
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ monitoring of additives during CO2 gas hydrate formation
M.
Schwenk
a,
A.
Katzir
b and
B.
Mizaikoff
*a
aInstitute of Analytical and Bioanalytical Chemistry, Ulm University, Albert-Einstein-Allee 11, Ulm 89081, Germany. E-mail: boris.mizaikoff@uni-ulm.de
bThe School of Physics and Astronomy, Tel Aviv University, Tel Aviv 69978, Israel
First published on 5th July 2016
Abstract
Research activities on the reduction of carbon dioxide emissions via effective carbon capture and storage (CCS) techniques are steadily increasing with the concept of storing CO2 as hydrates among the most prominently discussed strategies. The present study utilizes mid-infrared (MIR) fiber-optic evanescent field sensing techniques as a promising in situ monitoring tool for investigation of molecular changes occurring during CO2 hydrate formation. The identification and evaluation of characteristic IR absorption features associated with additive molecules (here, THF and SDS) and their changes during hydrate formation were pronounced via studies in D2O next to H2O as the hydrate-forming matrix. By correlating IR-spectroscopic data with continuously recorded pressure and temperature traces, hypotheses on the involvement and promoting effect of such additives during carbon dioxide gas hydrate formation were experimentally consolidated.
1 Introduction
Gas hydrates are crystalline compounds comprising structured water cavities hosting small guest molecules. Different hydrate structures result from combinations of individual cages. In the case of natural gas hydrates, three distinctive structures have been identified: cubic sI, cubic sII, and hexagonal sH. The capability of trapping increasingly larger guest molecules is associated with increasing cage dimensions from sI to sH.1,2CO2 gas hydrates are known to form the cubic structure sI characterized by a unit cell comprising two smaller dodecahedral (512) cages, and six larger tetrakaidecahedral (51262) cages formed by 46 water molecules.1,2 At full occupation, this leads to a composition of CO2 × 5.75 H2O. However, it is commonly agreed that nearly all the large cages are being occupied along with a fraction of the small cages, thereby leading to a hydrate composition of CO2 × 5.75–7.66 H2O.3,4
Research interest in CO2 hydrates is frequently associated with the potential storage of CO2 as gas hydrates, and/or the simultaneous production of methane during a hydrate exchange process (i.e., methane stored in gas hydrates is retrieved, while sequestering CO2 into the ice structure).5–8 Another process of significant interest relates to reducing greenhouse gas emission by formation of gas hydrates as a novel strategy for capturing/separating CO2 from synthesis or flue gases.9–12 The usage of specific additives in hydrate research to influence the formation conditions is widely accepted in this context. Generally, such additives are divided into thermodynamic and kinetic promoters. Tetrahydrofurane (THF) has proven to be an effective thermodynamic promoter via its ability to alter the equilibrium conditions of hydrate formation towards higher temperatures and/or lower pressures.13,14 In the field of kinetic promotors, the anionic tenside sodium dodecyl sulphate (SDS) positively influences the hydrate/water conversion ratio, as well as the growth rate for hydrocarbon hydrates.15,16 Also, the combined use of both additives affecting CO2 hydrate formation has led to promising results.17–19 However, the underlying mechanisms of the promoting effect remain under debate requiring further research to elucidate the governing processes. Commonly, two main principles of additive-assisted hydrate formation are discussed, especially for kinetic additives such as SDS: (i) capillary-driven mechanisms,15,20 and (ii) promotion via surfactant micelle formation.16
In order to gain further insight into the process of gas hydrate formation, innovative monitoring techniques are demanded. Despite their exquisite molecular selectivity, IR-based spectroscopic and sensing techniques have to date rarely been applied in hydrate research, as reported within only a few publications.21–29 Bertie and Othen pioneered this field by applying IR spectroscopy to investigate ethylene oxide hydrate in the spectral window of 4000–20 cm−1.21,22 Later, Fleyfel and Devlin proved that two separate peaks for CO2 in the small and large cages of sI hydrate structures may be identified within thin films of the CO2 hydrate, which was epitaxially grown on ethylene oxide hydrate. The absorption signals were evident at 2347 cm−1 and 2338 cm−1 at a temperature of 135 K for the small dodecahedral and the large tetrakaidecahedral cages, respectively.25 Later, Kumar et al. applied infrared attenuated total reflection (IR-ATR) spectroscopy to investigate thin films of CO2 hydrate, and consolidated the results reported at that time via absorption signals at 2347 cm−1 and 2336 cm−1 at −50 °C for small and large cages, respectively.26 In order to gain in situ access, while actually monitoring hydrate formation in real time, the group of Mizaikoff successfully applied for the first time IR fiber-optic evanescent field spectroscopy (FEFS) based on the principles of IR-ATR in order to investigate hydrocarbon gas hydrate formation.30,31 These studies revealed that the observed IR-spectroscopic changes of water could be associated with gas hydrate formation processes. The evaluation of the infrared absorption signatures of SDS lead to the suggestion of a mechanism for the promotion of propane hydrate formation via SDS.31 Although IR spectroscopy remains a rarely applied analytical technique, the achieved results clearly indicate that its potential is underrated in gas hydrate research and that IR-based techniques are indeed viable options for fundamentally understanding and molecularly deciphering gas hydrate formation processes.
The present study focuses on the application of fiber-optic IR-ATR sensing technologies for in situ monitoring of CO2-containing gas hydrates formed in water and deuterated water in the presence of relevant additives (i.e., THF and SDS). The obtained spectral data were correlated with simultaneously recorded pressure and temperature traces, which add additional physical information to the chemical data derived from the IR-spectra.
2 Results and discussion
CO2 hydrates produced during the present study were analyzed both non-spectroscopically, by recording pressure/temperature (pT) traces, and via continuous in situ MIR-FEFS monitoring. Accordingly, thus obtained IR spectra were correlated with the information provided via the pT traces.2.1 Pressure/temperature monitoring of CO2 gas hydrates
Initial hydrate formation is indicated via a sharp temperature peak due to the exothermic nature of the crystallization process. Fig. 1 shows typical pT-traces versus the time for CO2 gas hydrate experiments. For clarity, only the first 50 hours are displayed for an experiment using H2O (Fig. 1a) and D2O (Fig. 1b) as the background matrix, respectively. | ||
| Fig. 1 Pressure/temperature vs. time for (a) CO2 hydrate experiment in H2O and (b) in D2O as the bulk matrix. | ||
The initial pressure reduction can be attributed to increased gas solubilization with decreasing temperature during the cooling phase. After approx. 3 hours, the experiment in H2O (Fig. 1a) showed a sharp temperature peak (1) of 7.6 °C, thereby indicating initiation of gas hydrate nucleation. Subsequently, the pressure dropped (2) due to the incorporation of gas into the clathrate structure. The gas supply was activated several times throughout the experiment in order to replenish the already consumed gas (3) in the system until a characteristic pressure drop was not observable anymore. The temperature was kept at approx. 5 °C during the entire experiment. The experiment using D2O as the matrix revealed similar behaviour (Fig. 1b). The sharp temperature peak (1) occurred here after approx. 2.8 hours at 8.5 °C. The subsequent pressure drop (2) was more pronounced compared to that of the H2O experiment. The lattice constant for deuterated methane hydrate is up to 0.15% larger than that for the hydrogenated species. This increase may lead to higher cage occupancy in the case of deuterated CO2 hydrates.32 This would give rise to an increased uptake of gas, and a lower resulting pressure during the formation (Fig. 1b). Subsequently, both experiments show additional temperature peaks, which indicate further nucleation (not shown in Fig. 1). Gas consumption during the first nucleation period ceased after approx. 15 and approx. 10 hours for H2O and D2O, respectively (Fig. 1a and b). During the present experiments, first nucleation occurred nearly after the same time even though in general gas hydrate nucleation is supposed to be a stochastic event. Using in situ injection of THF, Ricaurte et al. could successfully eliminate the stochasticity of hydrate crystallisation enforcing nucleation on the system.33 In order to facilitate hypothesizing on the hydrate species forming during the first nucleation phase, pressure vs. temperature data of both experiments (Fig. 1a and b) are presented in comparison with equilibrium curves for pure CO2 hydrate derived from the literature (Adisasmito et al. 199134), and CO2 hydrate in the presence of 3.8 wt% THF (Delahaye et al. 200613) in Fig. 2.
 | ||
| Fig. 2 Pressure vs. temperature data obtained during the present study in comparison with equilibrium data reported in the literature (pressure and temperature data have been converted to bar and °C, respectively).13,34 | ||
For clarity, Fig. 2 only shows the pressure vs. temperature traces for part of the cooling phase and the first nucleation phase observed during the experiments depicted in Fig. 1. It is evident that the finally achieved pressure value is lower in the case of D2O, as previously stated. In a system with THF and CO2, the initially formed hydrate is apparently a mixed species comprising THF and CO2.17 The fact that for both experiments the initial nucleation occurred at the boundary of the equilibrium curve for pure CO2 hydrate (Fig. 2; Adisasmito et al. 199134), and that the subsequently observed gas consumption occurred deep within the mixed hydrate regime (Fig. 2; Delahaye et al. 200613) leads to the well-founded assumption that indeed primarily mixed species are formed. This hypothesis is further substantiated in relation to the obtained IR-spectroscopic data in the presence of THF (see Section 2.2.2). To which grade pure CO2 hydrate forms during the remaining time of the experiment remains an open question. However, based on the continuous gas consumption after replenishing gaseous CO2 (i.e., an increase in pressure to approx. 27 bar; Fig. 1a and b: 3) it is assumed that also pure species are formed, as the system is clearly driven into the pure CO2 hydrate regime (Fig. 2).
2.2 In situ MIR-FEFS monitoring of CO2 gas hydrates
Next to recording conventional pressure/temperature traces, the main focus of this study was to correlate for the first time such physical observations with molecularly discriminative data on CO2 gas hydrate formation via mid-infrared fiber optic evanescent field spectroscopy (MIR-FEFS). During gas hydrate formation, the present molecules are responsible for specific spectroscopic changes.30,31Fig. 3a and b exemplarily display such obtained IR-spectra with and without gas hydrate being present in H2O (a) and in D2O (b). | ||
| Fig. 3 IR-spectra recorded in situ via MIR-FEFS on sample solutions enriched with CO2 (i.e., the liquid phase with the dissolved gas) in comparison to solid gas hydrate spectra in (a) H2O and (b) D2O; the insets magnify the spectral region where SDS provides characteristic IR absorption bands within the hydrate spectra. | ||
In the MIR spectral regime water (i.e., aqueous species including H2O and D2O) is characterized by several distinctive absorption features. The broad absorption band ranging from 3750–2750 cm−1 represents the OH stretching vibration (νOH). Between 2250 and 1950 cm−1, the rather weak 3rd overtone of the libration (3νL) mode is evident. The HOH bending vibration (ν2(HOH)) absorbs in the range of 1850–1520 cm−1. Limited by the cut-off frequency of the applied mercury–cadmium–telluride (MCT) detector, the fundamental libration band is just evident <1000 cm−1. Due to the isotopic effect, these absorption features are accordingly shifted to lower wavenumbers for D2O. The broad νOD absorption band is evident at 2800–2000 cm−1, and the DOD bending vibration (ν2(DOD)) at 1350–1145 cm−1. The weak absorption feature between 1680 and 1535 cm−1 may be assigned to the 3rd overtone of the libration (3νL) mode. The libration mode (νL) of D2O is not detectable given the frequency cut-off of the applied detector. Due to the impurities by water/vapour, the mixed species HDO is ubiquitously present during D2O experiments, which is evident due to the associated absorption features between 3620 and 3200 cm−1 (νOH), and between 1520 and 1385 cm−1 (ν2(HOD)).35,36 Next to H2O and D2O, the used additives THF and SDS also provide specific vibrational absorption features in the MIR. The magnifications of the obtained MIR-FEFS spectra represent the region of interest for the νasCHSDS (centered at 2920 cm−1 in H2O and 2917 cm−1 in D2O), and the νsCHSDS (centered at 2851 cm−1 in H2O and 2849 cm−1 in D2O) CH2 bands37 in the hydrate spectra. For THF, the pronounced asymmetric C–O–C vibration νasC–O–CTHF is evident (centered at 1041 cm−1 in H2O and 1039 cm−1 in D2O) in the sample spectra.38 In the region between 1270 and 1100 cm−1 absorption features of the used poly(tetrafluoroethylene) (PTFE) ferrules for sealing the IR fiber into the feed-through of the pressure vessel are evident. In the D2O experiment, these are being convoluted by the DOD absorption feature (ν2(DOD)). CO2 is characterized by the strong asymmetric stretching vibration centered at 2342 cm−1 in H2O and at 2341 cm−1 in D2O (for sample solution spectra), which appears convoluted by the νOD feature. The νasCO2 appears substantially increased in the hydrate spectra, which is attributed to the formation of gas hydrate and/or to the increase in dissolved CO2 in the remaining intermediate water phase. A more detailed description of this band is given in Section 2.2.3. During hydrate formation, H2O/D2O associated features are subject to specific changes that are qualitatively similar to the changes occurring during cooling and crystallization of pure water.39,40 While in the present study these changes are qualitatively similar for both H2O and D2O, they appear more clearly evident during H2O experiments, due to the absence of HOD and other interferences (CO2 and PTFE). While the νOH (νOD) features are subject to a red-shift, the 3νL and νL features shift to the blue. For the bending absorption (i.e., ν2(HOH) (ν2(DOD))), the intensity decreases with increased hydrate formation. In general, the evaluation of such features was achieved via peak area integration or peak fitting methods for the different absorption features, respectively. A more detailed description on the evaluation of CO2-containing gas hydrate IR-spectra has been published elsewhere.41
The main focus of the present study was to provide further data on spectroscopic changes associated with additives present during the gas hydrate formation. Due to the interference of the IR-signature associated with the water background matrix, D2O was considered as a viable alternative providing a similar chemical environment, yet it facilitates monitoring of these species by the shifted background absorptions of the bulk matrix. As evident in the magnified spectral regime in Fig. 3b, the region of interest for SDS is not convoluted by any water absorbances, which particularly facilitates spectral studies of this system.
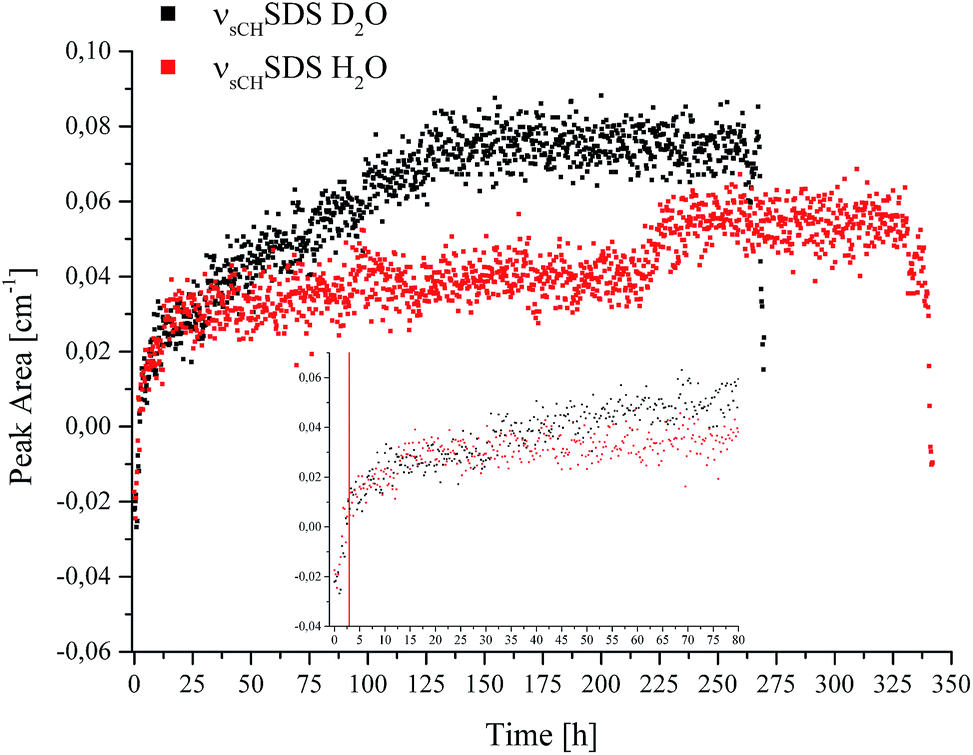 | ||
| Fig. 4 Peak area of the νsCHSDS feature vs. time recorded during CO2 hydrate formation in D2O (black) and H2O (red). | ||
The borders of the applied peak area integration method were selected to cover the range of the evolving peak for all spectra in both experiments; the first spectra do not show any signal characteristic for SDS. Gas hydrate nucleation occurred after approx. 2.8 hours in D2O, and after approx. 3 hours in H2O, as evident in Fig. 1. For clarity, this is illustrated by a red line at 2.9 hours in the inset of Fig. 4. The inset in Fig. 4 shows the development of νsCHSDS during the first 80 hours of the experiments. The peak area for both measurements tends to increase over several hours following nucleation, until reaching equilibrium. For D2O, after approx. 25–30 hours the peak area gradually increases again, which can be correlated to a pressure drop (see Fig. 1). This increase continues until approx. 130 hours, where again a pressure drop occurred. Thereafter, no further increase in the peak area was observed. Thereafter, the pressure was increased two more times, however, with rather slow observable decreases as compared to the previous pressure drops. Hence, the formation process was considered complete or to continue only very slowly. The observed increases and decreases in pressure did not lead to any further detectable changes regarding SDS. In the case of H2O, the gradual increase in peak area was not observed. However, such behaviour is evident in the data between approx. 218 and 230 hours. A decrease in pressure occurred in the corresponding pT trace during that time, after which the pressure was increased once more. The peak area only decreased again upon inducing decomposition of the gas hydrate at the end of the data recording for both experiments. For interpreting the involvement of SDS during gas hydrate nucleation/formation, the attraction of surfactant molecules to the surfaces at the solid/liquid interface is of importance. Since the peak area rises gradually over time during the experiment (in the case of D2O), and since a distinguishable influence via a decrease or increase of pressure appears absent except for the events discussed above, it is hypothesized that the increase in peak area is likely due to an accumulation of SDS at the waveguide surface. Furthermore, it is known that dodecyl sulphate anions adsorb at the hydrate/liquid interface of THF hydrate particles,42 which may in turn affect the detection of SDS. An inclusion of SDS molecules in the hydrate cages is unlikely due to the size limitation of the cages.2 Luzinova et al. were able to correlate a sharp rise in the peak area of SDS, and changes of IR water features with the nucleation of gas hydrate at the surface of the fiber.31 Even though the nucleation of the present experiments started after approx. 2.8/3 hours and a rise in the peak area during the following period was observed, it was not possible attributing this behaviour unanimously to gas hydrate formation at the surface of the fiber, as previous experiments did not reveal sharp instantaneous changes for the spectral features of water simultaneously with the hydrate nucleation,41 as shown by Luzinova et al. Additionally, the concentration of SDS in the present work was considerably higher than in the experiments by Luzinova et al.31 The comparison of the two systems studied so far (i.e., propane gas hydrate and CO2 gas hydrate) is therefore challenging, as the solubility of CO2 in water is significantly higher compared to that of short-chained hydrocarbons. Hence, the initially proposed mechanism31 that SDS increases the transport of gas within the liquid phase is not likely to be applicable for CO2 gas hydrate formation. Therefore, based on the data obtained herein the question whether SDS leads to the nucleation/formation of gas hydrate in the vicinity of the waveguide surface for CO2/THF hydrate remains debatable. While the measurement technique used herein is in general highly reproducible, in the specific case of SDS the shape of the molecule-specific absorption features revealed some variance apparently resulting from parameters associated with the hydrate growth.
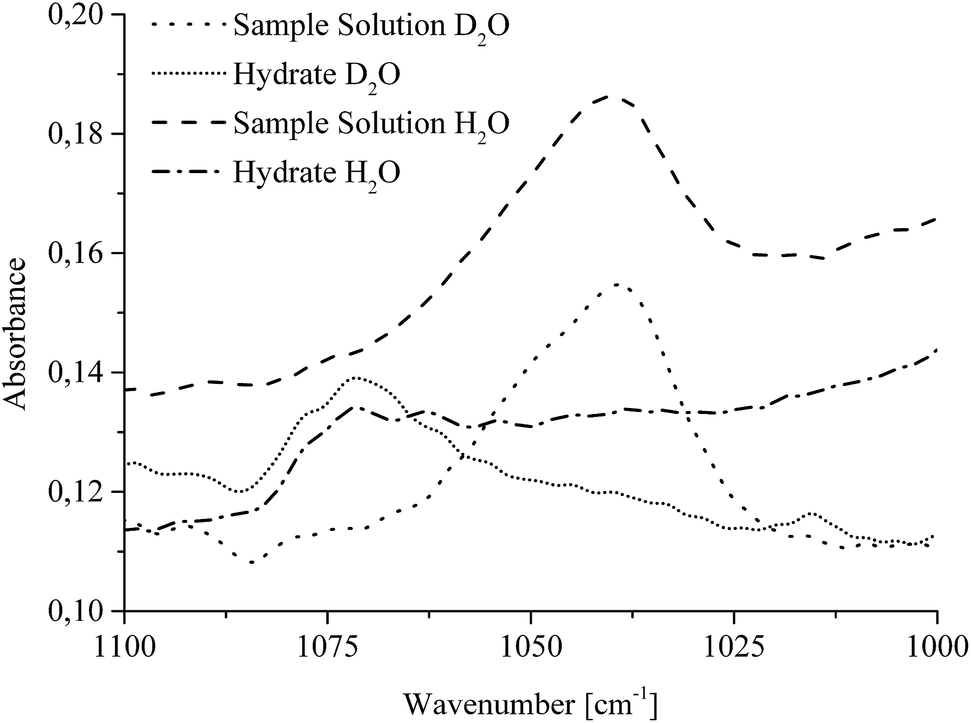 | ||
| Fig. 5 MIR-FEFS spectra illustrating the spectral shift of the νasCOCTHF absorption band during CO2 hydrate formation in H2O and D2O, respectively. | ||
IR spectroscopic studies on thin films of THF hydrate by Fleyfel and Devlin assigned an absorption feature at 1074 cm−1 at 90 K to the asymmetric C–O–C stretch of THF in the large cages of the sII hydrate structure. Furthermore, they observed a doublet of the THF absorption feature at very low temperatures (13 K), which was attributed to THF in two unequal positions within the hexakaidecahedral cages of the hydrate structure.25 Recent publications observed the feature at 1073 cm−1 for a double clathrate hydrate consisting of THF and HCN at 130 K,43 and at 1073 cm−1 for a 90% deuterated THF and HCN binary clathrate hydrate at 170 K.44 The weak absorption feature centered at 1072 cm−1, which is present in the H2O hydrate spectrum in Fig. 4 and evident in more detail in Fig. 5, may therefore be attributed to THF in the large cages of the hydrate structure. The minor deviation of the frequency position compared to the literature values is within the spectral resolution (i.e., 2 cm−1) selected for the present study. For the D2O experiment, the absorption feature of hydrated THF is much more clearly evident (Fig. 3 and 5), which is attributed to the absence of interferences by the νL absorption of H2O in this spectral region (Fig. 5). This observation further consolidates the utility of D2O facilitating IR spectroscopic investigations on the effects of additives. However, the assignment of this absorption feature to THF inside the clathrate cages remains ambiguous, as the presented data were collected at considerably higher temperatures. A more detailed evaluation of this feature remains limited due to possible influences by spectral features from SDS, which is also present in the solution. The observed species at 1041/1039 cm−1 present in the sample spectra of H2O/D2O (Fig. 3 and 5) may be assigned to THF connected with more than three water molecules.45 To obtain further evidence on the assignment of an enclathrated species, a control experiment with D2O and only THF as the additive was performed. In the absence of SDS, hydrate growth commences much more slowly17 without filling the gas hydrate cell even within weeks, in contrast to the presented experiments. In order to provide a more distinct assignment, a pressure cell with a significantly reduced volume is currently developed. Hence, for the purpose of the present study only the THF absorption feature in solution was evaluated. Similar to SDS, the evaluation of the νasCOCTHF-related feature was executed via peak area integration between 1058 and 1020 cm−1 (Fig. 6).
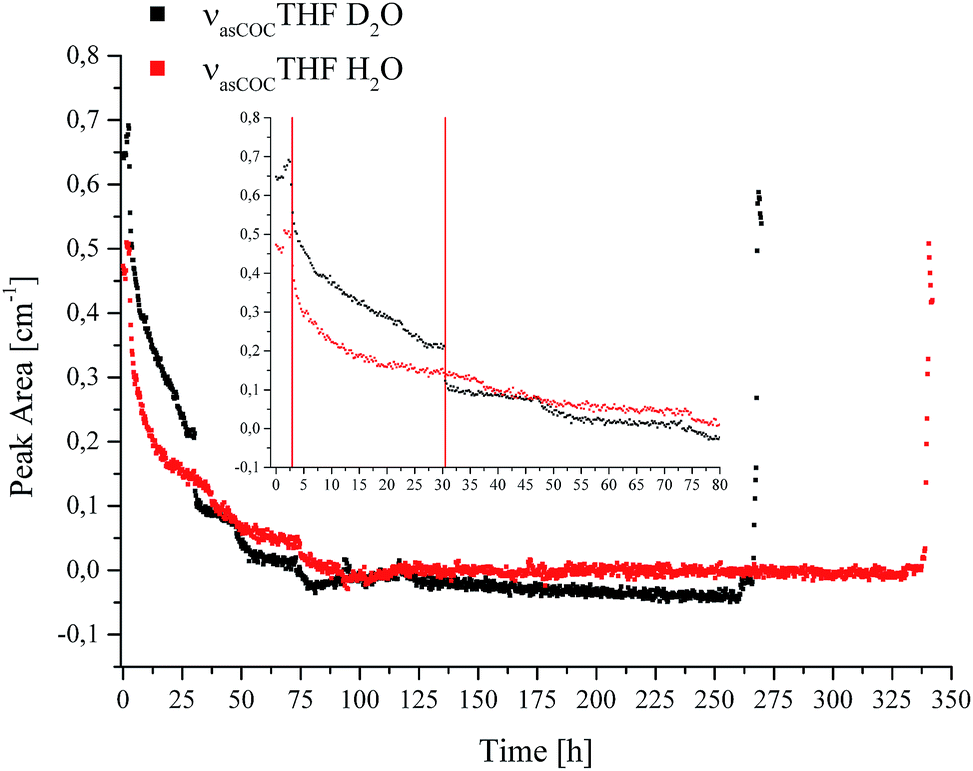 | ||
| Fig. 6 Peak area for the νasCOCTHF absorption in solution vs. time during CO2 hydrate formation in D2O (black) and H2O (red). | ||
The inset shows the first 80 hours of the experiments in detail. For comparability, the data was evaluated using the same method for all spectra. The first red line in the inset of Fig. 6 indicates the initiation of nucleation (Fig. 1a and b, in Fig. 6 represented by a single line at 2.9 hours). It is evident that the absorption feature, i.e., the peak area of the non-hydrated THF signal dropped sharply after the first nucleation occurred and continued to decrease with time due to the advancing formation of the hydrate accompanied by the enclathration of THF. The resulting structure of the THF/CO2 hydrate is known as sII with the large cavities occupied by THF, and the small cavities by CO2.46 In the case of D2O another drop is evident at 30.5 hours, indicated by the second red line in the inset of Fig. 6, which can be correlated with a pressure drop (Fig. 1b). The signals of both measurements decrease further until detection is not possible anymore. This implies that there is none or only very little residual THF present in solution in the evanescently probed sampling volume around the fiber after this time. It is assumed that the majority of THF is enclathrated in the hydrate structure at this point. After this point there are still pressure drops evident in the data, which indicate that hydrate formation is not yet completed. Subsequent dissociation of gas hydrate reversed these changes, as anticipated. Torré et al. performed CO2 hydrate experiments under quiescent conditions at various concentrations of THF and SDS, and hypothesized that next to pure CO2 hydrate a mixed hydrate comprising THF and CO2 is also formed.17,18 It should be noted that during the present studies the incorporated gas was discontinuously replenished via gas supply several times after the first nucleation occurred ensuring complete formation of gas hydrate within the pressure cell, and especially within the evanescent field surrounding the fiber acting as the ATR-waveguide. The results obtained for THF indicate that the formation of the mixed hydrate in fact commences during the initial phase of the experiment indicated via the steadily decreasing peak area for THF in solution. Based on the observation of the absorbance feature of the hydrated THF in Fig. 5, it is assumed that the solid hydrate formed around the fiber consisted of THF and CO2 with THF being present within the large, and CO2 within the small cavities of the resulting structure sII. While an undisputable statement on the mechanism of hydrate nucleation cannot be made yet, the applied MIR-FEFS methodology provides a more pronounced sensitivity towards the present additives (i.e., SDS and THF) than any other method reported to date. In the case of water, no distinct changes in the peak area or position could be observed during the initial hydrate formation.41 An explanation of this behaviour may be attributed (i) to the presence of considerable amounts of non-hydrated water during the initial formation in the vicinity of the fiber, which renders the detection of structured water species difficult or (ii) based on the interpretation of the obtained results for SDS, that the nucleation is not primarily occurring at the waveguide surface for this system. However, with advancing hydrate formation, spectral changes of water were clearly observed until an equilibrated state was reached (i.e., no further changes were evident in the IR spectra).41 An explanation for the high efficiency of the combined application of both additives (i.e., THF and SDS) during CO2 hydrate formation may be the creation of a porous hydrate structure, which in turn leads to a high permeability for CO2 during the formation.17 This capillary/pore-based growth mechanism was also proposed for another hydrate system, i.e., gas hydrates of CH2F2 with SDS. The authors confirmed that the reaction solution is continuously sucked into the porous hydrate layer at a partially submerged solid interface, which they attributed to the dominating mechanism behind the promoting effect of SDS during hydrate formation.20 Furthermore, it should be noted that during the present study the existence of micelles is excluded, since the applied concentration of SDS is below the critical micelle concentration (8 mmol L−1).47 Additionally, the existence of micelles at temperatures below the Krafft point (TK) of SDS is debatable under hydrate forming conditions.20,48 In contrast to the studies on SDS, the obtained results for THF appear less affected by the occurring processes and more reproducible from the point of view of molecule-specific spectral characteristics. Yet, common to all executed experiments is a sharp decrease in the peak area after nucleation, evident in the pT – trace along with highly reproducible spectral characteristics for THF.
3 Experimental
3.1 MIR-FEFS gas hydrate setup
The MIR-FEFS gas hydrate setup comprises four main components: a pressure cell, a FTIR spectrometer, optical coupling components for guiding the IR radiation, and temperature/pressure sensors along with switchable gas/liquid supply connections. Detailed schemes and features of the pressure cell are described elsewhere.50,51 In brief, the pressure cell provides a fiber feed-through specifically adapted for IR-transparent silver halide fibers enabling in situ spectroscopic access to monitoring hydrate formation/dissociation processes. To prevent the silver halide fiber from degradation by contact with base metals the tube fittings of the feed-through are equipped with custom-made PTFE ferrules.51Fig. 7 schematically shows the MIR-FEFS gas hydrate setup along with a view into the pressure vessel and the mounted silver halide fiber. | ||
| Fig. 7 Schematic of the MIR-FEFS setup and view into the pressure vessel with the mounted silver halide sensing fiber. | ||
A Bruker IFS/66s model FTIR spectrometer (Bruker Corporation, Billerica, MA, USA) was used during the present studies. The parallel MIR beam was coupled out of the spectrometer and directed via a planar 2′′ gold coated mirror (Fig. 7 #1) onto a first 90° (2′′ focal length) gold-coated off-axis parabolic mirror (OAPM) (Fig. 7 #2), which focuses the radiation onto the facet of the 900 μm diameter silver halide fiber (composition: AgCl0.3Br0.7). Inside the fiber, radiation is propagated via total internal reflection. After passing the pressure cell (Fig. 7 #3), the emanating light at the distal end of the fiber is collected by a second 2′′ 90° OAPM (Fig. 7 #2), and the provided parallel beam is directed onto a third OAPM (Fig. 7 #2) (Edmund Optics Inc., Barrington, NJ, USA) focusing the radiation onto the detector element of a Stirling-cooled mercury–cadmium–telluride (MCT) detector (Model K508, Infrared Associates, Stuart, FL, USA) (Fig. 7 #4). Two optic positioners (Newport Corporation, Irvine, CA, USA) with custom-made PTFE fiber chucks flexibly position the fiber on either side of the pressure cell. Additional parts such as posts and the XYZ translation stage were purchased from Thorlabs Inc (Newton, NJ, USA). The MCT detector was connected to the IFS/66s spectrometer via an impedance-matched MCT-1000 pre-amplifier (Infrared Associates, Stuart, Florida USA) via an external port. In order to record temperature and pressure traces, the cell was equipped with appropriate sensors mounted at the rear of the pressure vessel: (i) a thermocouple (TEM01.08N.6.0020.T.1KA.0, PKP Prozessmesstechnik, Wiesbaden, Germany) serving as a temperature sensor, and (ii) a stainless steel diaphragm pressure transducer (AGS4003-60-G-E082, Althen Mess-und Sensortechnik GmbH, Kelkheim, Germany) for pressure tracing. Both were connected to a data logging system (midi LOGGER GL200A, Graphtec, Yokohama, Japan) for directly correlating physical and chemical parameters. Several connectors and drains for liquid and gas supply are implemented on the top and on the rear side of the pressure cell. Gas and liquid connectors are equipped with Swagelok back-pressure valves to avoid leakage (Swagelok, Solon, OH, USA). Cooling of the cell is realized via two cooling coils, an internal stainless steel coil (see Fig. 7 view into the pressure vessel), as well as an external copper coil wrapped around the cell. Both coils are connected to a cooling-bath (Lauda RE206, Lauda GmbH & Co. KG, Lauda-Königshofen, Germany) equipped with a thermostat (Lauda E200, Lauda GmbH & Co. KG, Lauda-Königshofen, Germany). To fill the cell with the sample solution, a high performance liquid chromatography pump (HPLC Pump 64, Knauer GmbH, Berlin, Germany) was used. In order to protect the silver halide fiber from degradation,52 the entire measurement system is surrounded by an acrylic glass box covered with black anodized aluminium foil. To minimize the background fluctuation during the long-term studies, this enclosure is flushed with compressed dry air. IR spectra have been collected every 15 min at a spectral resolution of 2 cm−1 between 4000 and 600 cm−1 with 500 scans averaged per spectrum. The pT traces have been recorded at intervals of 2 s. For recording and evaluation of the IR spectra, the software packages OPUS 6.5 (Bruker Corporation, Billerica, MA, USA) and Essential FTIR 3.50.047 (Operant LLC, Madison, WI, USA) were used.
3.2 Measurement procedure
The analyzed solution contained 1.5 g L−1 SDS and 40 g L−1 THF in demineralized water, and was mixed and stirred for approx. 1 hour before use. Demineralized water was replaced by D2O for the corresponding alternative experiment. Prior to collecting a background spectrum, the box was flushed with dry air for 1.5 hours. Subsequently, the pressure cell was filled with approx. 300 mL of sample solution via the HPLC pump system. To remove supernatant air, the cell was purged three times with approx. 10 bar CO2 and vented down to approx. 3 bar. To obtain a CO2-enriched solution, the system was then pressurized for 3 hours at approx. 27 bar CO2 at 20 °C. Pressure, temperature, and IR-spectroscopic data recording were initiated after 2 hours into this initial step. The gas supply was closed after completion of the enrichment step (i.e., 3 hours), and the cooling system was initiated to obtain a temperature of approx. 5 °C. To replenish enclathrated gas in order to entirely fill the cell with hydrate, the gas supply was reopened under pressure several times after the initial nucleation throughout the experiment. The procedure and solution composition have been selected as derived from the literature.18 Once the absence of a characteristic pressure decrease was determined, the cell was considered filled with hydrates. Dissociation of the hydrate was initiated via slow depressurization of the cell by the opening of a bleeding valve. This protocol was applied for all experiments reported herein using H2O or D2O as the hydrate matrix. The experiment using H2O as the matrix was independently repeated three times. Using D2O as the matrix, only one experiment was performed, as the only purpose was enabling qualitative insight into the spectral window usually obstructed by water absorption by taking advantage of the isotopic shift within the H2O vs. the D2O spectrum.3.3 Principles of MIR-FEFS
MIR-FEFS is based on the commonly applied principles of infrared attenuated total reflection (ATR) spectroscopy. In conventional IR-ATR spectroscopy a crystal is used as a waveguide (e.g., ZnSe, Ge, Si, etc.), whereas for FEFS an IR-transparent fiber is applied, here made from polycrystalline silver halides. Propagation of the IR radiation is achieved via total internal reflection in the fiber, which occurs at an interface of an optically denser (n1) to an optically rarer (n2) medium at angles exceeding the critical angle Θc. At each reflection an evanescent field is induced emanating from the waveguide surface. The penetration depth (dp) of this field into the surrounding medium is defined as the distance at which the field has decreased to 1/e of its initial intensity, and depends on the wavelength (λ), the refractive indices (n1 and n2 with n2 < n1), and the angle of incidence (Θ).53

The underlying principles of both techniques (ATR and FEFS) were first introduced by Harrick as internal reflection spectroscopy.54 Attenuation of the evanescent field by interaction with IR active molecules present within the penetration depth gives rise to the analytical signal. Fig. 8 schematically visualizes a cross-section of the fiber inside the pressure cell surrounded by the measurement solution containing THF, SDS, and CO2.
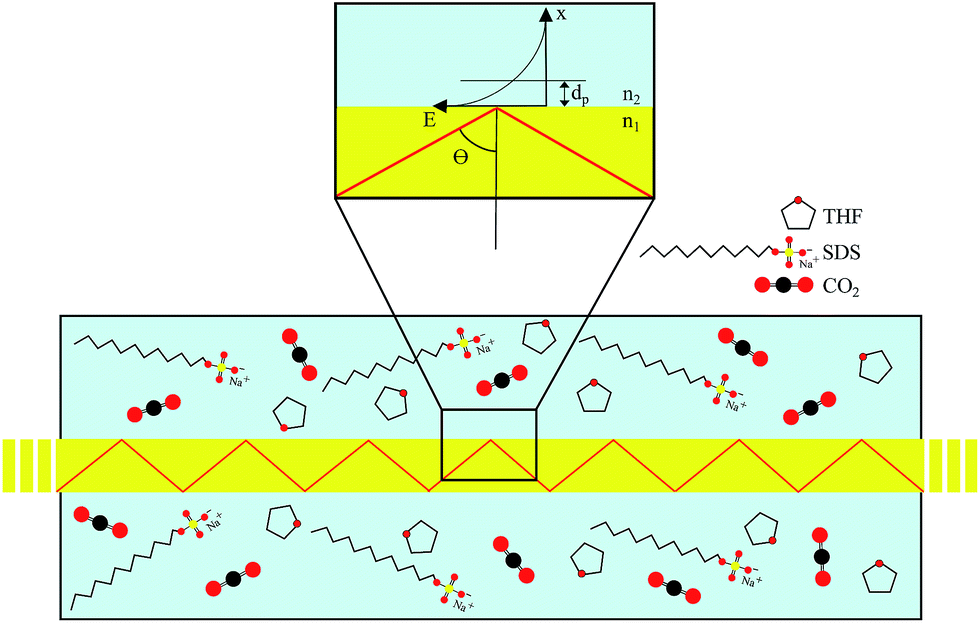 | ||
| Fig. 8 Principles of MIR-FEFS conceptualized at a cross-section of the scenario within the pressure cell; zoom-in visualizes the exponentially decaying evanescent field emanating from the fiber surface. | ||
3.4 Materials
SDS was purchased from Sigma Aldrich, St. Louis, MO, USA with ≥98.5% purity. THF was purchased from Merck KGaA, Darmstadt, Germany with a purity of ≥99.9%. D2O was purchased from Deutero GmbH, Kastellaun, Germany with 99.9% purity. CO2 gas was purchased from MTI IndustrieGase AG, Neu-Ulm, Germany with 99.8% purity. All chemicals were used as provided and not further purified. The silver halide fiber was fabricated by the team of Abraham Katzir at Tel-Aviv University, Israel.4 Conclusions
The present study applies for the first time mid-infrared fiber optic evanescent field spectroscopy for in situ studying of CO2 hydrate formation in the presence of various additives. Evidently, insight into complex hydrate formation processes is provided at the molecular-level detail. The spectroscopic evaluation of additives (i.e., THF and SDS) provides additional information for elucidating the promoting effects of these additives during hydrate formation. Finally, studies in D2O as the hydrate-forming matrix assisted in detailing the molecular signatures with yet unprecedented clarity by avoiding the convolution with vibrational absorbances of water in the spectral regions of interest.The spectroscopic method developed herein enables monitoring gas hydrate formation and dissociation processes associated with additives in situ rendering MIR-FEFS a promising technique for the fundamental investigation of bulk gas hydrates. In the next step, in situ monitoring of the exchange process from CH4 hydrate to CO2 hydrate is anticipated, which is of substantial significance in alternative energy retrieval, energy storage, and carbon capture/sequestration processes. Finally, in-field studies may be envisaged given the current efforts in miniaturizing and harnessing IR-spectroscopic equipment for deep-sea deployment.53,55
Acknowledgements
The authors greatly acknowledge the collaboration with A. Katzir and his research team at Tel-Aviv University (Israel) for providing customized silver halide fibers. Furthermore, the machine shop at Ulm University is acknowledged for assistance in establishing the MIR-FEFS gas hydrate setup. Partial financial support of this work by the European Union FP7 Project SCHeMA – Integrated in situ Chemical Mapping Probes (Grant Agreement Number 614002) is gratefully acknowledged.Notes and references
- E. D. Sloan and C. A. Koh, in Clathrate Hydrates of Natural Gases, CRC Press, Boca Raton, 3rd edn, 2008, ch. 2, p. 45 Search PubMed.
- E. D. Sloan and C. A. Koh, in Clathrate Hydrates of Natural Gases, CRC Press, Boca Raton, 3rd edn, 2008, ch. 2, p. 55 Search PubMed.
- K. A. Udachin, C. I. Ratcliffe and J. A. Ripmeester, J. Phys. Chem. B, 2001, 105, 4200–4204 CrossRef CAS.
- J. A. Ripmeester and C. I. Ratcliffe, Energy Fuels, 1998, 12, 197–200 CrossRef CAS.
- F. Qanbari, M. Pooladi-Darvish, S. Hamed Tabatabaie and S. Gerami, Energy Procedia, 2011, 4, 3997–4004 CrossRef CAS.
- J. Zhao, K. Xu, Y. Song, W. Liu, W. Lam, Y. Liu, K. Xue, Y. Zhu, X. Yu and Q. Li, Energies, 2012, 5, 399–419 CrossRef CAS.
- H. Komatsu, M. Ota, R. L. Smith and H. Inomata, J. Taiwan Inst. Chem. Eng., 2013, 44, 517–537 CrossRef CAS.
- D. Bai, X. Zhang, G. Chen and W. Wang, Energy Environ. Sci., 2012, 5, 7033–7041 CAS.
- L. C. Elwell and W. S. Grant, Power, 2006, 150, 60–65 CAS.
- P. Linga, R. Kumar and P. Englezos, J. Hazard. Mater., 2007, 149, 625–629 CrossRef CAS PubMed.
- P. Linga, R. Kumar, J. D. Lee, J. Ripmeester and P. Englezos, Int. J. Greenhouse Gas Control, 2010, 4, 630–637 CrossRef CAS.
- H. J. Lee, J. D. Lee, P. Linga, P. Englezos, Y. S. Kim, M. S. Lee and Y. D. Kim, Energy, 2010, 35, 2729–2733 CrossRef CAS.
- A. Delahaye, L. Fournaison, S. Marinhas, I. Chatti, J.-P. Petitet, D. Dalmazzone and W. Fürst, Ind. Eng. Chem. Res., 2006, 45, 391–397 CrossRef CAS.
- J.-P. Torré, D. Haillot, S. Rigal, R. de Souza Lima, C. Dicharry and J.-P. Bedecarrats, Chem. Eng. Sci., 2015, 126, 688–697 CrossRef.
- K. Okutani, Y. Kuwabara and Y. H. Mori, Chem. Eng. Sci., 2008, 63, 183–194 CrossRef CAS.
- Y. Zhong and R. E. Rogers, Chem. Eng. Sci., 2000, 55, 4175–4187 CrossRef CAS.
- J.-P. Torré, M. Ricaurte, C. Dicharry and D. Broseta, Chem. Eng. Sci., 2012, 82, 1–13 CrossRef.
- J.-P. Torré, C. Dicharry, M. Ricaurte, D. Daniel-David and D. Broseta, Energy Procedia, 2011, 4, 621–628 CrossRef.
- N. Liu, G. Gong, D. Liu and Y. Xie, Proceedings of the 6th International Conference on Gas Hydrates (ICGH 2008), Vancouver, 2008 Search PubMed.
- K. Watanabe, S. Imai and Y. H. Mori, Chem. Eng. Sci., 2005, 60, 4846–4857 CrossRef CAS.
- J. E. Bertie and D. A. Othen, Can. J. Chem., 1972, 50, 3443–3449 CrossRef CAS.
- J. E. Bertie and D. A. Othen, Can. J. Chem., 1973, 51, 1159–1168 CrossRef CAS.
- J. E. Bertie and J. P. Devlin, J. Chem. Phys., 1983, 78, 6340–6341 CrossRef CAS.
- F. Fleyfel and J. P. Devlin, J. Phys. Chem., 1988, 92, 631–635 CrossRef CAS.
- F. Fleyfel and J. P. Devlin, J. Phys. Chem., 1991, 95, 3811–3815 CrossRef CAS.
- R. Kumar, S. Lang, P. Englezos and J. Ripmeester, J. Phys. Chem. A, 2009, 113, 6308–6313 CrossRef CAS PubMed.
- Y. Jin, H. Oyama and J. Nagao, Jpn. J. Appl. Phys., 2009, 48, 108001 CrossRef.
- C. Lo, J. Zhang, P. Somasundaran and J. W. Lee, J. Colloid Interface Sci., 2012, 376, 173–176 CrossRef CAS PubMed.
- R. Kumar, P. Englezos, I. Moudrakovski and J. A. Ripmeester, AIChE J., 2009, 55, 1584–1594 CrossRef CAS.
- G. T. Dobbs, Y. Luzinova, B. Mizaikoff, Y. Raichlin and A. Katzir, Proceedings of the 6th International Conference on Gas Hydrates (ICGH 2008), Vancouver, 2008 Search PubMed.
- Y. Luzinova, G. T. Dobbs, Y. Raichlin, A. Katzir and B. Mizaikoff, Chem. Eng. Sci., 2011, 66, 5497–5503 CrossRef CAS.
- A. Klapproth, E. Goreshnik, D. Staykova, H. Klein and W. F. Kuhs, Can. J. Phys., 2003, 81, 503–518 CrossRef CAS.
- M. Ricaurte, J.-P. Torré, J. Diaz and C. Dicharry, Chem. Eng. Res. Des., 2014, 92, 1674–1680 CrossRef CAS.
- S. Adisasmito, R. J. Frank III and E. D. Sloan Jr, J. Chem. Eng. Data, 1991, 36, 68–71 CrossRef CAS.
- D. Neubauer, J. Korbmacher, M. Frick, J. Kiss, M. Timmler, P. Dietl, O. H. Wittekindt and B. Mizaikoff, Anal. Chem., 2013, 85, 4247–4250 CrossRef CAS PubMed.
- J.-J. Max and C. Chapados, J. Chem. Phys., 2002, 116, 4626–4642 CrossRef CAS.
- K. V. Padalkar, V. G. Gaikar and V. K. Aswal, J. Mol. Liq., 2009, 144, 40–49 CrossRef CAS.
- H. Deng, Z. Shen, L. Li, H. Yin and J. Chen, J. Appl. Polym. Sci., 2014, 40503 Search PubMed.
- J.-B. Brubach, A. Mermet, A. Filabozzi, A. Gerschel and P. Roy, J. Chem. Phys., 2005, 122, 184509 CrossRef PubMed.
- A. Millo, Y. Raichlin and A. Katzir, Appl. Spectrosc., 2005, 59, 460–466 CrossRef CAS PubMed.
- M. Schwenk, Y. Raichlin, A. Katzir and B. Mizaikoff, Proceedings of the 8th International Conference on Gas Hydrates (ICGH8-2014), Beijing, 2014 Search PubMed.
- J. S. Zhang, C. Lo, P. Somasundaran, S. Lu, A. Couzis and J. W. Lee, J. Phys. Chem. C, 2008, 112, 12381–12385 CAS.
- I. A. Monreal, L. Cwiklik, B. Jagoda-Cwiklik and J. P. Devlin, J. Phys. Chem. Lett., 2010, 1, 290–294 CrossRef CAS.
- N. Uras-Aytemiz, F. M. Balcı, Z. Maşlakcı, H. Özsoy and J. P. Devlin, J. Phys. Chem. A, 2015, 119, 9018–9026 CrossRef CAS PubMed.
- K. Mizuno, Y. Masuda, T. Yamamura, J. Kitamura, H. Ogata, I. Bako, Y. Tamai and T. Yagasaki, J. Phys. Chem. B, 2009, 113, 906–915 CrossRef CAS PubMed.
- M. C. Martínez, D. Dalmazzone, W. Fürst, A. Delahaye and L. Fournaison, AIChE J., 2008, 54, 1088–1095 CrossRef.
- D. Rana, G. H. Neale and V. Hornof, Colloid Polym. Sci., 2002, 280, 775–778 CAS.
- P. Di Profio, S. Arca, R. Germani and G. Savelli, Chem. Eng. Sci., 2005, 60, 4141–4145 CrossRef CAS.
- M. Falk and A. G. Miller, Vib. Spectrosc., 1992, 4, 105–108 CrossRef CAS.
- N. A. Pennington, Master's Thesis, Georgia Institute of Technology, 2003.
- G. T. Dobbs, Ph.D. Thesis, Georgia Institute of Technology, 2007.
- R. Chen, A. Katzir, A. Levite, F. Moser and D. Weiss, J. Opt. Soc. Am. B, 1986, 3, 696–700 CrossRef CAS.
- B. Mizaikoff, Meas. Sci. Technol., 1999, 10, 1185–1194 CrossRef CAS.
- N. J. Harrick, Internal Reflection Spectroscopy, Interscience, New York, 1967 Search PubMed.
- M. Kraft, M. Jakusch, M. Karlowatz, A. Katzir and B. Mizaikoff, Appl. Spectrosc., 2003, 57, 591–599 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |