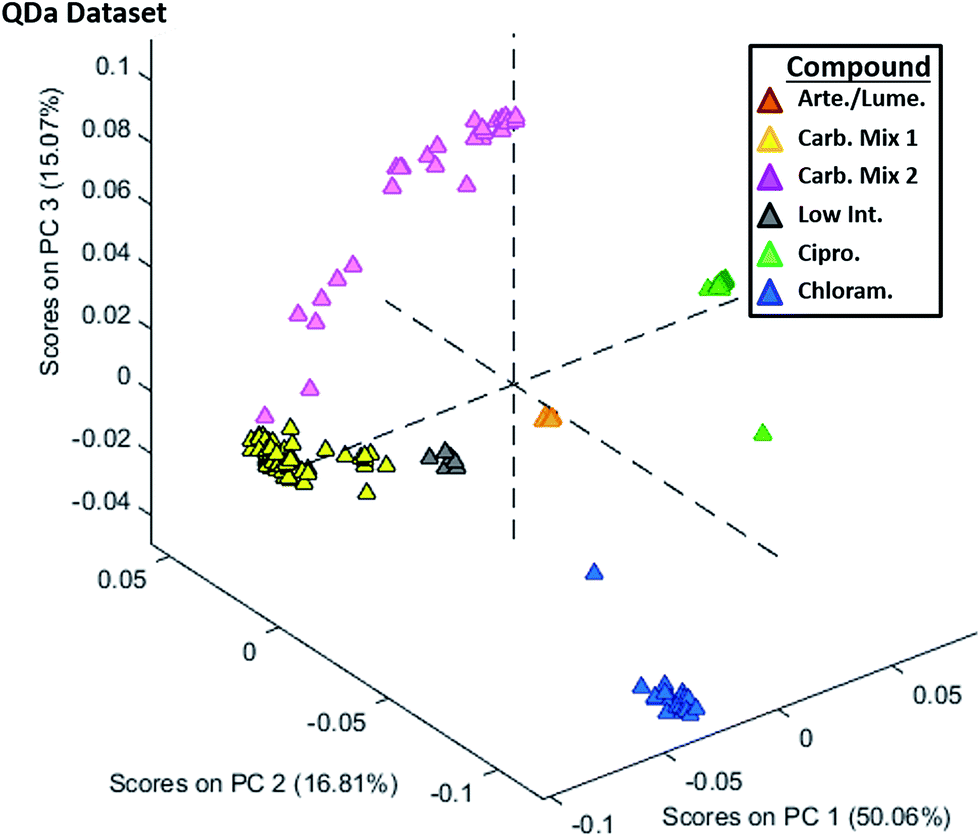 | ||
| Fig. 5 The second set of principal component analyses of the full set of anti-malarial therapy tablets via DART-MS from (a) a lower-resolution single-quad bench top instrument. The artemether/lumefantrine containing tablets are shown in the plot in orange, ciprofloxacin in green, chloramphenicol in blue, sugar mix 1 (standard sugars only) in yellow, sugar mix 2 (standard + alcohol sugars) in pink, and low intensity sugar tablets in gray. Low resolution single-quad data points are marked as triangles (△). | ||
3.5 In-source collision-induced dissociation (CID) QDa experiments vs. MS/MS QTOF experiments
Besides its lower mass resolving power, one major drawback regarding the use of the QDa for on-site analysis of suspect ACTs is the inability to perform “true” MS/MS experiments on previously unobserved wrong active ingredients. While the use of single quadrupole mass spectrometry would mainly be for triaging and categorizing falsified materials of already understood chemical make-up, in the hopes of avoiding adverse health effects and tracing their sources; the ability to perform fragmentation experiments to confirm putative identities is undoubtedly helpful upon the discovery of new “recipes” of falsified drugs. Fig. S17† shows the comparison of sugar alcohol (m/z 183.09, [C6H14O6 + H]+), chloramphenicol (m/z 323.02, [C11H12Cl2N2O5 + NH4]+) and ciprofloxacin (m/z 332.14, [C17H18FN3O3 + H]+) MS/MS spectra obtained using the QTOF compared to the in-source cone CID (i.e. without precursor isolation) performed with the QDa on the same tablet powders. For the sugar alcohol, it is seen that the fragment ion peaks match in five instances, but there is some interference with the −2 Da C6H12O6 fragments seen in the QDa spectrum. Less interferences and more clean matches were observed for chloramphenicol and ciprofloxacin, since the starting spectra were sparser.It should be understood that these tablet fragmentation comparisons are best case scenarios in that the ions subject to precursor isolation in the QTOF were the base peaks in the QDa spectra, and therefore the QDa in-source CID spectra matched well with each MS/MS spectrum. Fragmentation via cone activation could produce cluttered and indecipherable results when starting with more complex mass spectra. However, sequential ramping of the cone voltage from 5 to 70 V with a sampling rate of 10 Hz at each energy, would allow for obtaining more selective fragmentation patterns if precursors with different-enough activation energies were present. Temperature ramping of the DART ion source,40,41 could also be utilized as a method for selectively volatilizing chemical constituents of the examined samples.
4. Conclusions
Portable DART-QDa MS has been shown to be an efficient method for fingerprinting tablet pharmaceuticals and providing information consistent with bench-top high resolution DART-QTOF analysis, taking only minutes for both sample preparation and MS introduction. Use of a single-quadrupole mass analyzer, or other portable and low-cost MS instrument, in conjunction with data processing that enables rapid and simplified statistical sorting of complex falsified anti-malarial treatments showed the unique ability of such an instrument to balance laboratory costs for operation and rapid sampling with the identification of otherwise overlooked chemical species. The QDa does also suffer from some disadvantages in that its mass accuracy would not allow for confident identification of new compounds and it would not be portable or resource efficient enough to be taken to any location without the necessary electricity (possibly with a generator). While it is not a handheld device it offers an extremely simple set-up and interface, low energy requirements, and while the system as a whole would weigh approximately 100–200 pounds it or other similar single-quad instrument could be fixed to a cart for semi-portability as was provided for the model used in these studies.Based on the findings from the set of tablets investigated here, the presence of the wrong ingredients detected could have serious health implications. For example, chloramphenicol and ciprofloxacin both have mild antimalarial activity42,43 but are not efficacious antimalarial monotherapies, and will be grossly clinically inferior to ACTs. Furthermore, the cryptic ingestion of these antibiotics will engender antibacterial drug resistance, both through selection of resistance mutants in the patients' gastrointestinal tract and for bacteria, especially non-typhoidal Salmonella species, that not infrequently co-infect malarious patients.44 With the increased realization that antibacterial resistance is a global disaster,45 more attention is needed on preventing this cryptic and harmful fraudulent use of antibiotics, both for individual patients and for society. To target these and other possible illegitimate falsified ingredients quickly and efficiently would be a great boost to the health efforts of underdeveloped regions as a whole.
The fingerprinting of a larger sample set including this and future seized falsified anti-malarial treatments would provide a highly robust level of comparison and categorization of any falsified ACTs detected with primary detection methods. What would most efficiently facilitate the creation of such a database would be a targeted usage of this MS set-up where both genuine and falsified samples could be analyzed without any costly shipping of materials out of country or continent from seizure location with multiple portable/fieldable MS instruments of various analyzer set-ups compared. Additionally, future implementation of this method would include a proper interface for data processing where newly collected data could be added in a simple fashion allowing for routine sorting of new data into a PCA of previously collected samples providing a user friendly way of viewing spectral differences without needing extensive data manipulation experience. It is the hope that from the method described and discussion of the specific characteristics of analysis, that this procedure could be seen as a valuable one for achieving the long-term plan of the WHO to eradicate malaria in the coming decades.1
Acknowledgements
We would like to thank the ACT Consortium and Center for Chemical Evolution for access to high resolution mass spectrometry instrumentation.References
- World Health Organization, 2016. Fact Sheet No. 94, http://www.who.int/mediacentre/factsheets/fs094/en/, accessed April 20th, 2016 Search PubMed.
- World Health Organization, Top Ten Causes of Death 2012. Fact Sheet No. 310, http://www.who.int/mediacentre/factsheets/fs310/en/, accessed April 20th, 2016 Search PubMed.
- J. P. Renschler, et al., Estimated under-five deaths associated with poor-quality antimalarials in sub-Saharan Africa, Am. J. Trop. Med. Hyg., 2015, 92(6), 119–126 CrossRef PubMed.
- P. N. Newton, et al., Poor quality vital anti-malarials in Africa – an urgent neglected public health priority, Malar. J., 2011, 10, 352 CrossRef PubMed.
- S. Sengaloundeth, et al., A stratified random survey of the proportion of poor quality oral artesunate sold at medicine outlets in the Lao PDR – implications for therapeutic failure and drug resistance, Malar. J., 2009, 8, 172 CrossRef PubMed.
- S. Yeung, et al., Quality of Antimalarials at the Epicenter of Antimalarial Drug Resistance: Results from an Overt and Mystery Client Survey in Cambodia, Am. J. Trop. Med. Hyg., 2015, 92(6), 39–50 CrossRef CAS PubMed.
- P. N. Newton, et al., Characterization of “Yaa Chud” Medicine on the Thailand-Myanmar Border: Selecting for Drug-resistant Malaria and Threatening Public Health, Am. J. Trop. Med. Hyg., 2008, 79(5), 662–669 CAS.
- R. Bate and K. Hess, Anti-malarial drug quality in Lagos and Accra – a comparison of various quality assessments, Malar. J., 2010, 9, 157 CrossRef PubMed.
- L. K. Basco, Molecular epidemiology of malaria in Cameroon. XIX. Quality of antimalarial drugs used for self-medication, Am. J. Trop. Med. Hyg., 2004, 70(3), 245–250 CAS.
- P. Tabernero, et al., A Repeat Random Survey of the Prevalence of Falsified and Substandard Antimalarials in the Lao PDR: A Change for the Better, Am. J. Trop. Med. Hyg., 2015, 92(6), 95–104 CrossRef PubMed.
- T. Ehianeta, et al., Quality survey of some brands of artesunate–amodiaquine in Lagos drug market, Afr. J. Pharm. Pharmacol., 2012, 6(9), 636–642 CAS.
- H. Kaur, et al., Antimalarial drug quality: methods to detect suspect drugs, Therapy, 2010, 49–57 CrossRef CAS , 7 (Copyright (C) 2016 American Chemical Society (ACS)). All Rights Reserved.
- T. Kelesidis and M. E. Falagas, Substandard/Counterfeit Antimicrobial Drugs, Clin. Microbiol. Rev., 2015, 28(2), 443–464 CrossRef PubMed.
- G. M. L. Nayyar, J. G. Breman and J. E. Herrington, The Global Pandemic of Falsified Medicines: Laboratory and Field Innovations and Policy Perspectives: Summary, Am. J. Trop. Med. Hyg., 2015, 92(6), 2–7 CrossRef PubMed.
- F. M. Fernandez, M. D. Green and P. N. Newton, Prevalence and detection of counterfeit pharmaceuticals: a mini review, Ind. Eng. Chem. Res., 2008, 47(3), 585–590 CrossRef CAS.
- M. D. Green, F. Atieli and M. Akogbeto, Rapid colorimetric field test to determine levels of deltamethrin on PermaNet((R)) surfaces: association with mosquito bioactivity, Trop. Med. Int. Health, 2009, 14(4), 381–388 CrossRef CAS PubMed.
- R. Bate, et al., Pilot study comparing technologies to test for substandard drugs in field settings, Afr. J. Pharm. Pharmacol., 2009, 3(4), 165–170 CAS.
- J. S. Batson, et al., Assessment of the effectiveness of the CD3+ tool to detect counterfeit and substandard anti-malarials, Malar. J., 2016, 15(1), 119 CrossRef PubMed.
- C. Ricci, et al., Characterization of genuine and fake artesunate anti-malarial tablets using Fourier transform infrared imaging and spatially offset Raman spectroscopy through blister packs, Anal. Bioanal. Chem., 2007, 389(5), 1525–1532 CrossRef CAS PubMed.
- C. Ricci, et al., Assessment of hand-held Raman instrumentation for in situ screening for potentially counterfeit artesunate antimalarial tablets by FT-Raman spectroscopy and direct ionization mass spectrometry, Anal. Chim. Acta, 2008, 623(2), 178–186 CrossRef CAS PubMed.
- Y. L. Loethen, et al., Rapid screening of anti-infective drug products for counterfeits using Raman spectral library-based correlation methods, Analyst, 2015, 7225–7233 RSC , 140 (Copyright (C) 2016 American Chemical Society (ACS)). All Rights Reserved.
- M. Hajjou, et al., Assessment of the performance of a handheld Raman device for potential use as a screening tool in evaluating medicines quality, J. Pharm. Biomed. Anal., 2013, 74, 47–55 CrossRef CAS PubMed.
- M. de Veij, et al., Fast detection and identification of counterfeit antimalarial tablets by Raman spectroscopy, J. Raman Spectrosc., 2007, 38(2), 181–187 CrossRef CAS.
- M. D. Green, et al., A colorimetric field method to assess the authenticity of drugs sold as the antimalarial artesunate, J. Pharm. Biomed. Anal., 2000, 24(1), 65–70 CrossRef CAS PubMed.
- K. A. Hall, et al., Characterization of counterfeit artesunate antimalarial tablets from southeast Asia, Am. J. Trop. Med. Hyg., 2006, 75(5), 804–811 CAS.
- M. D. Green, et al., Integration of Novel Low-Cost Colorimetric, Laser Photometric, and Visual Fluorescent Techniques for Rapid Identification of Falsified Medicines in Resource-Poor Areas: Application to Artemether–Lumefantrine, Am. J. Trop. Med. Hyg., 2015, 92(6), 8–16 CrossRef CAS PubMed.
- F. Khuluza, et al., Use of thin-layer chromatography to detect counterfeit sulfadoxine/pyrimethamine tablets with the wrong active ingredient in Malawi, Malar. J., 2016, 15(1), 215 CrossRef PubMed.
- V. Habyalimana, et al., Analytical tools and strategic approach to detect poor quality medicines, identify unknown components, and timely alerts for appropriate measures: case study of antimalarial medicines, Am. J. Anal. Chem., 2015, 977–994 CrossRef CAS , 6 (Copyright (C) 2016 American Chemical Society (ACS)). All Rights Reserved.
- P. N. Newton, et al., A collaborative epidemiological investigation into the criminal fake artesunate trade in South East Asia, PLoS Med., 2008, 5(2), 209–219 Search PubMed.
- Y. L. Yong, Collaborative Health and Enforcement Operations on the Quality of Antimalarials and Antibiotics in Southeast Asia (vol 92, pg 105, 2015), Am. J. Trop. Med. Hyg., 2015, 93(4), 891 CrossRef PubMed.
- F. M. Fernandez, et al., Characterization of solid counterfeit drug samples by desorption electrospray ionization and direct-analysis-in-real-time coupled to time-of-flight mass spectrometry, ChemMedChem, 2006, 1(7), 702–705 CrossRef CAS PubMed.
- K. Karunamoorthi, The counterfeit anti-malarial is a crime against humanity: a systematic review of the scientific evidence, Malar. J., 2014, 13, 209 CrossRef PubMed.
- D. T. Snyder, et al., Miniature and Fieldable Mass Spectrometers: Recent Advances, Anal. Chem., 2016, 88(1), 2–29 CrossRef PubMed.
- L. Nyadong, et al., Combining Two-Dimensional Diffusion-Ordered Nuclear Magnetic Resonance Spectroscopy, Imaging Desorption Electrospray Ionization Mass Spectrometry, and Direct Analysis in Real-Time Mass Spectrometry for the Integral Investigation of Counterfeit Pharmaceuticals, Anal. Chem., 2009, 81(12), 4803–4812 CrossRef CAS PubMed.
- M. Zhou, J. F. McDonald and F. M. Fernandez, Optimization of a Direct Analysis in Real Time/Time-of-Flight Mass Spectrometry Method for Rapid Serum Metabolomic Fingerprinting, J. Am. Soc. Mass Spectrom., 2010, 21(1), 68–75 CrossRef CAS PubMed.
- L. Nyadong, et al., Direct quantitation of active ingredients in solid artesunate antimalarials by noncovalent complex forming reactive desorption electrospray ionization mass spectrometry, J. Am. Soc. Mass Spectrom., 2008, 19(3), 380–388 CrossRef CAS PubMed.
- G. A. Harris, C. E. Falcone and F. M. Fernandez, Sensitivity “Hot Spots” in the Direct Analysis in Real Time Mass Spectrometry of Nerve Agent Simulants, J. Am. Soc. Mass Spectrom., 2012, 23(1), 153–161 CrossRef CAS PubMed.
- K. Rajabi, M. L. Easterling and T. D. Fridgen, Solvation of electrosprayed ions in the accumulation/collision hexapole of a hybrid Q-FTMS, J. Am. Soc. Mass Spectrom., 2009, 20(3), 411–418 CrossRef CAS PubMed.
- Y. Wang, et al., The coupling effects of hexapole and octopole fields in quadrupole ion traps: a theoretical study, J. Mass Spectrom., 2013, 48(8), 937–944 CrossRef CAS PubMed.
- J. M. Nilles, T. R. Connell and H. D. Durst, Thermal separation to facilitate Direct Analysis in Real Time (DART) of mixtures, Analyst, 2010, 135(5), 883–886 RSC.
- C. M. Jones and F. M. Fernandez, Transmission mode direct analysis in real time mass spectrometry for fast untargeted metabolic fingerprinting, Rapid Commun. Mass Spectrom., 2013, 27(12), 1311–1318 CrossRef CAS PubMed.
- A. E. T. Yeo and K. H. Rieckmann, The in vitro Antimalarial Activity of Chloramphenicol against Plasmodium falciparum, Acta Trop., 1994, 56(1), 51–54 CrossRef CAS PubMed.
- Y. F. Falajiki, et al., Amodiaquine–Ciprofloxacin: a potential combination therapy against drug resistant malaria, Parasitology, 2015, 142(6), 849–854 CrossRef CAS PubMed.
- J. Church and K. Maitland, Invasive bacterial co-infection in African children with Plasmodium falciparum malaria: a systematic review, BMC Med., 2014, 12, 31 CrossRef PubMed.
- A. H. Holmes, et al., Understanding the mechanisms and drivers of antimicrobial resistance, Lancet, 2016, 387(10014), 176–187 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ay01418f |
| This journal is © The Royal Society of Chemistry 2016 |