
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrochemical aptamer-based biosensors as potential tools for clinical diagnostics
Marta
Jarczewska
,
Łukasz
Górski
* and
Elżbieta
Malinowska
Institute of Biotechnology, Department of Microbioanalytics, Faculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: lukegor@ch.pw.edu.pl; Fax: +48 226 282741; Tel: +48 222 347573
First published on 8th April 2016
Abstract
Aptamers are single-stranded DNA and RNA sequences that belong to the group of “functional nucleic acids”, which exhibit both catalytic and receptor properties. Aptamers change their spatial conformation upon binding to a target analyte, and so far more than 200 sequences selective for large variety of molecules such as metal ions, organic dyes, proteins and bacteria cells have been identified. Aptamers can be applied in several fields including diagnostics, therapy and as the recognition layers for biosensors, which is one of the most promising areas. The utilization of aptamers as receptor elements in electrochemical assays requires not only the choice of an appropriate immobilization method with respect to the detected target, but also experimental conditions including the manner of analytical signal generation. The latter issue might affect the efficiency of binding between the aptamer-modified surface and analyte, and consequently the sensitivity of target molecule quantification. Herein, a review of electrochemical aptasensors dedicated to the determination of target analytes crucial in clinical diagnostics developed during the past 10 years is presented. It contains a short characterization of aptamers and their application as sensing layers in electrochemical assays, which is followed by a description of the examples of the use of aptasensors.
 Marta Jarczewska | Marta Jarczewska obtained her MSc degree from the Faculty of Chemistry (Warsaw University of Technology, WUT). She is a PhD student at the same University; she works on the development of DNA sensors, including aptasensors. |
 Łukasz Górski | Łukasz Górski received his MSc degree in chemistry from Warsaw University of Technology (WUT, Poland) and PhD from the same University in 2006. His research is focused on ion-selective electrodes, flow-injection analysis and the application of self-assembled monolayers for the preparation of biosensors. |
 Elżbieta Malinowska | Elżbieta Malinowska received her PhD (1984) and DSc (2002) titles in Analytical Chemistry from Warsaw University of Technology (WUT), Poland. She has been in the Faculty of Chemistry, WUT since 1984, and currently she is a professor at the Institute of Biotechnology. She was on leave during the period 1985–1986 to work with Prof. Wilhelm Simon at the Swiss Federal Institute of Technology (ETH) and during 1993–1995 to work with Prof. Mark E. Meyerhoff at the University of Michigan. Her primary research interest is the development of new types of chemical sensors and biosensors. |
1. Introduction
The discovery of the double-stranded DNA structure forming a three-dimensional helix presented by Watson and Crick1 is known as one of the most important findings in the research concerning nucleic acids, their function in living organisms and potential application in areas such as molecular biology, genetic engineering and clinical chemistry. The description of the double-helix structure was followed by the detailed investigation of the role of DNA and RNA with respect to the processes including cell division and apoptosis as well as gene expression.2 Although nucleic acids are acknowledged as genetic information storage units, further research demonstrated other unique features of DNA and RNA molecules. Accordingly, it was shown that some nucleic acids might play catalytic and receptor roles, and therefore form a group of “functional nucleic acids” (FNAs).3,4 One of the most significant types of FNAs are aptamers (the term originates from the Latin words aptus – to fit and meros – part), which were characterized for the first time in 1990 by Ellington and Szostak.5 Aptamers are short, single-stranded DNA and RNA oligonucleotides, which change their conformation upon interaction with a target analyte. Aptamer three-dimensional shapes can be defined as pseudoknots, hairpin-loops, G-quadruplexes etc. (Fig. 1).6 Binding of an aptamer to a particular molecule can result from several effects including spatial adjustment, electrostatic interactions, hydrogen bonds, van der Waals forces and interaction between aromatic groups (π–π stacking).6 The nature of aptamer–analyte binding depends predominantly on the type of target molecule.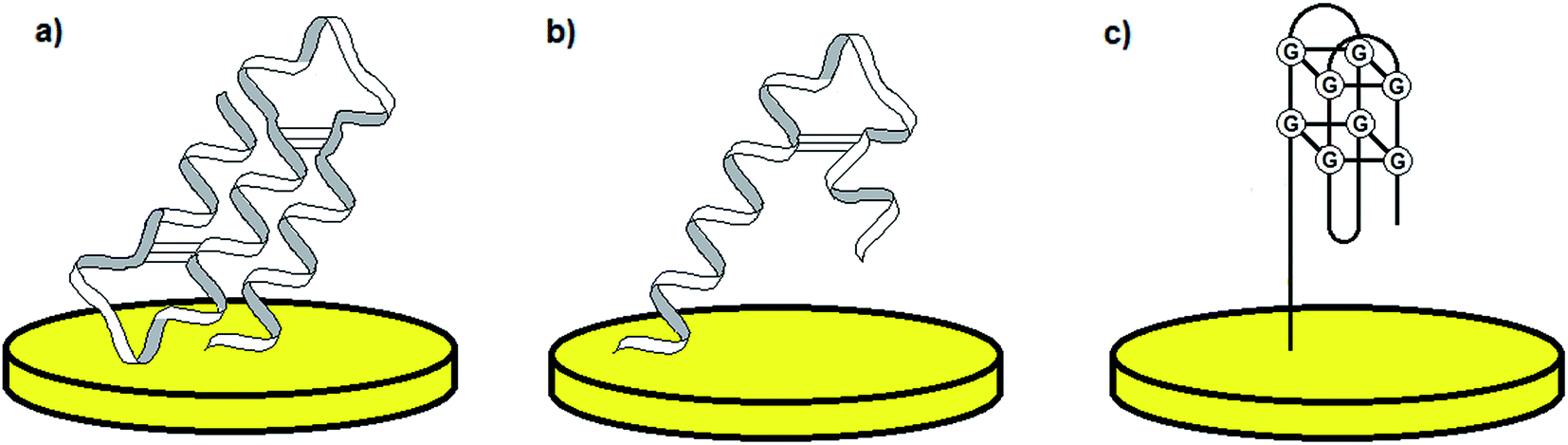 | ||
| Fig. 1 Examples of the most common aptamer conformations: pseudo-knot (a), hairpin (b), and G-quadruplex (c). | ||
Aptamers are artificial molecules, which are isolated in vitro from the pools of random nucleic acid sequences through the SELEX process. The collection of various DNA/RNA molecules is synthesized with the use of combinatorial chemistry and might consist of up to 1013 to 1015 various sequences.6–8 The first step of SELEX involves the incubation of the oligonucleotide library with an analyte, which is followed by the separation of unbound sequences from the strands that interact with a target molecule. Next, the aptamer–analyte complexes are disjoined, and the remaining pool of sequences is amplified via PCR. The subsequent steps of selection are repeated and the total number of cycles performed during the SELEX process varies from 7 to 20 (Fig. 2). The final relatively limited collection of oligonucleotides is cloned and sequenced. Furthermore, the secondary structure of aptamers is determined by the use of computer programs, whereas the three-dimensional structure is defined on the basis of NMR and X-ray measurements.6
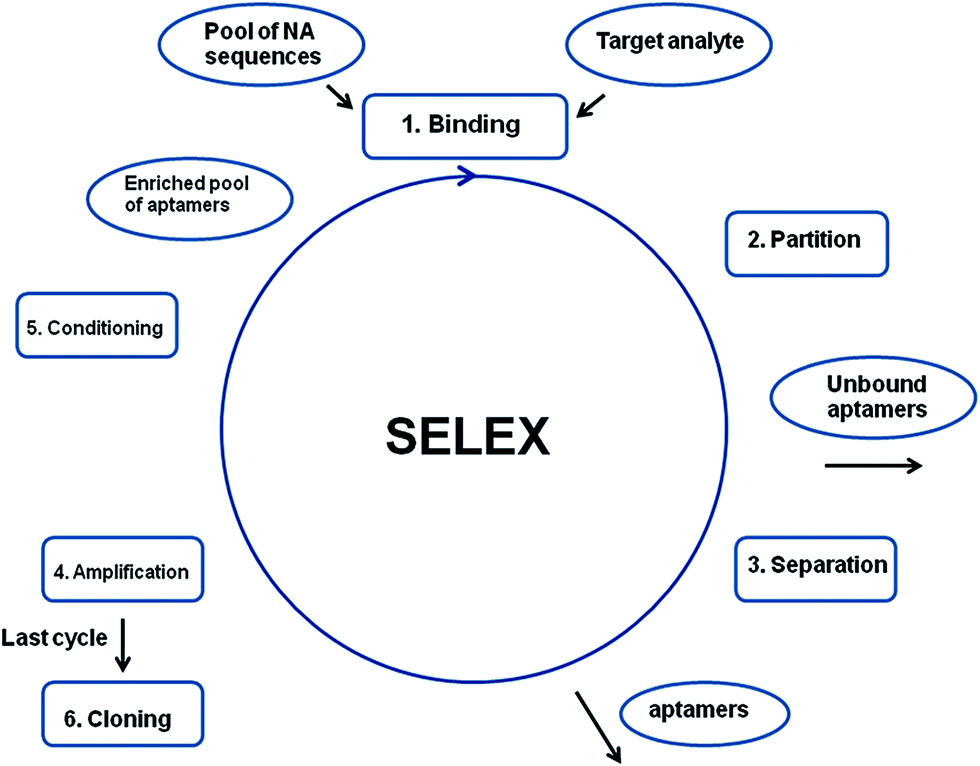 | ||
| Fig. 2 Schematic representation of the SELEX process. | ||
So far, more than 200 aptamer sequences have been identified, which are specific towards various types of targets including metal ions, organic dyes, peptides, proteins and bacteria cells.9 Most of the discovered aptamer probes show high affinity towards proteins in terms of dissociation constants – Kd that are at the micro-, nano- and even picomolar level, and thus are comparable with values exhibited by antibodies.6 In contrast to antibodies, aptamers are chemically synthesized with high efficiency and reproducibility, do not induce immunogenic response, and furthermore, their small dimensions provide the possibility of penetration into cells.10 It should be noted that the SELEX process requires the adjustment of experimental features for an individual target as the obtained strand might be sensitive to biological conditions. To enhance the aptamer resistance to nucleases, several modifications have been introduced, e.g. substitution of an –OH 2′ group in a ribose ring with –NH2 or fluorine. Further modifications refer to aptamer conjugation with fluorescence and electroactive labels or other functional groups enabling surface immobilization.11 As a result, aptamers are frequently applied in various areas including therapeutics, diagnostics, analytical chemistry and biosensors.6
The use of aptamers in therapeutics seems to be one of the most gripping fields and the first drug using PEGylated aptamers – Macugen (applied for the treatment of macular degeneration) was accepted by US FDA in 2004.12 Two more drugs containing aptamers selective towards thrombin and factor IX, respectively, are in the advanced stage of development by Archemix Corporation.13,14 Aptamers could also be utilized in diagnostics, mostly as an alternative to antibodies applied, e.g. in ELISA kits (termed ELONA tests in the case of antibody–aptamer sandwich assays)15 as well as for in vivo imaging due to their small dimensions and fast clearance rate.6 Aptamers might also be employed in analytical chemistry, as they improve the selectivity of detection systems through the molecular recognition of ligands shown by DNA/RNA molecules. Such attempts were already adopted in affinity chromatography, affinity capillary electrophoresis and flow cytometry.16 Finally, aptamers are frequently used for the preparation of recognition layers in biosensors specific for a wide range of analytes.17
Biosensors might serve as an alternative to traditionally applied detection techniques, since their main advantages are the ease of operation, fast response and portability, which is of special importance in the case of e.g. point-of-care systems (POC systems) for patients18,19 and monitoring of environmental samples.20–22 According to IUPAC definition, a biosensor is “a self-contained integrated device that is capable of providing specific quantitative or semi-quantitative analytical information using a biological recognition element (biochemical receptor), which is in direct spatial contact with a transducer element”.23 The recognition element might refer to biomolecules with catalytic activity such as enzymes, organelles and cells, or to the molecules capable of molecular recognition including antibodies, proteins, nucleic acids and biomimetics.24
Since huge interest was given to the application of aptamers as recognition layers, at present the aptamer-based sensors constitute one of the most significant groups of DNA-based sensors, which were traditionally applied as hybridization assays or for the assessment of NA damaging agents.25
Aptamers exhibit high affinity towards target molecules of low molecular weight and attempts of cell detection with the application of aptamer-based platforms were also reported. This refers to the possibility of analysis of pathogens such as Escherichia coli O157:H7,26Listeria monocytogenes,27Streptococcus pyogenes,28,29Mycobacterium tuberculosis,30Salmonella typhimurium,31 cancer cells including human liver hepatocellular carcinoma cells (HepG2)32,33 and SMMC-7721,34 cancer cell lines MCF-7, HL-60, K562,35 gastric cell-line HGC-27 (ref. 36) as well as tissue imaging of e.g. breast cancer cells MDA-MB-231 (ref. 37) and Ramos cells from human Burkitt's lymphoma.38 Nevertheless, the majority of the aptamer-based devices are dedicated to the detection of less complex target analytes.
Although aptamer-based recognition layers were successfully conjugated with optical and piezoelectric transducers, the application of electrochemical methods is more beneficial because of the low detection limits, high selectivity, simplicity and possibility of miniaturization.39,40 As in such platforms the receptor monolayer is attached to the solid surface, an appropriate tethering method must be employed, which not only provides stability of the sensing layer, but also maintenance of binding properties. Therefore, the most popular aptamer immobilization techniques are physical adsorption, covalent bonding with use of self-assembled monolayers (SAMs), e.g. thiol-based, streptavidin–biotin interaction and surface activation in the presence of EDC/NHS mixture (Fig. 3).41 The method of probe tethering has an influence on the aptamer surface coverage, which might affect the efficiency of binding of the sensing layer to a target analyte.
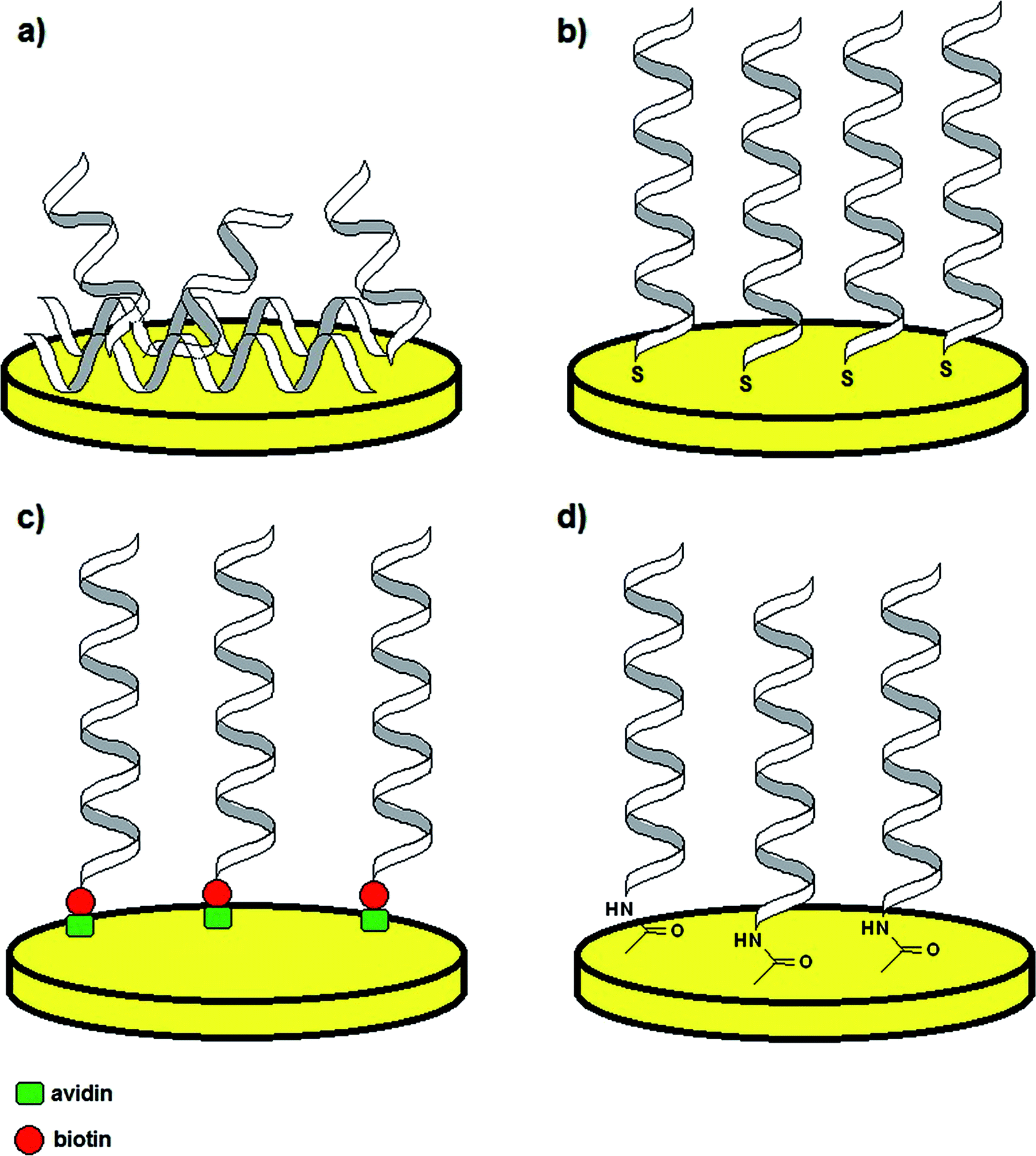 | ||
| Fig. 3 Examples of aptamer immobilization methods: via physical adsorption (a), thiol self-assembled monolayers (SAMs) (b), avidin–biotin complex formation (c) and surface activation with EDC/NHS (d). | ||
One of the most significant parts of the design of an electrochemical aptamer-based assay is the choice of the method of analytical signal generation. It should be emphasized that the direct electroanalysis of bound molecules is rarely exploited, since only few target analytes exhibit redox properties. In contrast, conjugation of electroactive labels such as methylene blue, ferrocene or AQMS is frequently utilized while preparing aptamer-based sensors.42,43 However, the main obstacle of such an approach is that the knowledge of conformational change is limited to the restrained number of aptamer sequences. Moreover, the conjugation of a redox active molecule to an aptamer is an elaborative and expensive process. Therefore, the aptamer–analyte binding might not be effectively evidenced with the use of electroactive labels. As an alternative, electroactive molecules might be introduced into the system as soluble indicators (Fig. 4).42 The nature of the interaction between an aptamer-modified surface and an electroactive indicator depends on the structure and charge of the redox molecule, and might be based on the electrostatic attraction/repulsion, intercalation into the double-stranded fragments of the receptor layer, interaction with minor grooves or with particular nucleobases. The use of a redox label/indicator is advantageous, as it leads to the enhancement of biosensor working parameters such as the detection limit, linear range of response and sensitivity. Nevertheless, the choice of electroactive species should be considered with respect to the experimental conditions, which might induce the rearrangement of the aptamer-sensing layer, and as a result have an influence on the degree of binding between the aptamer and target molecule. The introduction of a redox active molecule enables the application of several electrochemical techniques including voltammetry, chronoamperometry and impedance spectroscopy (EIS).25
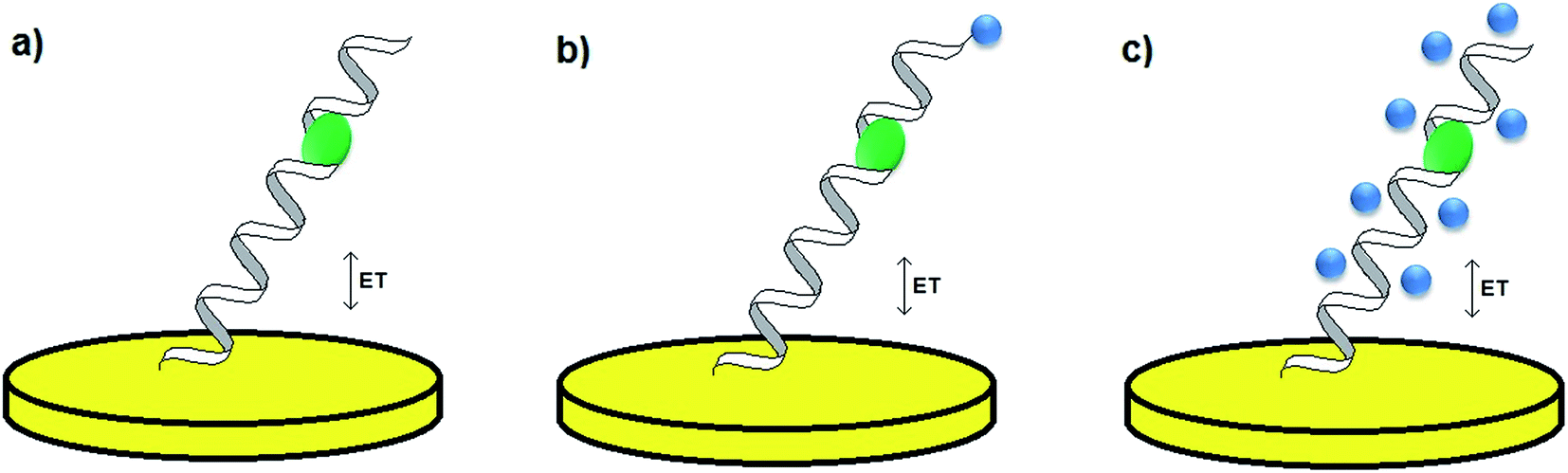 | ||
| Fig. 4 Methods of analytical signal generation: without a redox label/indicator (a), with a conjugated redox label (b), with a soluble redox indicator (c). | ||
Since the description of the first aptamer sequence, numerous aptamer-based biosensors have been developed with an immense emphasis on their potential application in clinical chemistry. The following sections will focus on the most representative examples of developed electrochemical aptamer-based platforms, which could serve as diagnostic tools.
2. Aptasensors in clinical diagnostics
2.1 Detection of potassium ions
Although more than 200 aptamer sequences have so far been identified, the number of aptamer probes that exhibit high affinity towards mono- and bivalent ions is limited. This could be attributed to the small dimensions (radius) and the structure of inorganic ions, which lead to the restrained possibility of spatial compatibility with the aptamer. This is in contrast to the mode of aptamer binding to more complex analytes such as proteins, as it results from various types of interactions. Moreover, the metal cation–aptamer interaction depends mostly on the electrostatic attraction, which might not provide high selectivity of the binding. Potassium ions are one of the rare examples of metal ions forming complexes with aptamers with high affinity. Determination of K+ level in physiological fluids (average level between 3.8 and 5.4 mmol L−1) is of special importance, as potassium plays a dominant role in the maintenance of nerve stimulation, hormone secretion and enzyme activation.44 Furthermore, its abnormal concentration may cause various diseases including cardiovascular disease, diabetes, kidney diseases and high blood pressure.45 For detection of K+ ions several DNA aptamers were applied, with the 15-mer thrombin binding aptamer (TBA) being the most frequently used probe.46Use of an electrochemical aptasensor elaborated by O'Sullivan is the first example of an aptamer-based assay dedicated to potassium.47 The recognition layer was formed on a gold surface with the use of a 3′-thiolated DNA TBA aptamer (5′-GGTTGGTGTGGTTGG-3′). The probe was additionally conjugated with a ferrocene label (Fc) at the 5′ end to generate the redox signal. The interaction of TBA and K+ caused a rearrangement of the recognition layer, leading to the formation and stabilization of a G-quadruplex structure. As a result, the distance between the redox label and electrode surface was reduced, which was evidenced by the increase of Fc current. The developed electrochemical beacon assay (e-beacon) can be distinguished with its simplicity of design and the working parameters including the linear range from 0.1 to 1 mmol L−1 and a lower detection limit of 0.015 mmol L−1. The aptasensor response was also analyzed by the use of impedance spectroscopy, which confirmed the high selectivity of the proposed sensor, with only a minor signal change in the presence of Ca2+ ions.
One of the main disadvantages of the formerly presented aptasensor was a narrow range of linear response. Therefore, further studies were focused on the enhancement of the working parameters of the aptamer-based assay. One of the possibilities is the use of gold nanoparticles for the modification of the gold surface, which could enhance the conductivity of the sensing monolayer and increase the aptamer surface density.48 A 21-mer DNA aptamer (TBA flanked with a triple thymine tag (T3) at the 3′ and 5′ ends) was tethered to a AuNPs/gold electrode and the electrochemical measurements were conducted in the presence of the ferri/ferrocyanide redox couple. The impedimetric spectra confirmed the effectiveness of subsequent steps of electrode surface modification, whereas the chronocoulometry experiments revealed the increase of aptamer surface coverage on the AuNPs/Au electrode in comparison to the bare Au electrode. On the basis of voltammetric measurements the analytical parameters of the aptasensor were determined including linear response referring to two ranges from 10 pmol L−1 to 1 μmol L−1 and from 0.5 μmol L−1 to 1 mmol L−1 as well as the LOD of 0.13 pmol L−1. The proposed aptasensor also showed high selectivity towards potassium, as the signal change was negligible in the presence of interferents.
A 21-mer aptamer sequence containing thymine T3 tags was also utilized by Chen for the preparation of the recognition layer for potassium ions.49 The aptamer probe was tethered to a gold surface via an Au–S linkage and the disulfide bond at the 3′-end was reduced with TCEP prior to the immobilization step. As the guanine residues exceeded 40% of all bases in the sequence, the formation of G-quadruplex and further stabilization upon addition of K+ ions was likely to happen. The rearrangement of aptamer probes into G-quadruplexes caused alteration of the sensing layer thickness and a decrease of its permeability. Furthermore, the G-quadruplex structure could be distinguished with a denser local charge in comparison to ssDNA probes. As a consequence, the Fe(CN)63−/4− couple, introduced into the system as a redox indicator, was repulsed from the surface upon creation of a steric hindrance. The designed aptasensor showed a linear response within the range from 0.1 nmol L−1 to 1 mmol L−1 with a lower detection limit of 0.1 nmol L−1. Moreover, the proposed assay exhibited high selectivity towards potassium and repetitive response while analyzing the K+ spiked urine samples.
Chen also elaborated an e-beacon assay with the use of a 21-mer aptamer labelled with ferrocene at the 5′ end.50 Although such an approach was similar to the design described by O' Sullivan,39 the application of a longer aptamer probe improved the working parameters of the sensor. The binding of potassium with the aptamer led to the rearrangement of the monolayer from the random-coil shape to the folded structure, hence ferrocene was in closer proximity to the electrode surface. The developed aptamer-based platform operated within the linear range from 0.1 to 50 nmol L−1 with a LOD of 0.1 nmol L−1. It also showed good selectivity towards K+ ions and a negligible signal change in the presence of interfering ions that was 50 times smaller than for potassium ions.
In another approach, a ssDNA-specific exonuclease RecJf was introduced into the aptasensor dedicated to K+ ions.51 The receptor layer consisted of 25-mer probes (5′-AAAGGTTGGTGTGGTTGGAACCTTT-3′), which were labelled with MB at the 5′ end and tethered to a gold surface via a thiol group at the 3′ end. The central 11-mer part of the sequence was responsible for binding to potassium, whereas the flanking 7-mer parts enabled the formation of a stem-loop structure in the absence of K+. After incubation with potassium, a G-quadruplex was formed, which enabled the binding of the ssDNA 5′ end with exonuclease. As a consequence, the aptamer probe was subjected to cleavage by the action of RecJf exonuclease and the number of MB molecules, that were at a close distance to the electrode surface, was reduced. The cleavage of the aptamer probe caused a decrease of MB current signal. The exonuclease-induced damage was possible only upon the rearrangement of the aptamer strand in the presence of K+, which was confirmed by electrophoretic experiments. The proposed system showed a linear response within the range from 50 nmol L−1 to 1 mmol L−1 and a LOD of 50 nmol L−1. The elaborated aptasensor exhibited selectivity towards potassium, as after incubation with other mono- and bivalent ions a G-quadruplex structure was not formed. As a result, the exonuclease could not have induced a strand cleavage, which resulted in a minor decrease of MB current.
Although a 15-mer TBA was utilized as a sensing layer in most of the electrochemical aptamer-based biosensors for potassium ions, other DNA aptamers – AG3 (5′-GGGTTAGGGTTAGGGTTAGGG-3′) and PS2M (5′-GTGGGTAGGGCGGGTTGG-3′) were applied in the development of several optical aptamer-based assays. To specify which aptamer provides the most efficient binding to potassium ions, three aptamer sequences were utilized for the modification of gold transducers.52 The aptamer probes were immobilized on a gold surface via an Au–S bond and the analysis was performed in the presence of redox indicators – MB, AQMS and Na4Fe(CN)64− (label-free assay) (Fig. 5). The studies conducted with the application of methylene blue showed that the TBA–K+ complex exhibited the highest affinity with a Kd of 1.13 μmol L−1, whereas Kd values for AG3 and PS2M were 5.03 and 1.49 μmol L−1, respectively. Furthermore, the TBA-based biosensor was distinguished with a linear response from 10−8 to 10−5 mol L−1, LOD of 2.31 nmol L−1 and good selectivity towards K+.
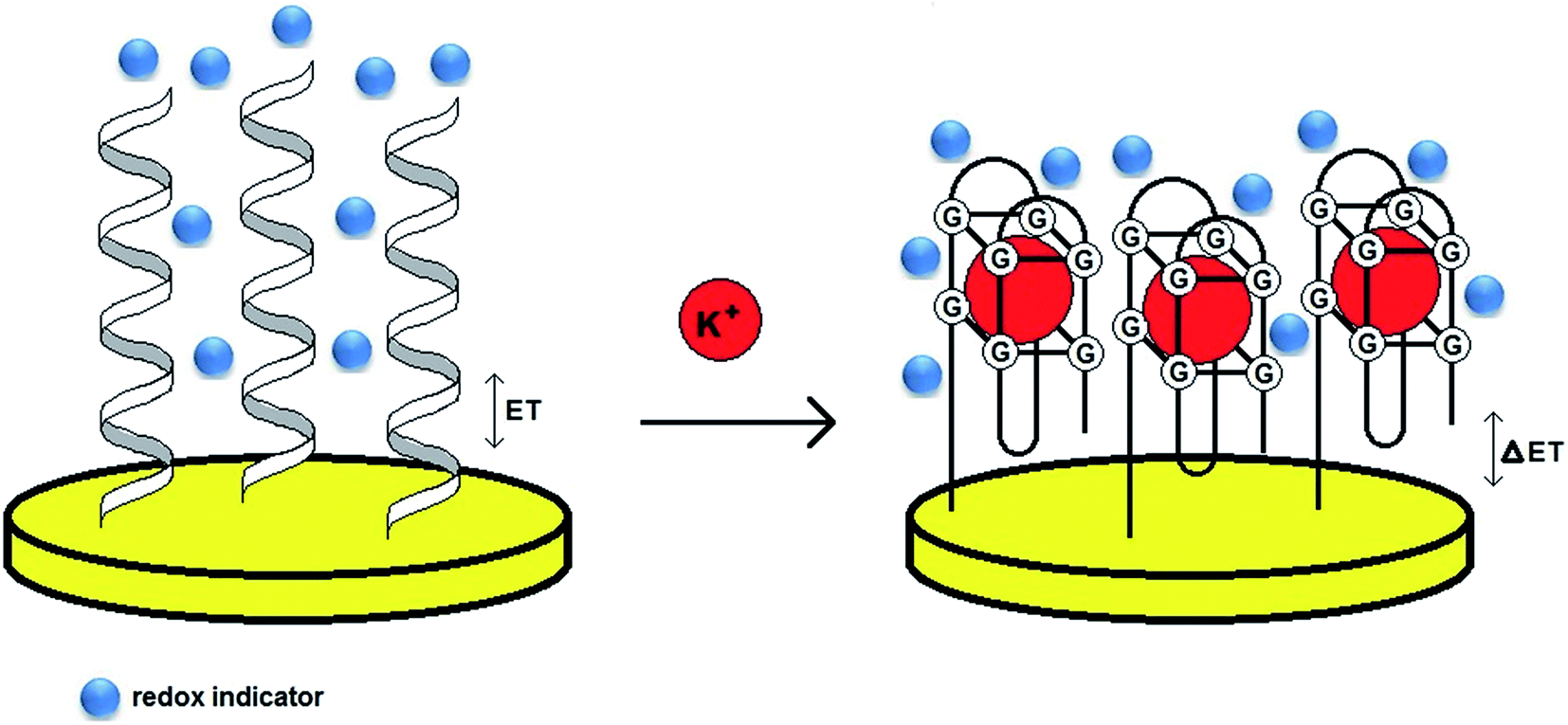 | ||
| Fig. 5 Schematic representation of the behavior of a label-free aptasensor with the formation of a G-quadruplex structure after binding to potassium ions. | ||
There have been several attempts to develop aptamer-based sensors, which are advantageous as they are simple in preparation and show better working parameters than other detection systems such as potentiometry. Since the TBA probe seems to show the highest affinity towards K+, further studies on the aptamer-based biosensors should focus on the enhancement of analytical response while testing real samples, utilization of novel nanomaterials for their preparation and miniaturization of sensors.
2.2 Determination of neurotransmitters – dopamine
As already mentioned, the majority of aptamer sequences were identified for complex analytes such as proteins. Nevertheless, a number of strands specific towards simple organic molecules were also found, for instance RNA aptamer binding to dopamine. As various neurodegenerative and psychiatric diseases are dependent on the level of this neurotransmitter, it is vital to monitor its concentration in physiological fluids. Dopamine level in the blood does not exceed 10−8 to 10−6 mol L−1 and an abnormal DA concentration might result in neural diseases such as Parkinson's, Huntington's, ADHD and schizophrenia. As a 57-mer RNA aptamer specific for DA was discovered, much effort was given to the development of aptamer-based assays for the determination of that neurotransmitter.One of the crucial issues concerning the design of an aptasensor is the choice of the method of probe tethering that could enable efficient binding between the aptamer and target molecule as well as the stability of the recognition layer. In the case of RNA aptamers the introduction of e.g. alkanethiol or protein at one of the distal ends might be a laborious and inefficient process. Concerning the need for steric freedom for the aptamer–analyte interaction, one of the possibilities is the formation of an additional layer of positive charge at the electrode surface. Such an attempt was performed during the preparation of an RNA aptasensor for dopamine described by Farjami.53 In this construct an intermediate cysteamine layer of positive charge was formed on a gold surface and the aptamer was immobilized through electrostatic attraction (Fig. 6). Since dopamine possesses electroactive properties, the analytical signal was related directly to the DA oxidation current, hence it was recognized as a label/indicator free assay. The proposed sensor allowed DA determination within the concentration range from 100 nmol L−1 to 5 μmol L−1. The studies also revealed that for an RNA aptamer/cysteamine Au electrode the current signals of other neurotransmitters, which used to overlap with each other prior to the electrode modification, were separated. Furthermore, the aptamer-based sensor exhibited a rapid response upon addition of dopamine to the solution and minor current signal after addition of ascorbic and uric acids.
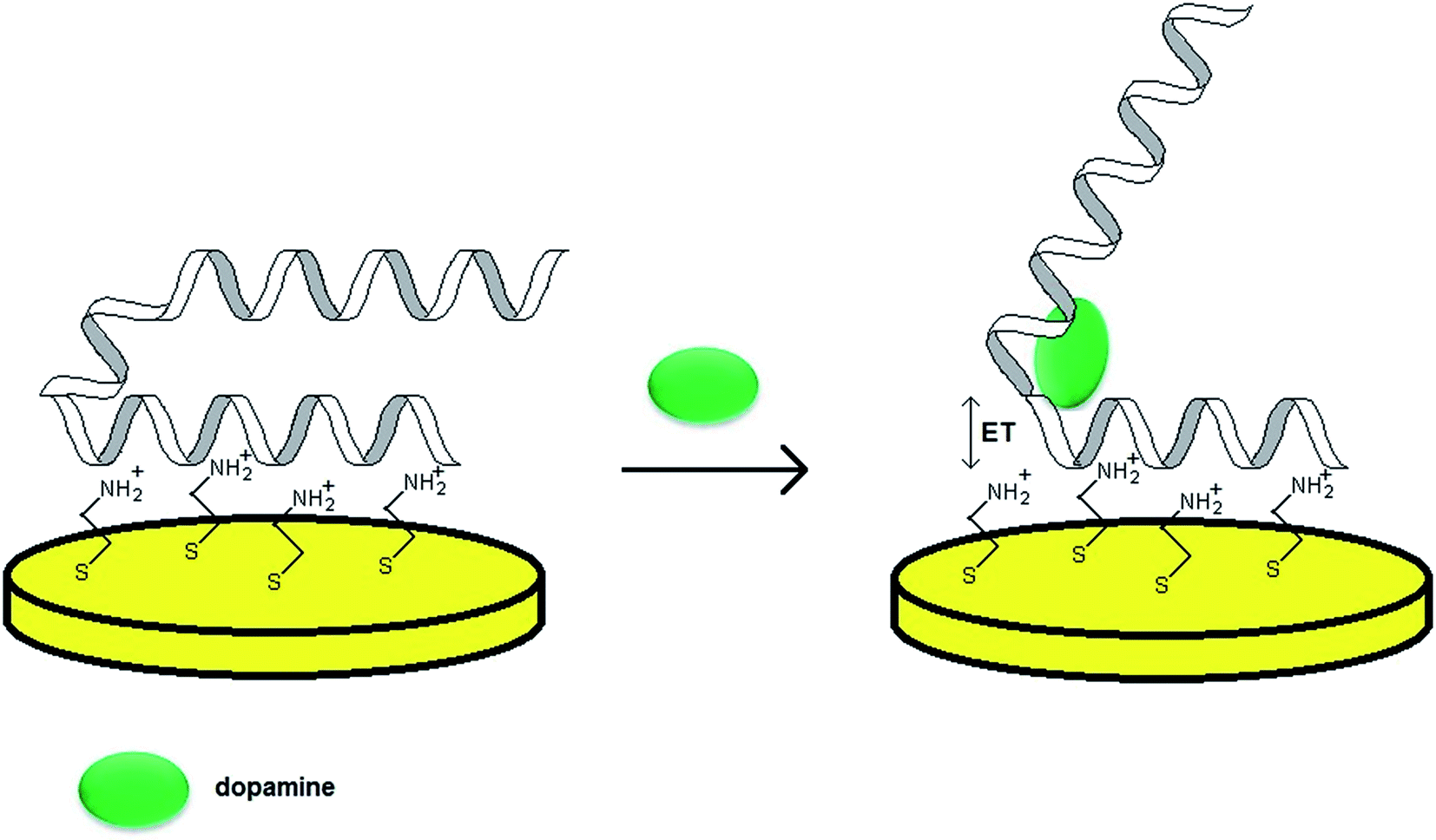 | ||
| Fig. 6 Mechanism of performance of a biosensor towards dopamine with an aptamer sensing layer formed by electrostatic attraction to a cysteamine-modified Au electrode. | ||
Further research concerned the influence of the arrangement of an RNA aptamer receptor layer on the efficiency of aptamer–dopamine interaction.54 The maximal binding was observed for the surface coverage of 3.5 pmol cm−2 for the aptamer placed horizontally on the cysteamine-modified Au surface after 30 min immobilization. The calculated dissociation constant for such an aptamer surface density was 0.12 μmol L−1 and the relationship of DA oxidation current versus dopamine level was linear within the range from 0.1 to 2 μmol L−1 with an LOD of 0.1 μmol L−1.
It was shown that a 58-mer DNA sequence exhibited higher affinity towards dopamine than the RNA sequence, as Kd values for both strands were 0.7 and 1.6 μmol L−1, respectively.55 Therefore, a DNA aptamer was also utilized for the preparation of a sensing layer for DA. In one of the examples, a thiol linkage was introduced at the 5′ end of the sequence and the Au–S covalent bond was formed.56 The electrochemical measurements were performed in the presence of redox indicators – ferri/ferrocyanide couple and methylene blue. The impedimetric studies confirmed the effectiveness of subsequent electrode modification steps, which were related to the increase of electron transfer resistance. It was also observed that for 1 h incubation in dopamine solution and the use of MB as the electroactive indicator, the aptasensor exhibited a linear response within the range from 5 to 150 nmol L−1 with an LOD of 1 nmol L−1. Moreover, the aptamer-based assay showed a minor response in the presence of interferents and maintained its stability after two weeks storage.
Several attempts were undertaken to improve the working parameters such as the linear range of response, mostly by the use of nanomaterials. In one of the approaches, glassy carbon electrode was incubated in a mixture of graphene oxide/poly(3,4-ethylenedioxythiophene) (PEDOT) solution, and the resulting layer was electrochemically reduced.57 Then, the electrode surface was activated with the use of the EDC/NHS mixture, which enabled the attachment of an amine-functionalized 58-mer DNA dopamine aptamer. The proposed label-free assay exhibited the highest efficiency after 60 min incubation in dopamine solution, for which the linear response was in the range from 0.001 to 160 nmol L−1, while the LOD was 0.078 pmol L−1. The proposed aptasensor also showed high selectivity with negligible current signal in the presence of interfering molecules including glucose, ascorbic and uric acids.
An aptamer-based sensor dedicated to dopamine was also developed by simple tethering of a 5′-thiolated 57-mer DNA aptamer probe to the gold electrode.58 The dopamine concentration was determined without the use of any external redox indicator and based on the DA oxidation current. The elaborated aptasensor operated within the linear range from 0.05 to 1 mmol L−1 and with an LOD of 26.8 μmol L−1 after 5 min incubation in dopamine solution under stirring conditions. For further improvement of the aptasensor performance, the glassy carbon electrode was modified with reduced graphene oxide and AuNPs, which was followed by deposition of the thiol-modified DNA aptamer. Such a construct showed a linear response from 5 to 75 μmol L−1, lower limit of detection of 3.36 μmol L−1 and good selectivity towards dopamine.
The application of RNA and DNA aptamers as recognition layers allowed the detection of dopamine at significantly low concentrations. A substantial improvement of analytical parameters of aptamer-based assays including the sensitivity and linear range of response was achieved by the use of nanomaterials including gold nanoparticles and graphene, which constituted the intermediate layer of aptamer-based sensors. As the dopamine aptamer is a relatively long strand, an appropriate method of tethering must be applied to allow efficient binding of the recognition layer and DA. To further explore the feasibility of aptamer-based biosensors for dopamine, the studies should focus on the miniaturization of formerly elaborated constructs, so that they could be applied as standard diagnostic tools.
2.3 Detection of protein biomarkers
One of the first protein – specific aptamers was identified for thrombin, which is a serine protease that takes part in thrombosis and haemostasis processes. Since its discovery, a 15-mer DNA TBA aptamer (5′-GGTTGGTGTGGTTGG-3′),46 which forms a G-quadruplex structure upon binding to thrombin, was frequently applied for the preparation of recognition layers conjugated with optical, mass and electrochemical transducers.
The first attempt of the development of an electrochemical aptasensor dedicated to thrombin was performed by Radi.59 A 3′-thiolated probe was immobilized on a gold electrode surface, which was followed by deposition of 2-mercaptoethanol to eliminate nonspecific interactions at the Au surface. Moreover, the use of a relatively short blocking agent also facilitated efficient interfacial electron transfer. The impedimetric measurements conducted in the presence of the ferri/ferrocyanide redox couple revealed a linear response of the aptasensor within the range from 5 to 35 nmol L−1 and a LOD of 2 nmol L−1. The proposed sensor was successfully regenerated with the use of 2 mol L−1 NaCl and applied for 15 subsequent measurements, moreover it retained its stability for 90 days.
Radi also elaborated an e-beacon assay for thrombin detection by the use of a TBA aptamer conjugated with a ferrocene label at the 5′ end.60 Upon incubation with thrombin, a G-quadruplex structure was formed, which led to the reduction of the distance between the redox label and electrode surface. As a result, the ferrocene oxidation signal increased linearly within the thrombin concentration range from 5 to 35 nmol L−1 with a lower limit of detection of 0.5 nmol L−1. The cyclic voltammograms revealed a ferrocene peak broadening and increase of peak separation, which probably resulted from the interaction between electroactive groups, location of the redox active centers as well as heterogeneity of the aptamer layer. The binding of thrombin to an aptamer-modified surface was also studied with EIS measurements, which showed a decrease of electron transfer resistance after incubation in thrombin solution. The aptamer-based assay exhibited high selectivity towards thrombin and a minor response after addition of proteins such as IgG, BSA and streptavidin. Interestingly, it was proved that the aptamer conformational change was highly dependent on the presence of counterions including K+ and Mg2+, hence the choice of experimental conditions was of special importance. Finally, the aptamer-based sensor was regenerated with the application of 0.1 mol L−1 HCl and utilized for 25 subsequent measurements.
As thrombin contains two active sites for aptamer binding, a sandwich assay to detect thrombin was also elaborated.61 Firstly, the thiolated TBA aptamer was immobilized on a gold surface, which was followed by incubation with thrombin and AuNPs-functionalized secondary TBA aptamer. As a single gold nanoparticle was conjugated with several aptamer probes, the construct contained a high negative charge that strongly repulsed the ferri/ferrocyanide redox indicator. The comparison of two blocking agents – mercaptoethanol and mercaptohexanol – showed a distinct manner of response, as for the shorter thiol a decrease of electron transfer, whereas for the latter a Ret increase were recorded, respectively. After formation of the sandwich construct Au/TBA aptamer/MCE/thrombin/AuNPs/TBA aptamer electron transfer resistance was enhanced and a linear response within the range from 0.05 to 18 nmol L−1 and a LOD of 0.02 nmol L−1 were derived from voltammetric experiments. To provide the stability of gold nanoparticles, rhodamine 6G was conjugated to aptamer-functionalized AuNPs, which contributed to the increase of Ret after creation of the sandwich assay.
A sandwich aptamer-based assay was also developed by Deng.62 In this approach a thiolated 15-mer TBA was directly deposited on the gold surface, which was also modified with mercaptohexanol to eliminate nonspecific interactions at the electrode surface. The aptamer/Au electrode was incubated in thrombin solution, which was followed by binding to the aptamer conjugated with AuNPs. The gold nanoparticles were catalytically enlarged by incubation in HAuCl4/NADH solution and additionally stabilized by the use of a negatively charged SDS. The subsequent steps of electrode modification resulted in the increase of electron transfer resistance evidenced by impedimetric measurements. The proposed aptasensor exhibited a dynamic response from 100 fmol L−1 to 100 nmol L−1, with a linear part in the range from 0.05 to 35 nmol L−1, and a limit of detection of 100 fmol L−1. The aptamer-based sandwich platform showed high selectivity towards thrombin, as the signal change in the presence of BSA, IgG and fibrinogen was minor. It was also proved that in the absence of thrombin the adsorption of the AuNPs/TBA aptamer as well as the enlargement of AuNPs did not occur.
Another aptamer-based sandwich assay was elaborated by Wang.63 The construct was prepared by tethering thiolated TBA aptamer to the gold surface, followed by the attachment of dithiothreitol to eliminate nonspecific interactions. After binding to thrombin, a secondary 29-mer aptamer was introduced, which was conjugated with a graphene nanocomposite consisting of reduced graphene oxide and gold nanoparticles. A 29 nucleotide sequence (5′-AGT CCG TGG TAG GGC AGG TTG GGG TGA CT-3′) exhibited a Kd of 0.5 nmol L−1, which was higher than the Kd for a 15-mer sequence (Kd = 100 nmol L−1).64 The EIS studies performed with the use of the Fe(CN)63−/4− indicator showed enhancement of electron transfer resistance after the electrode modification. A linear increase of Ret was recorded after incubation in thrombin solution within the range from 0.3 to 50 nmol L−1, and the calculated limit of detection was 0.01 nmol L−1. The proposed aptasensor exhibited a slight response in the presence of BSA, IgG and trypsin and a linear increase of Ret for the thrombin concentration from 0.3 to 35 nmol L−1 during the analysis in diluted serum.
In the studies performed by Mir, three various strategies of thrombin detection with the use of aptasensors were proposed.65 In the first construct, a thiolated 20-mer aptamer was immobilized on the gold surface, which was followed by the attachment of mercaptoethanol that would block the remaining free sites. As thrombin catalyses the hydrolysis of b-Ala-Gly-Arg-p-nitroaniline to electroactive p-nitroaniline, its concentration was related to the current signal of the formed compound. The voltammetric curves showed a current peak at a potential of −0.45 V, which was attributed to the thrombin substrate injected into the system. This was followed by the occurrence of a current decrease and the appearance of current signal at a potential of −0.7 V that referred to the p-nitroaniline. The second assay was a sandwich platform, which was formed by a thiolated-aptamer, tethered to an Au surface, and a secondary aptamer conjugated with biotin (Fig. 7). In the presence of thrombin a sandwich construct was formed, which was bound with streptavidin-labelled HRP that enabled the catalysis of nonspecific oxidation in the presence of H2O2. Moreover, a freely diffusing osmium complex was applied as a secondary mediator. In the third assay, thrombin was adsorbed on the mercaptoethanol-modified Au surface. The surface was additionally covered with 1% BSA to avoid the nonspecific adsorption. The analytical signal was generated in a similar manner, as in the former construct and its limit of detection was 3.5 nmol L−1. However, the main disadvantage of the proposed sensor was the nonspecific adsorption of thrombin on the electrode surface.
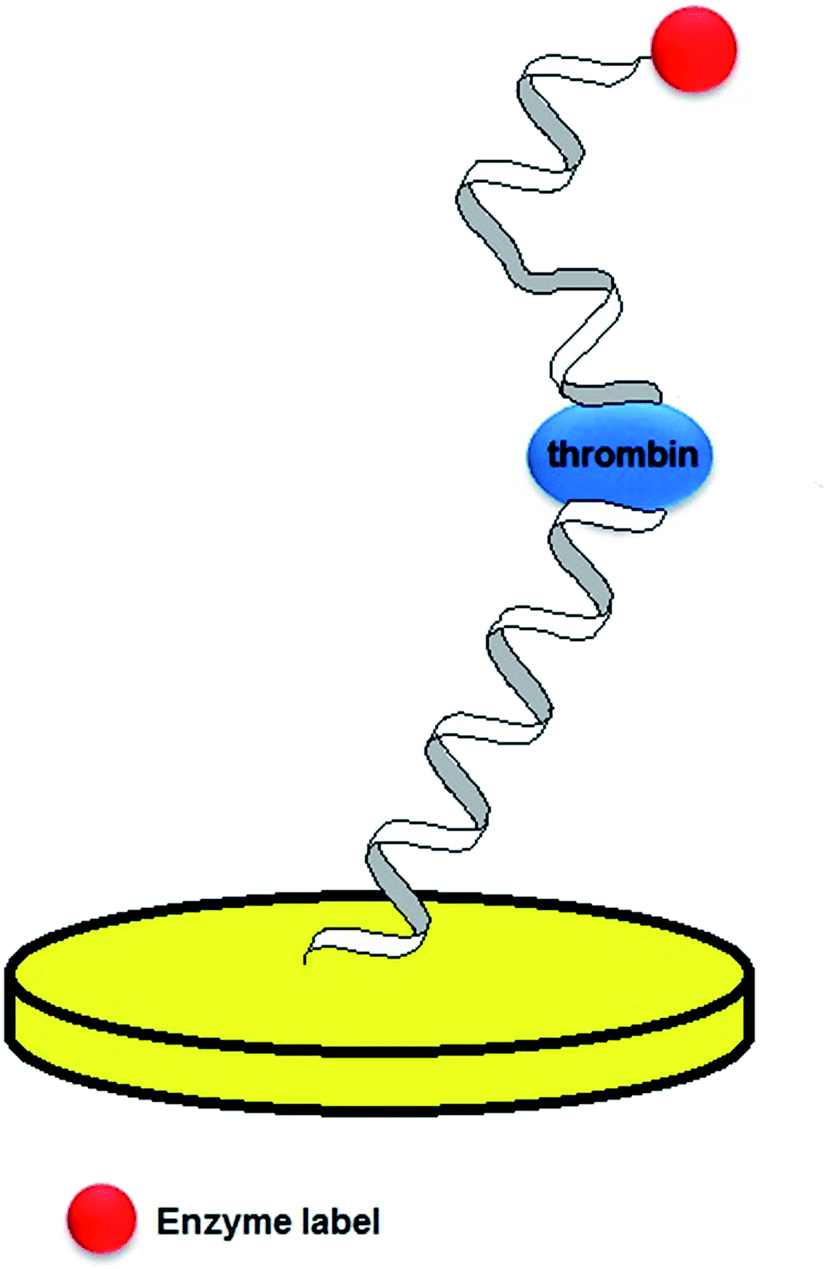 | ||
| Fig. 7 Schematic representation of an aptamer sandwich assay with a conjugated enzyme label. | ||
As the aptamer surface coverage might affect the sensitivity of detection of analyte–aptamer binding, a number of studies were focused on the enlargement of the working electrode surface. In one of the examples Au nanoparticles were attached to a gold electrode via a hexanedithiol linker.66 Then, a 5′-thiolated 27-mer aptamer (a longer version of the 15-mer TBA probe) was deposited on the AuNPs/Au surface. The chronocoulometric experiments for the AuNPs-functionalized surface showed 6-fold increase of the aptamer surface density in comparison to the simple TBA/Au electrode. The interaction of thrombin with the aptamer-modified surface was analyzed with the application of the impedimetric technique, which revealed a linear increase of Ret within 0.1–30 nmol L−1 of thrombin concentration and with a LOD of 0.013 nmol L−1. The elaborated assay showed a negligible response after incubation in a 1 mmol L−1 mixture of lysozyme, bovine haemoglobin and bovine serum albumin. The use of AuNPs significantly improved the aptasensor working parameters, as for a simple aptamer/Au electrode a linear response from 0.1 to 10 nmol L−1 and limit of detection of 0.05 nmol L−1 were recorded.
The miniaturization of biosensors is one of the most important issues in terms of their feasibility as standard diagnostic tools. Such devices should provide high sensitivity along with the possibility of operation with the use of samples of small volumes. Accordingly, aptamer-based microelectrode arrays on a gold surface were elaborated to detect thrombin.67 The gold arrays were fabricated on silicon wafers through the growth of a thermal oxide layer, followed by the photolithography and metalization steps. Then a thiolated 29-mer aptamer was tethered to Au microelectrodes and the surface was covered with mercaptohexanol to eliminate nonspecific interactions. The subsequent steps of fabrication caused an increase of charge transfer resistance evidenced by the impedimetric measurements. Further analysis of aptamer–thrombin binding was performed with the use of methylene blue as a redox indicator. A decrease of MB, which was observed after the incubation in thrombin, could be attributed to the specific binding between the protein and aptamer. As a result, the rearrangement of the monolayer and repulsion of MB molecules from the surface occurred. The developed system showed a linear response for 10−12 to 10−5 mol L−1 thrombin concentration and 0.143 pmol L−1 LOD. The aptasensor exhibited a minor response after incubation in BSA and casein, and was successfully utilized for the determination of thrombin in a spiked serum sample.
The aptamer-based sensors dedicated to thrombin were also developed with the use of nanomaterials, which might contribute to the improvement of analytical parameters. In one of the examples, a gold electrode was modified with an amino-terminated PANAM dendrimer through glutaraldehyde coupling, followed by attachment to an NH2-TBA aptamer via the glutaraldehyde coupling and reduction with NaBH4.68 Addition of the dendrimer caused a pronounced increase of Ret, which could be explained by the presence of numerous aptamer binding sites for thrombin. The impedimetric data revealed a linear increase of electron transfer resistance within the range from 0.01 to 50 nmol L−1 and 0.01 nmol L−1 LOD. The proposed assay exhibited a minor response after contact with BSA, and was successfully applied for six subsequent measurements. Moreover, it retained its stability after 1 month storage and was used for the analysis of a spiked sample of bovine plasma.
In another attempt, the gold electrode was covered with Nafion-graphene-nickel hexacyanoferrate nanoparticles (NiHCFNPs) to expand the surface and introduce several active sites.69 Next, the AuNPs were electrodeposited on a nanocomposite layer, which was followed by immobilization of the 5′-thiolated TBA probe and hexanethiol spacer. CV studies showed a significant increase of current signal, which could be attributed to the presence of electroactive NiHCFNPs and graphene that enlarged the active surface area and conductivity of the electrode. The proposed aptasensor exhibited linear response within two concentration ranges: 1 pmol L−1 to 1 nmol L−1 and 1–80 nmol L−1 and minor response to 10 nmol L−1 BSA, Hb and L-cysteine.
Detection of thrombin was also conducted with the use of intermediate multilayer film of AuNPs/thionine and Nafion.70 Firstly, the gold electrode was covered with a Nafion layer, which was followed by layer-by-layer (LBL) self-assembly of negatively charged AuNPs and positively charged thionine. The outer layer of gold nanoparticles enabled the tethering of the thiolated 15-mer TBA probe and hexanethiol spacer. A well-defined current signal was recorded due to the presence of electroactive thionine as well as the conductive properties of AuNPs. Moreover, the optimization studies showed the highest current for the electrode containing 4 thionine/AuNPs layers and Au nanoparticles of 16 nm in diameter. Both aptamer tethering and deposition of thrombin caused the formation of steric hindrance, which was evidenced by the decrease of current signal and the enhancement of charge transfer resistance. The proposed sensor showed a linear response within the range from 0.12 to 46 nmol L−1, 40 pmol L−1 LOD and a slight response after incubation with BSA and hemoglobin.
In another approach, an intermediate layer of aptasensor was composed of a toluidine–graphene nanocomposite.71 Gold nanoparticles were electrodeposited on the surface of a glassy carbon electrode, which was followed by several immersions in toluidine–graphene and in Au nanoparticles solution. Finally, a thiolated TBA probe and hexanethiol spacer were deposited on the outer layer of the gold nanoparticles. Each step of electrode modification was controlled by the use of cyclic voltammetry, and the recorded current resulted from the electroactive properties of the toluidine–graphene nanocomposite. The highest current was observed for the electrode containing the 4 Tb-Gra/nano-Au layer. The proposed assay showed a linear response within the range from 0.001 to 80 nmol L−1, the limit of detection of 0.33 pmol L−1 and minor signal change in the presence of hemoglobin, L-cysteine and BSA.
It is evident that in majority of examples a 15-mer TBA aptamer was utilized for the elaboration of the recognition layer for thrombin detection. Several attempts were performed for the improvement of the working parameters of the aptasensors, particularly with the use of graphene or gold nanoparticles as the intermediate layers as well as the application of sandwich constructs and introduction of electroactive and enzyme labels or indicators. The future projects should be aimed at miniaturizing the developed platforms and at minimizing the fouling of the transducer surface during the analysis of serum and blood samples.
One of the first examples of an electrochemical aptasensor for lysozyme was proposed by Cheng.72 The 5′-thiolated probe was immobilized on the gold surface, which was additionally covered with mercaptohexanol to eliminate nonspecific interactions at the surface. The specific binding between lysozyme and the aptamer was analyzed in the presence of ruthenium hexammine chloride and ferricyanide used as a redox indicator. A Fe(CN)63− current decrease was observed both after the surface modification with the aptamer and incubation with lysozyme. Such behaviour was attributed to the formation of a negatively charged layer on the electrode, moreover, the interaction of protein with the aptamer layer resulted in the creation of steric hindrance for the electroactive indicator. Both factors had an influence on the repulsion of ferricyanide from the electrode surface, as the negative charge of the monolayer was not completely neutralized after binding to lysozyme due to the strand length and a limited degree of protein–aptamer interaction. On the basis of cyclic voltammetry experiments performed in the presence of Ru(NH3)63+, the dissociation constant of 610 nmol L−1 was calculated for lysozyme bound to the surface-tethered aptamer. The studies also revealed the optimal aptamer surface coverage of 1.6–1.8 × 1012 molecules per cm2 for an efficient interaction.
In another approach an aptamer–antibody sandwich assay was developed for the determination of lysozyme.73 For that purpose, a screen printed electrode was modified with a diazonium salt, which enabled the conjugation of a 5′-amine terminated 40-mer aptamer via NHS/EDC chemistry. The electrode surface was also covered with BSA to reduce the nonspecific interactions. Lysozyme interacted with the aptamer and the secondary antibody bound to biotin, which enabled the interaction with avidin-modified alkaline phosphatase (ALP). The analytical signal was generated by the addition of 1-naphthyl phosphate, which was transformed into an electroactive 1-naphthol in the presence of ALP. The 1-naphthol oxidation current changed linearly for the lysozyme concentration from 5 fmol L−1 to 5 nmol L−1 and the calculated limit of detection was 4.3 fmol L−1. Moreover, the proposed assay exhibited small response in the presence of BSA, casein and cytochrome Cat5.
Another aptamer-based sensor dedicated to lysozyme operated via the conformational change of the aptamer – complementary ssDNA layer.74 In this construct a 3′-thiolated cDNA, conjugated with ferrocene at the 5′ end, was immobilized on a glassy carbon electrode covered with gold film and hybridized with a 30-mer DNA aptamer specific to lysozyme (Kd = 31 nmol L−1). The sensing layer formed a three helices structure in the absence of lysozyme, which changed its conformation after addition of the protein. As a result, the conductive pathway of duplex DNA was damaged, which was revealed by the decrease of Fc current. The developed aptasensor exhibited a linear response from 0 to 100 nmol L−1 lysozyme concentration and a minor response after incubation in BSA, hemoglobin, IgG and cytochrome C.
Since two aptamer sequences of high affinity towards lysozyme were identified, a comparison between both types of aptamer monolayers was performed.75 5′-NH2-modified strands were immobilized on GCE via a diazotization reaction, which was followed by surface activation by the use of NHS and EDC. The unoccupied succinimide groups were deactivated with the application of ethanolamine and the electrode surface was additionally covered with BSA to reduce the nonspecific interactions. The impedimetric data revealed a linear change of Ret in the range from 0.1 to 0.8 μmol L−1 and 100 nmol L−1 LOD for a 30-mer sequence (AptLysCOX). For a 40-mer probe (AptLysTRAN) the Ret increase was linear from 0.025 to 0.8 μmol L−1 of lysozyme concentration and a lower limit of detection of 25 nmol L−1. The longer AptLysTRAN probe could also be distinguished with higher selectivity towards lysozyme in comparison to the AptLysCOX-based sensing layer.
2.3.3.1 Platelet-derived growth factor (PDGF). Platelet-derived growth factor is a protein crucial in vascular proliferation and cell transformation, which might play a key role in tumor progression. A 35-mer PDGF-specific DNA aptamer was identified and frequently applied as a recognition layer in sensing constructs. In one of the first approaches, a 3′-thiolated aptamer, conjugated with methylene blue at the 5′ end, was tethered to a gold surface, hence operated as an e-beacon sensor.76 The mechanism of the proposed sensor relied on aptamer conformational change upon binding to PDGF-BB, which led to the reduction of the distance between the electrode surface and redox label. The voltammetric measurements conducted in undiluted serum allowed obtaining 1 nmol L−1 limit of detection and 50 pmol L−1 LOD for a 50% diluted serum sample. Moreover, the elaborated sensor showed high selectivity towards PDGF-BB with a 52% current increase and 22% and 7% current enhancement for PDGF-AB and PDGF-AA, respectively.
The specificity of a DNA aptamer towards PDGF was compared by the use of two reference sequences by Degefa.77 The probes were tethered to a gold surface via an Au–S bond at the 5′ end of a ssDNA probe and the interaction between the sensing layer and protein was analyzed with the use of the ferri/ferrocyanide redox couple. After incubation in PDGF, a current decrease and the enhancement of electron transfer resistance were recorded. The experiments revealed a minor change in CV and impedimetric data for random sequences (35 and 27-mer long), which indicated their negligible affinity towards PDGF. In contrast, the aptamer-based sensor showed a linear response towards the protein within the range from 1 to 40 nmol L−1 evidenced by DPV measurements.
Another aptasensor was dedicated to PDGF detection based on the interaction of the protein with a pair of aptamer probes.78 The design concerned the immobilization of short 8-mer single-stranded DNA molecules on a gold surface via an Au–S linkage. The aptamer probes were extended by the 3′ ferrocene-labelled 8-mer nucleotide tail to generate the electrochemical signal. The proposed platform enabled the determination of PDGF-BB homodimer concentration, as the aptamer did not hybridize with the surface-tethered DNA in the absence of the target protein. This ‘signal-on’ assay exhibited a linear response towards PDGF within the range from 1 pg mL−1 to 20 ng mL−1 and a detection limit of 1 pg mL−1. Moreover, the aptasensor showed a minor signal change after incubation with PDGF-AB, BSA, insulin, IgG and thrombin.
Nanomaterials were also utilized for the development of aptamer-based sensors dedicated to PDGF. In one of the approaches a GCE was modified with a composite of poly(diallyldimethylammonium chloride) (PDDA)-protected graphene and nanoparticles, which can be characterized by good conductivity, simplicity of functionalization and high surface area.79 Next, the electrode was covered with glucose oxidase (GOD), which was followed by the electrodeposition of gold nanoclusters and tethering of a PDGF-selective aptamer. In the absence of protein, well-defined redox peaks referring to the FAD/FADH2 forms of GOD active centre were recorded. After incubation with PDGF, a current decrease was observed, which was explained by the formation of steric hindrance by a protein–aptamer complex that limited the efficiency of electron transfer. The current decrease was linear within the range from 0.005 to 60 nmol L−1 of PDGF-BB concentration and the limit of detection was 1.7 pmol L−1. The proposed assay showed a minor response after incubation in thrombin, BSA, Hb, AFP and CEA solutions and retained its activity after 20 days of storage.
Another example of an aptamer-based sensor for PDGF-BB was a sandwich assay containing selenide (MoS2)/carbon aerogel nanocomposites.80 The GC electrode was incubated in chitosan aqueous solution containing MoS2/CA nanocomposites, which was followed by attachment of AuNPs and immobilization of a 45-mer thiolated DNA aptamer (Ap1). BSA was used as a blocking agent to eliminate nonspecific interactions at the electrode surface. The final two steps included 2 h incubation with PDGF-BB and immersion in the solution containing a ferrocene–AuNPs–aptamer (Ap2) conjugate. Impedimetric studies revealed the enhancement of the efficiency of electron transfer at GCE after modification with the MoS2/CA composite and AuNPs. The optimization studies concerned the choice of an adequate amount of MoS2/CA composite, ferrocene–aptamer1 molar ratio as well as incubation time in the solutions of PDGF-BB and the secondary aptamer. The response of the sensor derived from the relationship of Fc current–PDGF-BB concentration was linear in the range from 0.001 to 10 nmol L−1, whereas the LOD was 0.3 pmol L−1. Moreover, the aptamer sandwich assay showed a minor response towards BSA, CEA, Hb, and IgG, good reproducibility and remained stable after 1 month storage.
Another aptasensor dedicated to PDGF-BB was prepared by the deposition of a molybdenum selenide–graphene composite (MoS2–Gr) on the GCE, which was followed by attachment of AuNPs, thiolated capture DNA (iDNA) and BSA, used as a blocking agent.81 Then the electrode was incubated in the mixture of PDGF-BB, aptamer, complementary DNA (cDNA), auxiliary biotinylated DNA (aDNA) and ExoIII nuclease. In the absence of PDGF, the aptamer strand hybridized with cDNA, which enabled the binding of iDNA with biotinylated-aDNA at the electrode surface. The subsequent addition of avidin–HRP resulted in a significant current response. In contrast, after the introduction of PDGF-BB, the aptamer formed a complex with a protein, which allowed the hybridization of cDNA and aDNA. As a consequence, the cDNA–aDNA duplex was subjected to digestion by ExoIII, and a current decrease was recorded. The proposed aptasensor exhibited a linear response within 0.0001–1 nmol L−1 concentration range and a lower limit of detection of 20 fmol L−1. The aptamer-based assay was also characterized by the high selectivity towards PDGF-BB and was tested by the analysis of a spiked serum sample.
A similar mechanism of response was also utilized in another construct, where GCE was modified with a AuNPs/VS2–graphene composite followed by immobilization of tetrahedral DNA.82 The absence of PDGF-BB enabled the hybridization of the aptamer and cDNA, which prevented the signal DNA strand from digestion by ExoIII and provided a high current response after addition of avidin–HRP. In contrast, the introduction of PDGF-BB allowed the cDNA – signal DNA duplex formation and a subsequent digestion by ExoIII leading to the decrease of current response. The developed sensor provided a linear response from 0.0001 to 1 nmol L−1, LOD of 30 fmol L−1 and good selectivity towards PDGF-BB.
PDGF-BB belongs to one of the most frequently studied protein biomarkers in terms of the development of aptamer-based sensors. Accordingly, DNA aptamer strands varying in sequence length and content were applied for the preparation of recognition layers. One of the major disadvantages of the majority of the presented sensors is the elaborative process of surface modification as well as the multi-step mechanism of aptamer-based sensor response. In contrast, the proposed assays provided a significant improvement of working parameters and the limitation of nonspecific interactions at the electrode surface.
2.3.3.2 Prostate-specific antigen (PSA). Prostate specific antigen is one of the most well-known cancer biomarkers that is usually detected with the use of immuno-based assays. For that glycoprotein the aptamer-based sensors were also developed with the application of a 32-mer DNA sequence as a receptor layer. In one of the examples, two different aptamer immobilization methods were tested.83 The first one concerned the tethering of a thiolated aptamer probe to a gold electrode, followed by the attachment of mercaptohexanol. The second protocol compromised the incubation of the Au electrode in the mixture of thiols: mercaptoundecanoic acid and thiol-terminated sulfobetaine. Then, an amine-terminated aptamer was tethered to mercaptoundecanoic acid chains through the activation of MUA carboxylic groups with the use of EDC/NHS and the surface was covered with ethanolamine to block the unreacted groups of MUA. For both assays a decrease of electron transfer resistance for Fe(CN)63−/4− was recorded after incubation in PSA. This might be attributed to the reduction of a negative charge at the electrode surface, as prostate specific antigen is positively charged. The surface charge played a dominant role in the interaction between the sensing layer and redox indicator, as the occurrence of steric hindrance through the aptamer–PSA complex formation had a negligible effect on signal change. In the case of a mixed MUA/sulfo-betaine monolayer it was possible to detect prostate-specific antigen at the concentration below 1 ng mL−1 (the level above 4 ng mL−1 indicates the progression of prostate cancer). It was also observed that the nonspecific adsorption of human bovine albumin was prevented with the use of MUA/sulfo-betaine surface modification, which could not be inhibited at the aptamer/MCH modified surface. The binding of DNA aptamer to PSA was also verified by SPR measurements, which enabled estimation of the dissociation constant of the complex that was 9.50 ng mL−1.
In another approach, a PSA-specific DNA aptamer was immobilized on the pyrolytic graphite electrode.84 Firstly, gold nanoparticles covered with graphitized mesoporous carbon nanoparticles were deposited on the electrode surface and this was followed by incubation in a streptavidin solution. The electrode was also immersed in BSA solution to eliminate nonspecific adsorption and in the final step a biotin-conjugated DNA aptamer was tethered to the electrode surface. It was shown that the subsequent steps of electrode modification and the incubation in PSA caused a decrease of ferri/ferrocyanide current along with the enhancement of Ret. The proposed assay exhibited a linear response towards PSA within the range from 0.25 to 200 ng mL−1 and a detection limit of 0.25 ng mL−1. Moreover, the aptasensor showed a minor signal change in the presence of BSA, hemoglobin and thrombin and maintained its stability after 30 days of storage.
A label/indicator-free aptamer-based sensor for PSA was developed by Souada.85 The GCE was modified with 3-(5-hydroxy-1,4-dihydro-1,4-dioxonaphthalen-2(3)-yl) propionic acid poly(JUG-co-JUGA) film containing quinone groups via electropolymerization. The amino-terminated aptamer probe was attached to the electrode through surface activation by EDC/NHS. PSA binding to the aptamer-modified surface caused a decrease of current signal, as the conducting properties of a polymer tethered to the electrode were limited. The SWV studies revealed a linear relationship of PSA concentration versus current from 1 ng mL−1 to 10 μg mL−1. A dissociation constant of 2.6 nmol L−1 for the aptamer–PSA complex was calculated on the basis of the calibration curve, which resembled the Langmuir binding isotherm. The designed aptasensor also exhibited a minor signal change in the presence of BSA. Interestingly, after removal of PSA and hybridization of complementary ssDNA, the initial current was regained to some extent.
2.3.3.3 Vascular endothelial growth factor (VEGF). Vascular endothelial growth factor is a signalling protein responsible for the promotion of angiogenesis for vascular development. It constitutes a biomarker for various diseases including rheumatoid arthritis, retinopathy and cancer. For this protein, a 58-mer DNA aptamer was identified (Kd of 315 nmol L−1) and was applied to the development of VEGF sensing layers. In one of the examples the first step concerned the preparation of gold IDE capacitors on SiO2 wafers by the use of photolithography.86 Then, mercaptopropionic acid was deposited on the gold surface and an amino-terminated DNA aptamer was attached by the EDC/NHS surface activation. Additionally, BSA was placed on the surface to reduce the nonspecific interactions. Two aptamer constructs were tested – a simple aptamer–VEGF complex assay and an aptamer–antibody sandwich platform. As a negligible change of capacitance was recorded for the aptamer–VEGF assay, a sandwich-based system was utilized for further measurements. For the frequencies of 65, 90, 120 and 212 MHz, a dynamic response within the range from 5 to 1000 pg mL−1 was observed, and the lowest limit of detection of 401 pg mL−1 was achieved for the frequency of 65 mHz.
In another approach an aptasensor dedicated to the simultaneous detection of VEGF and MUC1 cancer biomarkers was elaborated.87 The recognition layer was prepared by tethering a 5′-thiolated single-stranded 32-mer with a ferrocene at the 3′ end to the gold surface. The Au electrode was also covered with mercaptohexanol to reduce nonspecific adsorption. A cDNA strand was partially complementary to VEGF and MUC1 aptamers, hence a dual protein recognition was possible. It was observed that in the absence of proteins, cDNA was hybridized with both aptamers and the duplex DNA was orientated upright to the Au electrode. As a consequence, the long distance of the ferrocene label to the electrode surface prevented efficient charge transfer. In the presence of one of the proteins, an increase of ferrocene current was observed, which was further enhanced after the addition of both proteins. The current increase was attributed to the dehybridization of the duplex, which provided a more flexible cDNA chain and a reduction of the distance between the redox label and the gold surface. In the presence of MUC1 protein, an aptamer-based sensor showed a linear response from 1 to 20 nmol L−1 and a detection limit of 0.33 nmol L−1. Moreover, at the constant 20 nmol L−1 MUC1 concentration, a linear current increase was recorded from 1 to 20 nmol L−1 of VEGF. The SWV measurements were consistent with impedimetric data, which revealed a decrease of electron transfer resistance after binding to both target proteins.
2.3.3.4 Osteopontin. One of the novel cancer biomarkers is osteopontin – a phosphorylated glycoprotein, which might affect the tumor formation, progression and metastasis, e.g. in breast cancer. Several immunoassays and an aptamer-based sensor88 were developed for the detection of osteopontin. The gold electrode was covered with 3,3-dithiodipropionic acid (DPA), which was followed by the activation of the carboxyl groups by EDC/NHS. Next, a streptavidin layer was deposited on the electrode and a 5′-biotinylated 40-mer RNA aptamer (Kd = 18 nmol L−1) was attached to the surface. Upon the increase of osteopontin concentration a ferri/ferrocyanide current decreased in the range from 25 to 2402 nmol L−1 and a maximum signal was observed for 800 nmol L−1. The proposed aptasensor exhibited a limit of detection of 3.7 nmol L−1 and an average selectivity towards osteopontin, as the signal change was significant in the presence of thrombin.
2.3.3.5 Urokinase plasminogen activator (uPA). Urokinase plasminogen activator is another novel prognostic biomarker of ovarian and breast cancer. For this protein 79-mer and 33-mer RNA aptamers of high affinity were identified.89 An aptamer-based array was elaborated by the immobilization of phosphorothiolated polyadenine-terminated RNA probe on a gold surface.90 For the analysis of the aptamer–uPA binding, ferricyanide and MB were used as redox indicators. It was shown that the urokinase–aptamer complex could be distinguished with a dissociation constant of 0.003 nmol L−1 in the presence of BSA as a counter protein and MB as the electroactive molecule. The proposed sensor exhibited a dynamic response towards uPA within the range from 1 pmol L−1 to 100 nmol L−1.
2.3.3.6 HER2. Avian erythroblastosis oncogene B (HER2) is one of the most significant biomarkers of breast cancer. An 86-mer DNA aptamer was identified for that protein and utilized for the preparation of the aptamer-based assay.91 A gold electrode was covered with gold nanoparticles, which was followed by the deposition of 3-mercaptopropionic acid (MPA). The amine-terminated aptamer was attached to the electrode surface by the activation of carboxylic groups of MPA by the use of EDC/NHS. Finally, BSA was tethered to the surface to eliminate the nonspecific interactions. The analysis of formation of a HER2–aptamer complex was performed in the presence of the ferri/ferrocyanide redox couple. After incubation of the aptamer-modified electrode with HER2, a linear increase of electron transfer resistance was observed from 10−5 to 102 ng mL−1 of HER2 and the detection limit was 10−5 ng mL−1. The elaborated assay showed a minor Ret change after incubation in IgG, glucose, RNA and DNA. The aptamer-based sensor was regenerated for 4 subsequent measurements and remained stable after 30 days of storage.
Recently, a number of aptamer-based assays dedicated to various cancer biomarkers have been elaborated. One of the challenges in the preparation of recognition layers is the choice of an appropriate method of tethering of long aptamer strands, as it should enable efficient binding to a target protein. Although the proposed sensors could serve as alternative tools in early cancer diagnostics, their main obstacle is the limited stability of aptamer-based monolayers as well as the possibility of surface fouling during the analysis of serum or blood samples.
2.3.4.1 Cardiac troponin I (cTnI). Cardiac diseases are one of the main causes of death and therefore much effort has been given to identify potential biomarkers. One of the recently discovered protein biomarkers is cardiac troponin I, which is released into the blood right after acute myocardial infarction (AMI). For this protein a 40-mer DNA sequence was chosen through the SELEX process, which exhibited higher affinity than cTnI antibody, as their dissociation constants were 270 pmol L−1 and 20.8 nmol L−1, respectively.92 The aptamer probe was immobilized on the gold electrode formed on the PET substrate, and the Au surface was covered with mercaptohexanol to eliminate nonspecific interactions. Negatively charged ferrocene-modified silica nanoparticles (Fc-SiNPs) were utilized to generate the analytical signal. The binding of cTnI with the aptamer caused a linear decrease of the Fc-SiNPs anodic current in the range from 1 pmol L−1 to 10 nmol L−1 and the calculated LOD was 1 pmol L−1. Moreover, the proposed aptasensor showed a minor signal change after incubation with CtnT, cTnC, human serum albumin, myoglobin and B-type natriuretic peptide, and was used for the analysis of spiked human serum samples.
2.3.4.2 C-Reactive protein (CRP). C-Reactive protein is one of the crucial biomarkers of inflammatory states and its normal blood concentration is below 5 mg L−1. For the determination of CRP, several immunoassays were developed, however further studies showed that a 44-mer RNA aptamer exhibited higher affinity towards CRP (Kd = 125 nmol L−1). In one of the first approaches, C-reactive protein was detected with the use of an aptamer–antibody assay (ELONA test).93 A 5′-biotinylated F-pyrimidine RNA probe with a triethylene glycol tail was immobilized on streptavidin-modified magnetic beads that were placed on a screen-printed electrode by the use of a magnet. Prior to that step, the aptamer-modified beads were incubated with CRP, which was followed by the addition of biotinylated anti-CRP human immunoglobulin. Finally, the streptavidin-conjugated alkaline phosphatase was introduced (Fig. 8). The analytical signal was generated in the presence of the electroactive product of an enzymatic reaction of α-naphthyl phosphate catalyzed by ALP. The elaborated assay exhibited a dynamic response from 0 to 1000 mg L−1 CRP and the limit of detection was 0.054 mg L−1.
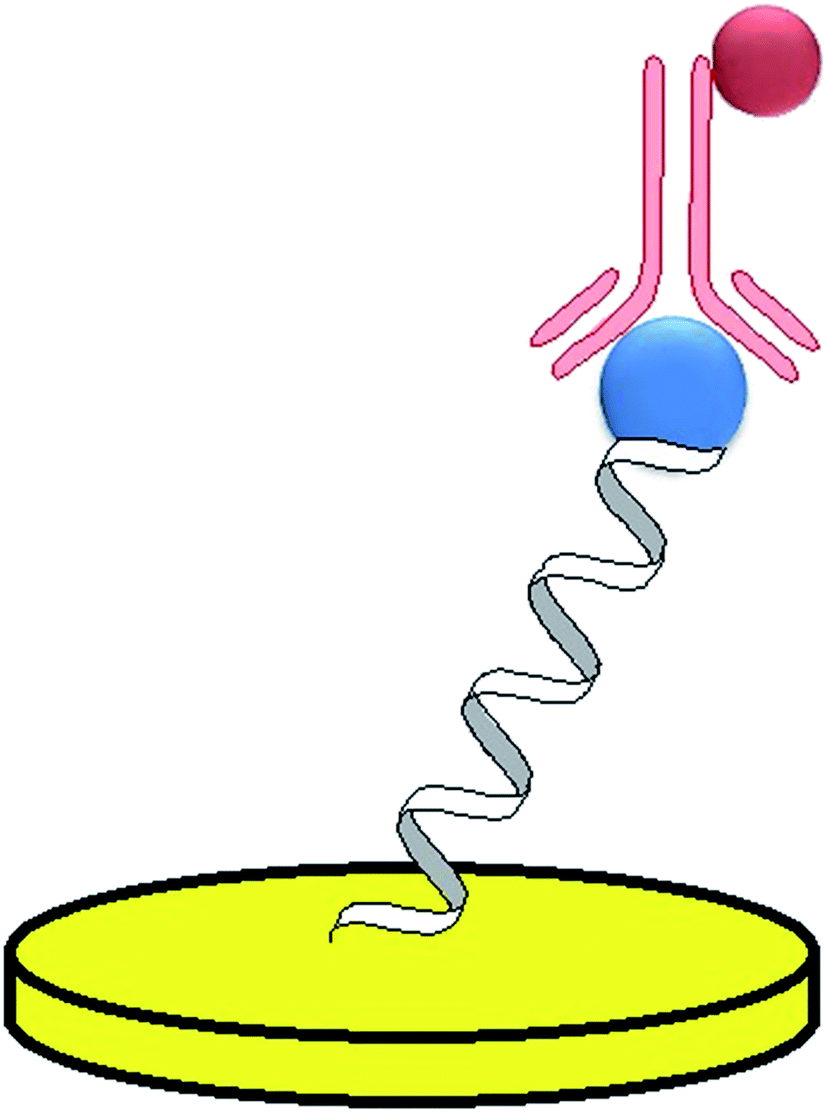 | ||
| Fig. 8 Example of an aptamer–antibody sandwich assay. | ||
The aptamer-based sensor was also developed with the use of non-faradaic impedance spectroscopy as the detection technique.94 The gold-interdigitated capacitors were formed on a SiO2 surface and the 5′-thiolated RNA probe was immobilized on the surface. The dielectric signal referring to the change of relative capacitance was generated by the aptamer–CRP complex formation. The proposed assay exhibited a linear response within the range from 100 to 500 pg mL−1 CRP. It was also observed that the highest affinity of C-reactive protein–aptamer binding with Kd = 1.6 μmol L−1 was recorded for the 208 mHz frequency.
The number of aptamer-based sensors for cardiac biomarkers is lower than for cancer biomarkers, which is explained by the identification of specific aptamer sequences for fewer proteins. Therefore, much effort should be given to the selection of aptamers specific to other biomarkers of cardiovascular disorders. Moreover, improvement of the working parameters of sensors could be achieved with the use of nanomaterials and by the choice of the method of generation of electrochemical signals. Eventually, such devices could substitute the commonly applied immuno-based assays.
3. Summary
This review focuses on the examples of electrochemical aptamer-based sensors dedicated to the detection of analytes significant in clinical diagnostics. The use of aptamers as the recognition elements is advantageous, as they exhibit high affinity towards their target molecules, especially to proteins.Although the elaborated sensors are characterized by low detection limits and good selectivity, a number of issues should be addressed during their development. Firstly, selection of the aptamer probe is a laborious process, as the SELEX conditions must be adjusted to a defined analyte. Moreover, several post-modifications must be often performed to decrease the susceptibility to enzymatic degradation and to introduce functional groups for surface tethering or analytical signal generation.
One of the limiting factors in the preparation of aptamer-based sensors is the choice of the immobilization method, which should provide efficient binding of the recognition layer to the target analyte. It is evident that self-assembled thiol monolayers (SAM) and biotin–streptavidin complexes are used in the most frequently applied immobilization protocols. However, the conjugation of functional groups can be time-consuming and might lead to a lower reaction yield, or even cause the contamination of the probe. The application of nanomaterials can contribute to the enlargement of the electrode surface and improvement of the interface conductivity, however it usually requires a multi-step process of surface modification.
The mode of analytical signal generation is also vital to evidence the interaction between an aptamer and analyte. Since the manner of aptamer conformational alteration upon binding to a target molecule was verified for a limited number of aptamers, the utilization of redox labels might cause an ambiguity of the sensor response. Furthermore, preparation of the probe for e-beacon assays requires a laborious and expensive synthesis. Therefore, the majority of aptasensors operate with the use of electroactive indicators. The redox active molecule should also be carefully selected, as the experimental conditions, such as the applied potential, might induce rearrangement of the sensing layer. As a result, the degree of interaction of the aptamer and target analyte can vary with time and while changing the electroactive indicator. Moreover, the mechanism of aptasensor response should not be complicated so that the output signal could be easily reproduced. Thus, sophisticated multi-step actions during the analysis are the main defect of some of the presented aptamer-based devices.
Although aptasensors were developed for the detection of potassium ions and dopamine, the vast majority of molecules detected with the use of aptasensors are proteins-growth factors (PDGF, VEGF), anticoagulant factors (thrombin), markers of cardiac diseases (CRP) and cancer (PSA, HER2, uPA). As the proposed sensors are aimed at testing blood or serum samples, one of the main challenges is the limitation of nonspecific adsorption at the transducer surface. This can be achieved by the selection of a blocking agent or adjustment of the potential range, however in most cases the serum or blood samples must be diluted. As a consequence, the output signal has to be related to the actual physiological conditions.
As the complete regeneration of the aptamer-based layer is difficult to achieve, the proposed aptasensors are single-use assays. The enhancement of their performance might be accomplished by the fabrication of multi-array platforms, which was already conducted for DNA hybridization assays.95 Further miniaturization of the aptamer-based devices is necessary for their adaptation as point-of-care systems. One of the possibilities is the preparation of small cartridges, which after the sample deposition are inserted into the analyzers.
The clinical use of aptamer-based sensors is still limited, as they are generally fabricated with the use of sophisticated methods and require specific readout devices. In contrast, improvement of the preparation protocols of aptamer-sensing layers, along with the optimization of the working parameters and the miniaturization of the sensors might contribute to the enhancement of the role of aptasensors in chemical diagnostics.
Glossary
| ALP | Alkaline phosphatase |
| AMI | Acute myocardial infarction |
| AQMS | Anthraquinone-2-sulfonic acid |
| AuNPs | Gold nanoparticles |
| BSA | Bovine serum albumin |
| CRP | C-Reactive protein |
| cTnI | Cardiac troponin I |
| CV | Cyclic voltammetry |
| DA | Dopamine |
| DNA | Deoxyribonucleic acid |
| DPA | 3,3-Dithiodipropionic acid |
| EDC | 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride |
| EIS | Electrochemical impedance spectroscopy |
| ELISA | Enzyme-linked immunosorbent assay |
| ELONA | Enzyme-linked oligonucleotide assay |
| FAD | Flavin adenine dinucleotide |
| Fc | Ferrocene |
| FNA | Functional nucleic acid |
| GCE | Glassy carbon electrode |
| GOD | Glucose oxidase |
| GRA | Graphene |
| Hb | Hemoglobin |
| HRP | Horseradish peroxidase |
| IgG | Immunoglobulin G |
| JUG | Poly(hydroxy-1,4-naphthoquinone) |
| LOD | Lower limit of detection |
| LBL | Layer-by-layer |
| MB | Methylene blue |
| MCE | Mercaptoethanol |
| MCH | Mercaptohexanol |
| MPA | 3-Mercaptopropionic acid |
| MUA | Mercaptoundecanoic acid |
| NiHCFNPs | Nafion-graphene-nickel hexacyanoferrate nanoparticles |
| NHS | N-Hydroxysuccinimide |
| NMR | Nuclear magnetic resonance |
| PANAM | Poly(amidoamine) |
| PCR | Polymerase chain reaction |
| PDDA | Poly(diallyldimethylammonium chloride) |
| PDGF | Platelet-derived growth factor |
| PEDOT | Poly(3,4-ethylenedioxythiophene) |
| PEG | Polyethylene glycol |
| PET | Polyethylene terephthalate |
| POC | Point-of-care |
| PSA | Prostate-specific antigen |
| RNA | Ribonucleic acid |
| SAM | Self-assembled monolayer |
| SDS | Sodium dodecyl sulfate |
| SELEX | Systematic evolution of ligands by exponential enrichment |
| SiNPS | Silica nanoparticles |
| SWV | Square wave voltammetry |
| TBA | Thrombin binding aptamer |
| TCEP | tris(2-Carboxyethyl)phosphine |
| US FDA | US food and drug administration |
| VEGF | Vascular endothelial growth factor |
Acknowledgements
This work was financially supported by the National Science Centre, Poland (grant No. 2014/15/N/ST4/02980) and by Warsaw University of Technology.References
- J. D. Watson and F. H. C. Crick, Nature, 1953, 171(4356), 737–738 CrossRef CAS PubMed.
- B. Alberts, A. Johnson and J. Lewiset al., Garland Science, New York, 2002 Search PubMed.
- N. K. Navani and Y. Li, Curr. Opin. Chem. Biol., 2006, 10, 272–281 CrossRef CAS PubMed.
- Y. Lu and J. Liu, Curr. Opin. Biotechnol., 2006, 17, 580–588 CrossRef CAS PubMed.
- A. D. Ellington and J. W. Szostak, Nature, 1990, 346(6287), 818–822 CrossRef CAS PubMed.
- R. Stoltenburg, C. Reinemann and B. Strehlitz, Biomol. Eng., 2007, 24, 381–403 CrossRef CAS PubMed.
- T. Sampson, World Pat. Inf., 2003, 25, 123–129 CrossRef CAS.
- C. L. A. Hamula, J. W. Guthrie, H. Zhang, X.-F. Li and X. C. Le, Trends Anal. Chem., 2006, 25(7), 681–691 CrossRef CAS.
- S. Song, L. Wang, J. Li, J. Zhao and C. Fan, Trends Anal. Chem., 2008, 27(2), 108–117 CrossRef CAS , 2008.
- X. Ni, M. Castanares, A. Mukherjee and S. E. Lupold, Curr. Med. Chem., 2011, 18(27), 4206–4214 CrossRef CAS PubMed.
- M. Famulok, J. S. Hartig and G. Mayer, Chem. Rev., 2007, 107(9), 3715–3743 CrossRef CAS PubMed.
- E. W. M. Ng, D. T. Shima, P. Calias, E. T. Cunningham Jr, D. R. Guyer and A. P. Adamis, Nat. Rev. Drug Discovery, 2006, 5(2), 123–132 CrossRef CAS PubMed.
- S. M. Nimjee, C. P. Rusconi, R. A. Harrington and B. A. Sullenger, Trends Cardiovasc. Med., 2005, 15(1), 41–45 CrossRef CAS PubMed.
- E. L. Howard, K. C. D. Becker, C. P. Rusconi and R. C. Becker, Arterioscler., Thromb., Vasc. Biol., 2007, 27(4), 722–727 CrossRef CAS PubMed.
- C. K. O'Sullivan, Anal. Bioanal. Chem., 2002, 372, 44–48 CrossRef PubMed.
- S. Tombelli, M. Minunni and M. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- S. Song, L. Wang, J. Li, J. Zhao and C. Fan, Trends Anal. Chem., 2008, 27(2), 108–177 CrossRef CAS.
- B. McDonnell, S. Hearty, P. Leonard and R. O'Kennedy, Clin. Biochem., 2009, 42, 549–561 CrossRef CAS PubMed.
- G. J. Kos, et al. , Chest, 1999, 115(4), 1140–1154 CrossRef.
- J. Wang, et al. , Anal. Chim. Acta, 1997, 347, 1–8 CrossRef CAS.
- J. Kirsch, C. Siltanen, Q. Zhou, A. Revzin and A. Simonian, Chem. Soc. Rev., 2013, 42, 8733–8768 RSC.
- S. Rodriguez-Mozaz, M. J. Lopez de Alda and D. Barceló, Anal. Bioanal. Chem., 2006, 386, 1025–1041 CrossRef CAS PubMed.
- D. R. The Venot, K. Toth, R. A. Durst and G. S. Wilson, Pure Appl. Chem., 1999, 71(12), 2333–2348 CAS.
- A. P. F. Turner, Chem. Soc. Rev., 2013, 42, 3184–3196 RSC.
- F. R. R. Teles and L. P. Fonseca, Talanta, 2008, 77, 606–623 CrossRef CAS.
- W. Wu, S. Zhao, Y. Mao, Z. Fang, X. Lu and L. Zeng, Anal. Chim. Acta, 2015, 861, 62–68 CrossRef CAS PubMed.
- S.-H. Lee, J.-Y. Ahn, K.-A. Lee, H.-J. Um, S. S. Sekhon, T. S. Park, J. Min and Y.-H. Kim, Biosens. Bioelectron., 2015, 68, 272–280 CrossRef CAS PubMed.
- C. L. A. Hamula, H. Peng, Z. Wang, G. J. Tyrrell, X.-F. Li and X. C. Le, Methods, 2016, 97, 51–57 CrossRef CAS PubMed.
- C. L. A. Hamula, X. C. Le and X.-F. Li, Anal. Chem., 2011, 83, 3640–3647 CrossRef CAS PubMed.
- R. Aimaiti, L. Qin, T. Cao, H. Yang, J. Wang, J. Lu, X. Huang and Z. Hu, Appl. Microbiol. Biotechnol., 2015, 99, 9073–9083 CrossRef CAS PubMed.
- H.-C. Park, I. A. Baig, S.-C. Lee, J.-Y. Moon and M.-Y. Yoon, Appl. Biochem. Biotechnol., 2014, 174, 793–802 CrossRef CAS PubMed.
- D. Sun, J. Lu, Z. Chen, Y. Yu and M. Mo, Anal. Chim. Acta, 2015, 885, 166–173 CrossRef CAS PubMed.
- X. Chen, Y. Pan, H. Liu, X. Bai, N. Wang and B. Zhang, Biosens. Bioelectron., 2016, 79, 353–358 CrossRef CAS PubMed.
- Q. Xie, Y. Tan, Q. Guo, K. Wang, B. Yuan, J. Wan and X. Zhao, Anal. Methods, 2014, 6, 6809–6814 RSC.
- L. Liang, M. Su, L. Li, F. Lan, G. Yang, S. Ge, J. Yu and X. Song, Sens. Actuators, B, 2016, 229, 347–354 CrossRef CAS.
- X. Zhang, J. Zhang, Y. Mac, X. Pei, Q. Liu, B. Lu, L. Jin, J. Wang and J. Liu, Int. J. Biochem. Cell Biol., 2014, 46, 1–8 CrossRef CAS PubMed.
- X. Li, W. Zhang, L. Liu, Z. Zhu, G. Ouyang, Y. An, C. Zhao and C. J. Yang, Anal. Chem., 2014, 86, 6596–6603 CrossRef CAS PubMed.
- Y. Pu, Z. Zhu, H. Liu, J. Zhang, J. Liu and W. Tan, Anal. Bioanal. Chem., 2010, 397, 3225–3233 CrossRef CAS PubMed.
- J. Wang, Anal. Chim. Acta, 2002, 469, 63–71 CrossRef CAS.
- T. Mairal, V. C. Özalp, P. L. Sánchez, M. Mir, I. Katakis and C. K. O'Sullivan, Anal. Bioanal. Chem., 2008, 390, 989–1007 CrossRef CAS PubMed.
- A. Sassolas, B. D. Leca-Bouvier and L. J. Blum, Chem. Rev., 2008, 108, 109–139 CrossRef CAS PubMed.
- E. E. Ferapontova, Curr. Anal. Chem., 2011, 7, 51–62 CrossRef CAS.
- T. G. Drummond, M. G. Hill and J. K. Barton, Nat. Biotechnol., 2003, 21, 1192–1199 CrossRef CAS PubMed.
- A. Verdian-Doghaei, M. R. Housaindokht and K. Abnous, Anal. Biochem., 2014, 466, 72–75 CrossRef CAS PubMed.
- Z. Chen, J. Guo, H. Ma, T. Zhou and X. Li, Anal. Methods, 2014, 6, 8018–8021 RSC.
- L. C. Bock, L. C. Griffin, J. A. Latham, E. H. Vermaas and J. J. Toole, Nature, 1992, 355, 564–566 CrossRef CAS PubMed.
- A.-E. Radi and C. K. O'Sullivan, Chem. Commun., 2006, 3432–3434 RSC.
- Z. Chen, T. Zhou, C. Zhang, H. Ma, Y. Lin and K. Li, RSC Adv., 2014, 4, 48671–48675 RSC.
- Z. Chen, L. Chen, H. Ma, T. Zhou and X. Li, Biosens. Bioelectron., 2013, 48, 108–112 CrossRef CAS PubMed.
- Z. Chen, J. Guo, S. Zhang and L. Chen, Sens. Actuators, B, 2013, 188, 1155–1157 CrossRef CAS.
- P. Miao, Y. Tang, B. Wang, K. Han, X. Chen and H. Sun, Analyst, 2014, 139, 5695–5699 RSC.
- M. Jarczewska, Ł. Górski and E. Malinowska, Sens. Actuators, B, 2016, 226, 37–43 CrossRef CAS.
- E. Farjami, R. Campos, J. S. Nielsen, K. V. Gothelf, J. Kjems and E. E. Ferapontova, Anal. Chem., 2013, 85, 121–128 CrossRef CAS PubMed.
- I. Álvarez-Martos, R. Campos and E. E. Ferapontova, Analyst, 2015, 140, 4089–4096 RSC.
- R. Walsh and M. C. DeRosa, Biochem. Biophys. Res. Commun., 2009, 388, 732–735 CrossRef CAS PubMed.
- J. Zhou, W. Wang, P. Yu, E. Xiong, X. Zhang and J. Chen, RSC Adv., 2014, 4, 52250–52255 RSC.
- W. Wang, W. Wang, J. J. Davis and X. Luo, Microchim. Acta, 2015, 182, 1123–1129 CrossRef CAS.
- M. Jarczewska, S. R. Sheelam, R. Ziółkowski and Ł. Górski, J. Electrochem. Soc., 2016, 163, B26–B31 CrossRef CAS.
- A.-E. Radi, J. L. A. Sanchez, E. Baldrich and C. K. O'Sullivan, Anal. Chem., 2005, 77, 6320–6323 CrossRef CAS PubMed.
- A.-E. Radi, J. L. A. Sanchez, E. Baldrich and C. K. O'Sullivan, J. Am. Chem. Soc., 2006, 128, 117–124 CrossRef CAS PubMed.
- B. Li, Y. Wang, H. Wei and S. Dong, Biosens. Bioelectron., 2008, 23, 965–970 CrossRef CAS PubMed.
- C. Deng, J. Chen, Z. Nie, M. Wang, X. Chu, X. Chen, X. Xiao, C. Lei and S. Yao, Anal. Chem., 2009, 81, 739–745 CrossRef CAS PubMed.
- Q. Wang, Z. Zhou, Y. Zhai, L. Zhang, W. Hong, Z. Zhang and S. Dong, Talanta, 2015, 141, 247–252 CrossRef CAS PubMed.
- B. Deng, Y. Lin, C. Wang, F. Li, Z. Wang, H. Zhang, X.-F. Li and X. C. Le, Anal. Chim. Acta, 2014, 837, 1–15 CrossRef CAS PubMed.
- M. Mir, M. Vreeke and I. Katakis, Electrochem. Commun., 2006, 8, 505–511 CrossRef CAS.
- L.-D. Li, H.-T. Zhao, Z.-B. Chen, X.-J. Mu and L. Guo, Sens. Actuators, B, 2011, 157, 189–194 CrossRef CAS.
- H.-Y. Bai, F. J. Del Campo and Y.-C. Tsai, Biosens. Bioelectron., 2013, 42, 17–22 CrossRef CAS PubMed.
- Z. Zhang, W. Yang, J. Wang, C. Yang, F. Yang and X. Yang, Talanta, 2009, 78, 1240–1245 CrossRef CAS PubMed.
- L. Jiang, R. Yuan, Y. Chai, Y. Yuan, L. Bai and Y. Wang, Analyst, 2012, 137, 2415–2420 RSC.
- Y. Yuan, R. Yuan, Y. Chai, Y. Zhuo, Z. Liu, L. Mao, S. Guan and X. Qian, Anal. Chim. Acta, 2010, 668, 171–176 CrossRef CAS PubMed.
- S. Xie, R. Yuan, Y. Chai, L. Bai, Y. Yuan and Y. Wang, Talanta, 2012, 98, 7–13 CrossRef CAS PubMed.
- A. K. H. Cheng, B. Ge and H.-Z. Yu, Anal. Chem., 2007, 79, 5158–5164 CrossRef CAS PubMed.
- C. Ocaña, A. Hayat, R. Mishra, A. Vasilescu, M. del Valle and J.-L. Marty, Analyst, 2015, 140, 4148–4153 RSC.
- Y. Xia, S. Gan, Q. Xu, X. Qiu, P. Gao and S. Huang, Biosens. Bioelectron., 2013, 39, 250–254 CrossRef CAS PubMed.
- C. Ocaña, A. Hayat, R. K. Mishra, A. Vasilescu, M. del Valle and J.-L. Marty, Bioelectrochemistry, 2015, 105, 72–77 CrossRef PubMed.
- R. Y. Lai, K. W. Plaxco and A. J. Heeger, Anal. Chem., 2007, 79, 229–233 CrossRef CAS PubMed.
- T. H. Degefa and J. Kwak, Anal. Chim. Acta, 2008, 613, 163–168 CrossRef CAS PubMed.
- Y.-L. Zhang, Y. Huang, J.-H. Jiang, G.-L. Shen and R.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 15448–15449 CrossRef CAS PubMed.
- K. Deng, Y. Xiang, L. Zhang, Q. Chen and W. Fu, Anal. Chim. Acta, 2013, 759, 61–65 CrossRef CAS PubMed.
- L.-X. Fang, K.-J. Huang and Y. Liu, Biosens. Bioelectron., 2015, 71, 171–178 CrossRef CAS PubMed.
- K.-J. Huang, H.-L. Shuai and J.-Z. Zhang, Biosens. Bioelectron., 2016, 77, 69–75 CrossRef CAS PubMed.
- K.-J. Huang, Y.-J. Liu and Q.-F. Zhai, J. Mater. Chem. B, 2015, 3, 8180–8187 RSC.
- P. Jolly, N. Formisano, J. Tkác, P. Kasák, C. G. Frost and P. Estrela, Sens. Actuators, B, 2015, 209, 306–312 CrossRef CAS.
- B. Liu, L. Lu, E. Hua, S. Jiang and G. Xie, Microchim. Acta, 2012, 178, 163–170 CrossRef CAS.
- M. Souada, B. Piro, S. Reisberg, G. Anquetin, V. Noël and M. C. Pham, Biosens. Bioelectron., 2015, 68, 49–54 CrossRef CAS PubMed.
- A. Qureshi, Y. Gurbuz and J. H. Niazi, Sens. Actuators, B, 2015, 209, 645–651 CrossRef CAS.
- J. Zhao, X. He, B. Bob, X. Liu, Y. Yin and G. Li, Biosens. Bioelectron., 2012, 34, 249–252 CrossRef CAS PubMed.
- S. G. Meirinho, L. G. Dias, A. M. Peres and L. R. Rodrigues, Biosens. Bioelectron., 2015, 71, 332–341 CrossRef CAS PubMed.
- D. M. Dupont, J. B. Madsen, R. K. Hartmann, B. Tavitian, F. Duconge, J. Kjems and P. A. Andreasen, RNA, 2010, 16, 2360–2369 CrossRef CAS PubMed.
- M. Jarczewska, L. Kékedy-Nagy, J. S. Nielsen, R. Campos, J. Kjems, E. Malinowska and E. E. Ferapontova, Analyst, 2015, 140, 3794–3802 RSC.
- L. Chun, S.-E. Kim, M. Cho, W. Choe, J. Nam, D. W. Lee and Y. Lee, Sens. Actuators, B, 2013, 186, 446–450 CrossRef CAS.
- H. Jo, H. Gu, W. Jeon, H. Youn, J. Her, S.-K. Kim, J. Lee, J. H. Shin and C. Ban, Anal. Chem., 2015, 87, 9869–9875 CrossRef CAS PubMed.
- S. Centi, L. B. Sanmartin, S. Tombelli, I. Palchetti and M. Mascini, Electroanalysis, 2009, 21(11), 1309–1315 CrossRef CAS.
- A. Qureshi, Y. Gurbuz, S. Kallempudi and J. H. Niazi, Phys. Chem. Chem. Phys., 2010, 12, 9176–9182 RSC.
- C. Batchelor-McAuley, G. G. Wildgoose and R. G. Compton, Biosens. Bioelectron., 2009, 24, 3183–3190 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |