
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA disposable chemical heater and dry enzyme preparation for lysis and extraction of DNA and RNA from microorganisms
J. R.
Buser
 *a,
X.
Zhang
a,
S. A.
Byrnes
a,
P. D.
Ladd
a,
E. K.
Heiniger
a,
M. D.
Wheeler
a,
J. D.
Bishop
a,
J. A.
Englund
bc,
B.
Lutz
a,
B. H.
Weigl†
d and
P.
Yager
a
*a,
X.
Zhang
a,
S. A.
Byrnes
a,
P. D.
Ladd
a,
E. K.
Heiniger
a,
M. D.
Wheeler
a,
J. D.
Bishop
a,
J. A.
Englund
bc,
B.
Lutz
a,
B. H.
Weigl†
d and
P.
Yager
a
aDepartment of Bioengineering, University of Washington, Box 355061, Seattle, WA, USA. E-mail: buserj@uw.edu
bProgram in Infectious Diseases, Clinical Research Division, Fred Hutchinson Cancer Research Center, Seattle, Washington, USA
cDepartment of Pediatrics, Seattle Children's Research Institute, University of Washington, Seattle, Washington, USA
dPATH, Seattle, WA, USA
First published on 3rd March 2016
Abstract
Sample preparation, including bacterial lysis, remains a hurdle in the realization of complete point-of-care tests for many pathogens. Here, we developed a sample preparation methodology for enzymatic lysis and sample heating for low-resource, point-of-care applications. We show an instrument-free chemical heater system for rapid lysis of a Gram-positive bacterium (Staphylococcus aureus) and an RNA virus (human respiratory syncytial virus) using a dried lysis enzyme mixture (achromopeptidase) for S. aureus. After a lysis step (<1 minute), lysis enzymes are heat deactivated (<5 minutes) using a simple disposable chemical heater. We demonstrated that both DNA and RNA in the heat-treated sample could be directly amplified without purification, even in the presence of a clinically-obtained human nasal sample. This simple approach to dry enzyme storage and sample heating is adaptable to many applications where samples need to be lysed, including use in low-resource laboratories and in single-use or cartridge-based point-of-care diagnostic devices.
Introduction
According to the 2010 Global Burden of Disease study, four of the top ten causes of death world-wide are attributed to communicable diseases, which disproportionally affect low resource settings (LRS).1–3 Number four on the list is lower respiratory infections and number seven is diarrhea.1 These infections have known and available treatments but often lack accurate diagnosis, especially in LRS where there is severely reduced availability of healthcare.4,5 In many settings, including the US and other developed countries, symptomatic diagnosis is commonly used by healthcare providers. Although symptoms are important, they can vary between individuals and are often shared by multiple infections. Diarrhea, for example, can be caused by viruses, bacteria, or parasites.6–9 Each of these classes of pathogens require different treatments and within classes, treatments can vary due to different susceptibility to common drugs.Antimicrobial resistance has been increasing around the world;10 one method for curbing this trend is accurate molecular diagnosis which can lead to identification of specific pathogens and potential drug resistances. Methicillin-resistant Staphylococcus aureus (MRSA) is a significant pathogen causing hospital- and community-acquired infections in developing and developed regions;11,12 further advancement in diagnostics which can quickly identify methicillin resistance could aid in slowing the spread of this pathogen. Nucleic acid amplification tests (NAATs), which utilize a pathogen's DNA or RNA, are commonly used in pathogen identification: polymerase chain reaction (PCR) is a widely used example. The use of NAATs for disease diagnosis offers multiple advantages including increased sensitivity, the ability to multiplex, and epidemiological tracking of disease transmission and drift via nucleic acid (NA) sequencing. These approaches, however, often require expensive equipment and highly trained personnel.
Integrated microfluidic systems, such as the Cepheid GeneXpert, provide sample-to-result diagnostics using disposable cartridges that contains the assay reagents. These cartridges are coupled with automated instrumentation to process the sample and perform the bioassay, and have been shown to expedite treatment for pathogens including Mycobacterium tuberculosis.13 These systems, however, have significant cost, infrastructure, and maintenance commitments associated with them14 and are most appropriate for use in well-equipped laboratories with reliable electricity. Significant advances in NAATs are still required for lower-resource settings.15
Microfluidic bioassays have the potential to expand the reach of NAATs, but sample preparation, including pathogen lysis and nucleic acid extraction, remains an underdeveloped aspect of microfluidics-based bioassays, especially those designed for point-of-care use.16 Many commercially available systems that are marketed for the point-of-care are often missing sample preparation components. In 2011, Niemz et al. evaluated 13 commercially available point-of-care NAAT-based systems. All of these systems include an expensive, non-disposable component that requires mains electricity and, likely, a service contract. Additionally, less than half of these include on-device sample preparation thereby increasing the overall time and costs required for operation and limiting their usability as truly point-of-care systems.17
Enzymatic lysis has been shown to be effective in bacterial sample preparation. Some lytic enzymes are highly specialized for a specific cell type, such as lysostaphin which targets staphylococcus bacteria,18 while others are more generally applied. Lysozyme is a commonly used and well understood lytic enzyme which cleaves the peptide-disaccharide linkage of peptidoglycans in bacterial cell walls causing them to denature.19 Many Gram-negative bacteria are insensitive to lysozyme because their thick outer membrane prevents the enzyme from interacting with the inner cell wall; additionally, some Gram-positive bacteria, such as S. aureus, are also resistant to lysozyme treatment.20 Achromopeptidase (ACP), purified from Achromobacter, was found to have bacteriolytic activity as early as the early 1970s.21 Since then, it has been widely used for the lysis of lysozyme-resistant Gram-positive bacteria such as S. aureus. Some MRSA diagnostic assays on the market use ACP for lysis and downstream real-time PCR for pathogen identification (e.g., the BD Gene Ohm MRSA test). Most of the assays employ ACP lysis at 37 °C for 10–20 minutes, followed either by heat deactivation at a controlled temperature22 or KOH deactivation.23
Isothermal NAATs, such as loop-mediated isothermal amplification (LAMP)24,25 and recombinase-polymerase amplification (RPA)26 have received much attention recently due to their simple heating requirements. In addition, isothermal NAATs have been demonstrated on paper microfluidic platforms.27–29 Furthermore, paper microfluidics have been proven capable of automating multistep assays without external equipment.30 Precise electricity-free heaters have been developed for these applications:24 these are powered by exothermic chemical reactions and use phase change to regulate temperature. Our group has demonstrated the heating of flat-profile paper microfluidic networks using this technique.31,32
These technologies suggest GeneXpert-like molecular diagnostics are possible in low-cost, disposable devices,27,28 though many existing isothermal NAAT-based diagnostics still rely on off-device sample processing steps that will not be available in all settings. Fortunately, technologies are beginning to emerge that enable the operation of molecular diagnostics without the need for laboratory infrastructure.29,33 The multiplexable, autonomous, disposable nucleic acid amplification test (MAD NAAT) project31,34–39 aims to create a comprehensive isothermal NAAT platform that takes a biological sample as input, lyses cells, amplifies nucleic acid sequences from the lysate, and provides visual readout of the assay results. Non-electric heating options allow for regulated, elevated temperatures optimal for bioassays in environments without the need for external electrical power or batteries.25,31,32,40,41 A rapid, low-cost NAAT amenable to point-of-care diagnostics in lower-resource settings could significantly expedite identification of pathogens in people, food, and drinking water.
Here we demonstrate rapid lysis of a suspension of S. aureus using dry-stored ACP, followed by a rapid (<5 minutes) electricity-free heat deactivation step, the output of which can be tested directly using quantitative PCR (qPCR). In addition to lysis of S. aureus suspended in Tris buffer, we also demonstrate direct qPCR amplification of lysed S. aureus suspended in a human nasal sample. Further, human respiratory syncytial virus (RSV) virions suspended in human nasal sample are heat-treated without ACP and direct reverse transcriptase PCR (RT-PCR) used to quantify released RNA. In addition to the applications demonstrated, this methodology is easily adaptable to any assay that requires a temperature step at ∼100 °C.
The chemical heaters described here were produced from common laboratory supplies with minimal tooling and production steps and could be incorporated into more complex multi-step devices for sample pretreatment prior to a bioassay. This approach, when combined with advances in point-of-care nucleic acid amplification assays, could enable sample-to-result nucleic acid detection in lower-resource settings.
Materials and methods
Bacterial culture
Staphylococcus aureus (strain RN4220 obtained from the Ferric Fang laboratory at the University of Washington) was cultured in Tryptic Soy Broth (BD Bacto, Sparks, MD, USA) at 37 °C, shaking at 250 rpm. Overnight cultures were diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 in fresh Tryptic Soy Broth and grown to mid-log phase (OD600 = ∼2). Cells were spun down at 10000 g for 3 minutes at 20 °C after growth and resuspended in the same volume of Tris buffer (10 mM Tris, pH 8.0) or Tris–EDTA (TE: 10 mM Tris–HCl, 1 mM EDTA, pH 8.0) buffer. Cells dilutions used the same buffer.
:100 in fresh Tryptic Soy Broth and grown to mid-log phase (OD600 = ∼2). Cells were spun down at 10000 g for 3 minutes at 20 °C after growth and resuspended in the same volume of Tris buffer (10 mM Tris, pH 8.0) or Tris–EDTA (TE: 10 mM Tris–HCl, 1 mM EDTA, pH 8.0) buffer. Cells dilutions used the same buffer.
Virus source and preparation
Human respiratory syncytial virus (RSV, laboratory strain obtained from the University of Washington Clinical Virology Laboratory) was cultured by the UW clinical virology laboratory. Virions were aliquoted at 106 RSV copies per microliter and stored at −80 °C.ACP lysis
S. aureus cell suspensions (OD600 = 2) were diluted in Tris or TE 1:1000 to ∼106 cfu mL−1. The cell suspension was added to tubes pre-loaded with ACP (20 U μL−1 stock, Sigma A3547) to a 3 U μL−1 (Lot 041M1380V) or 0.5 U μL−1 (Lot 031M1468V) final ACP concentration and mixed by gently pipetting up and down. Due to lot-to-lot variation of the ACP, 3 U μL−1 or 0.5 U μL−1 concentrations were used. The reaction was incubated at room temperature (20 °C) or 37 °C. ACP was deactivated by placing the tubes in a heating block (for experiments varying the heat deactivation time and temperature) or in the chemical heater (thermal profiles shown in Fig. 3). Lysate tubes were briefly spun at 3000 g to pull condensation down from the tube walls before conducting qPCR.
ACP mixture dehydration
In a 0.2 mL PCR tube, 37% trehalose (TS1M-100, Life Sciences Advanced Technologies, St Petersburg, FL, USA) and 20 U μL−1 ACP were mixed together for a final trehalose concentration of 5% in the mixture and a final ACP concentration of 3 U μL−1 or 0.5 U μL−1 in the lysate (depending on the lot of ACP). The tubes were dried in a vacuum concentrator (miVac DNA, GeneVac, Stone Ridge, NY, USA) at 20 °C for 1.5 hours, and then stored in a desiccator at 20 °C. For cell lysis, the dried ACP was rehydrated with cell suspension following the protocol for ‘ACP lysis’.Chemical heaters
This type of heater is based on the exothermic reaction of a magnesium–iron alloy (MgFe) with a solution containing sodium chloride (NaCl). This approach is widely used to warm up portable meals42 and is capable of releasing heat very quickly. The chemical heaters feature a tube-in-tube design: the outer heater tube contains the elements necessary for the exothermic reaction; the inner tube contains the dried ACP and sample. The outer heater tube consists of a modified 1.5 mL snap-cap tube (89000-028, VWR, Radnor, PA, USA), separated from the cap, cut to 26 mm height as measured from the conical end, and with a 0.5 mm hole drilled in the side to serve as a vent. The separated cap was drilled with a ¼” hole for insertion of the sample tube. A 3 cm × 3 cm square of paper towel (Kleenex C-fold towels, Kimberly Clark Professional, Roswell, GA) was inserted into the heater tube, followed by 120 mg of the solid MgFe fuel (Luxfer Magtech, Cincinnati, OH, USA), 70 mg of NaCl, and a portion of cotton ball (100% cotton, Kroger, Cincinnati, OH, USA). The modified heater tube was then fitted with the modified cap and pressed into a salvaged section of a Styrofoam shipping container. The heater was activated by adding 300 μL of deionized water through the ¼” hole, into which the sample tube (981005, Qiagen, Hilden, Germany) was then inserted. Caution should be observed: the MgFe reaction reaches 100 °C quickly and produces hydrogen and steam, which should vent from the vent hole. Venting is critical to prevent pressure build-up and sample tube ejection. Sample temperature was measured with a type T needle thermocouple inserted into a hole drilled in the sample tube cap, recording temperature data over time with a data acquisition system (OMB-DAQ-54, Omega Engineering, Stamford, CT, USA).Bead beater
Samples of S. aureus cell suspension (800 μL) were added to 2 mL O-ring screw top tubes (02-682-558, Thermo Fisher Scientific, Waltham, MA, USA) with 800 mg beads (9830, Research Products International Corp., Mt. Prospect, IL, USA). Tubes were loaded into the bead beater (Mini-Beadbeater-8, Biospec Products, Inc., Bartlesville, OK, USA), set to “homogenize”, and run for three 1 minute cycles with a 1 minute pause between cycles.qPCR
For quantification of S. aureus DNA, a commercially available qPCR kit (ELITech Group, Bothell, WA, USA) was used. Samples (2 μL) from the bead beater or ACP lysate were used in 20 μL qPCR reactions (Rotor-Gene Q, Qiagen, Valencia, CA, USA or CFX96 Touch, Bio-Rad, Hercules, CA, USA) using: 50 °C hold for 2 minutes, 93 °C hold for 2 minutes, 45 cycles of 93 °C for 10 seconds, 56 °C for 30 seconds, and 72 °C for 15 seconds, ending with a final elongation step at 72 °C for 5 minutes. Fluorescence data were collected during the 56 °C step using the orange channel. Genomic DNA copy numbers were determined relative to standard curve analysis using purified DNA of known copy number using the qPCR device software. The assay was sensitive down to ∼10 copies of the target sequence.qRT-PCR
For quantification of RSV RNA, UltraSense quantitative RT-PCR assay mix (Life Technologies, Carlsbad, CA, USA) was used with primer and probe sequences published previously.43 The thermal protocol used was: 50 °C hold for 15 minutes, 95 °C hold for 2 minutes, 40 cycles of 95 °C for 15 seconds and 60 °C for 55 seconds using 20 μL reactions on a CFX96 Touch (Bio-Rad). Genomic RNA copy numbers were determined relative to standard curve analysis using control RSV RNA (American Type Culture Collection (ATCC), Manassas, VA, USA) of known copy number.Clinical sample collection
Clinical samples were collected from patients following signed parental informed consent from January to March 2015 from pediatric patients with suspected influenza infection. Nylon flocked mid-nasal swabs were collected in the hospital or emergency department for influenza and RSV testing. Each swab was placed into 750 μL of phosphate buffered saline, 0.05% Tween-20, and 0.01% sodium azide. The diluted nasal swab sample was used for RT-PCR detection of influenza or RSV. Samples that were negative for either influenza or RSV were spiked with laboratory strains of RSV and MSSA to determine the impact of the sample composition on direct amplification assays. Seattle Children's Hospital Institutional Review Board approved the sample collection and analysis of specimens. Written consent was obtained from a parent or legal guardian, as approved by the Seattle Children's Institutional Review Board, with paper copies given to parent and also maintained under secure storage by the research team.Statistics
A Student's t-test was performed comparing 5- and 15 minute ACP data for 20 and 37 °C (Fig. 2A) in Excel (Microsoft, Redmond, WA, USA), comparing temperatures ≤70 °C to 80–99 °C (Fig. 2B), comparing fresh and dried ACP results with 105S. aureus cells, comparing performance of the thermal cycler and chemical heater for Tris buffer samples (Fig. 4B), and comparing chemical heater and thermal cycler results for RNA recovery in Tris buffer and human nasal sample matrix (Fig. 5). Two-tailed comparisons were used, assuming the data were homoscedastic. | ||
| Fig. 1 Device schematic. (A) Exploded view. The paper towel was inserted into the heater tube, followed by the MgFe, NaCl, and cotton. Water was added to activate heating, after which the sample tube is inserted. (B) Assembled sectional view. The MgFe corrosion reaction heats the sample tube. Hot gasses exit the vent upon device activation. | ||
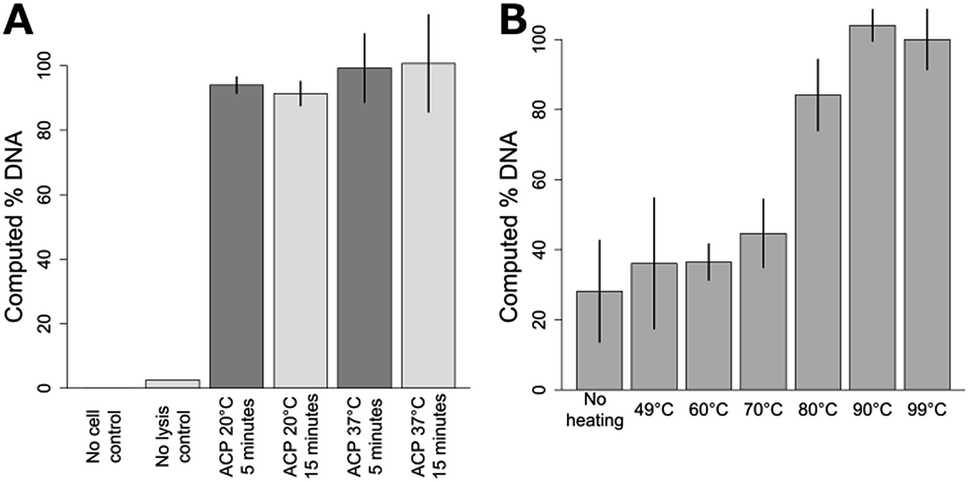 | ||
| Fig. 2 Optimization of ACP enzymatic lysis and heat-deactivation. (A) Influence of varying ACP lysis time and temperature on DNA recovery. All ACP lysis conditions resulted in similar DNA recovery (p > 0.05). The plotted data is the mean ± one standard deviation, with 37 °C ACP 15 minute lysis set to 100%, n = 3 for ACP lysis conditions, n = 2 for no cell and no lysis controls. (B) Comparison of heat deactivation temperatures for two minute ACP lysis at room temperature. Deactivation temperatures below 80 °C reduced performance (p < 0.0001). Data points are mean ± one standard deviation, n = 3. Data was normalized to the 99 °C data, which was set to 100%. | ||
 | ||
| Fig. 3 Chemical heater performance in varied ambient conditions. The chemical heater was run either in laboratory conditions (20 °C) or in a cold room (4 °C). The chemical heater was designed with enough MgFe fuel and NaCl to perform well even in cold ambient conditions. Solid lines are means, dotted lines are ± one standard deviation, n = 3. | ||
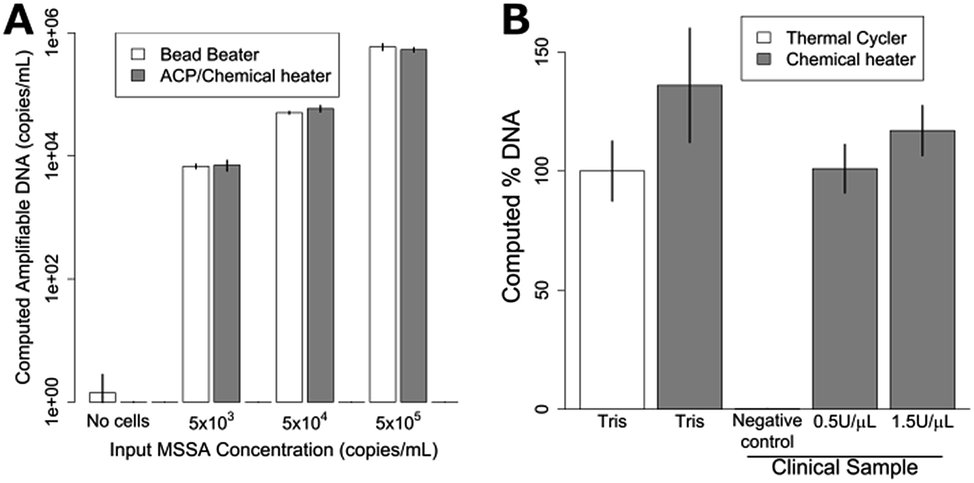 | ||
| Fig. 4 Lysis of MSSA with dry ACP and chemical heat deactivation in the device of Fig. 1. (A) Lysis of various concentrations of MSSA in Tris buffer. Bead beater DNA recovery is compared to dried ACP lysis at room temperature using a chemical heater for enzyme deactivation. The chemically-heated devices perform just as well as the bead beater for all concentrations tested. Data is mean ± one standard deviation, n = 3. (B) MSSA lysis using a conventional heater (thermal cycler) and chemical heat deactivation for samples in buffer and in human nasal sample matrix. Chemical heaters perform similarly (p = 0.08) to thermal cycler-heated tubes for Tris samples. MSSA added to human nasal sample matrix was also successfully amplified, with no significant difference compared to the Tris buffer thermal cycler-heated data. The negative control shows DNA recovery when no MSSA is spiked in the clinical sample. The data were normalized to the Tris thermal cycler, which was set to 100%. Plotted is the mean ± one standard deviation, n = 3. | ||
 | ||
| Fig. 5 Lysis of RSV in Tris and human nasal sample matrix with thermal cycler and chemical heater heat deactivation. Equal concentrations of RSV in Tris buffer or human nasal sample matrix were lysed for 5 minutes then analyzed by qRT-PCR. Output is reported by percent amplifiable RNA, with thermal cycler data set to 100%. Data is mean ± one standard deviation, n = 6 for Tris buffer, n = 5 for clinical sample. | ||
Results and discussion
As previously mentioned, NAATs that employ ACP lysis operated at 37 °C for 10–20 minutes, followed by heat deactivation at a controlled temperature, have been demonstrated previously. Here, we compare ACP lysis at 20 and 37 °C, for 5 and 15 minute incubations, followed by heat deactivation at 99 °C. As shown in Fig. 2A, the amount of recoverable DNA from S. aureus cells was similar for all conditions tested (p > 0.05). No DNA was detected for either the fresh or dried ACP no-cell control. Fig. 2B shows performance is dependent on the heat deactivation temperature, with temperatures less than 80 °C performing less well (p < 0.0001). Lot 031M1468v ACP only recovered 57% of the amplifiable DNA compared to lot 041M1380v at 3 U μL−1 (activity units were reported by the manufacturer, p = 0.005.) When lot 031M1468v was used at 0.5 U μL−1, the resulting lysis performance was similar to 041M1380v at 3 U μL−1 (p = 0.10). No difference in lysis efficiency was observed lysing cells in Tris or TE buffer (p = 0.45). Heat deactivation of ACP was also found to be robust, with times from one to ten minutes leading to similar performance.Based on these data, a chemical heater was designed to heat the sample to over 80 °C for over one minute from a range of initial ambient temperatures. Shown in Fig. 3 are temperatures measured in the lysis tube for a heater run either in our laboratory or cold room, with time–temperature profiles adequate for ACP deactivation in both cases. As shown in Fig. 3A, the chemical heater reliably exceeds 80 °C, and thereby deactivates the ACP enzymes in preparation for nucleic acid amplification, even in cold ambient conditions. The heater can be expected to reliably heat the samples in warmer ambient conditions due to the nature of the exothermic reaction.
Next, the enzymatic mixture was dried down into a form that can be stored easily and that can be rehydrated by the addition of the cell suspension. Fresh and dry ACP performs similarly at room temperature with a 5 minute 95 °C deactivation step for 105S. aureus cells (p = 0.16.). No DNA was detected for either the fresh or dried ACP no-cell control. This dried ACP does not lose activity when stored for 4 months compared to fresh ACP (p = 0.23).
For lysis with dried ACP and the chemical heater, the device shown in Fig. 1 was constructed by combining the separately-characterized chemical heater and dry ACP tube. S. aureus cells were lysed in the integrated device and the lysate was directly added to a PCR reaction for quantification of the ldh1 gene. Mechanical cell disruption is effective for difficult-to-lyse microorganisms,17 bead beating was therefore chosen as a technique for comparison with the prototype devices. Fig. 4A shows similar performance of the bead beater and the integrated device for each cell concentration tested. Fig. 4B reports the amount of amplifiable DNA recovered using ACP lysis with methicillin susceptible S. aureus (MSSA) in Tris buffer with chemical heat deactivation or thermal cycler, and also chemical heat deactivation for MSSA spiked into a patient nasal sample. Here, chemical-heater-powered deactivation of ACP performs similarly to deactivation using the thermal cycler (p = 0.16). The negative control shows very little target DNA is recovered when no MSSA is spiked into the patient sample. 1.5 U μL−1 ACP concentration was tested in addition to 0.5 U μL−1, to check whether higher concentrations of ACP would be beneficial to account for the additional complexity of the human nasal sample matrix.
With the chemical heater performing well for ACP lysis of bacteria, we wondered if it could also lyse RNA viruses. We selected RSV, an enveloped virus that contains a single segmented 15 kb genomic RNA fragment per virion.44 We compared lysis of RSV virions in Tris buffer or in an RSV-negative human nasal sample matrix by a thermal cycler to chemical heaters. The thermal cycler and chemical heater performed nearly identically when RSV was in Tris buffer (p = 0.94, Fig. 5). When RSV was spiked at a known concentration into a clinical sample that previously tested negative, the thermal cycler and chemical heater performed similarly as well (p = 0.62, Fig. 5). Overall, we conclude that the chemical heater is also an effective tool for RSV lysis.
Conclusions
Here we have demonstrated effective, rapid nucleic acid extraction from S. aureus and RSV. The method works in the presence of a clinically-obtained human nasal sample. S. aureus is a relatively hard-to-lyse Gram-positive pathogen, so these results highlight the robustness of the lysis method. RSV is a common childhood RNA virus, showing one potential application of the lysis method to a clinically-relevant diagnostic application. This method uses dried reagents compatible with non-refrigerated storage and a disposable chemical heat source for enzyme deactivation. No additional infrastructure or external processing was required before nucleic acid amplification. The total list price for the consumables (Eppendorf tubes, paper towel, cotton balls, NaCl, MgFe) used to construct the prototype heaters is less than $0.41. Purchasing these items at larger quantities will likely reduce the cost. The only tools used to build the prototype heater were a razor blade, a drill press, drill bits, a ruler, and a scale to weigh the components. In addition to being a laboratory tool that many could build themselves, it would be straightforward for a manufacturer to make modified tubes for this type of application.This work was performed with low-resource clinical settings in mind; however, this method is a generic tool compatible with any process (lysis or not) that requires heat. Such processes include the use of proteinase K or NaOH for relatively quick cell lysis in limited-resource laboratory settings, animal facilities, or for environmental sample analysis. Heat alone is sufficient to lyse many organisms,45 to perform heat-shock antigen–antibody dissociation,46 or to denature nucleic acid complexes or proteins. This method is ideal for thermal lysis47 of E. coli or other organisms that lyse at elevated temperature, which could enable quick screening of plasmid cultures.
We expect that this method and other sample preparation techniques in development by our group34,38,39 will help enable the expansion of the next generation of point-of-care diagnostics assays to areas without access to traditional diagnostic infrastructure.
Acknowledgements
Buser, Zhang, Byrnes, Heiniger, Wheeler, Bishop, Lutz, Weigl, and Yager were supported by DARPA DSO/BTO HR0011-11-2-0007, awarded to Yager. Ladd was supported by NIH 1 R01 AI 096184-01 awarded to Yager. We would like to acknowledge Anne Cent of the molecular virology laboratory for providing a laboratory RSV virus strain for experimentation, along with the team at Seattle Children's who collected the patient nasal samples, including Kirsten Lacombe, Bonnie Strelitz, Alastair Murray, and Catherine Bull, as well as all of the patients and their families who participated in the study. We would also like to acknowledge Traci Kinkel from the Ferric Fang lab at the University of Washington for providing a laboratory strain of Staphylococcus aureus. We wish to thank Shichu Huang, Tinny Liang, both of the University of Washington and Elain Fu, now at Oregon State University, for their help in obtaining patient samples. We would also like to thank Sujatha Ramachandran for the helpful guidance regarding ACP dry storage. We thank everyone in the Yager and Lutz labs, and collaborators at UW, PATH, GE Global Research, and the ELITech Group for the support and feedback.Notes and references
- R. Lozano, et al. , Lancet, 2013, 380, 2095–2128 CrossRef.
- H. Wang, L. Dwyer-Lindgren, K. T. Lofgren, J. K. Rajaratnam, J. R. Marcus, A. Levin-Rector, C. E. Levitz, A. D. Lopez and C. J. L. Murray, Lancet, 2012, 380, 2071–2094 CrossRef.
- C. J. L. Murray, et al. , Lancet, 2012, 380, 2197–2223 CrossRef.
- R. W. Peeling, D. Mabey, A. Herring and E. W. Hook, Nat. Rev. Microbiol., 2006, 4, S7–S19 CrossRef PubMed.
- D. Litaker, S. M. Koroukian and T. E. Love, Med. Care, 2005, 43, 531–540 CrossRef PubMed.
- S. Ramani and G. Kang, Curr. Opin. Infect. Dis., 2009, 22, 477–482 CrossRef PubMed.
- D. R. Hill and N. J. Beeching, Curr. Opin. Infect. Dis., 2010, 23, 481–487 CrossRef PubMed.
- Diarrhea Causes – Mayo Clinic, http://www.mayoclinic.org/diseases-conditions/diarrhea/basics/causes/con-200140258, accessed January 2016.
- K. Hodges and R. Gill, Gut Microbes, 2010, 1, 4–21 CrossRef PubMed.
- WHO, Antimicrobial Resistance Fact Sheet, 2015 Search PubMed.
- M. Z. David and R. S. Daum, Clin. Microbiol. Rev., 2010, 23, 616–687 CrossRef CAS PubMed.
- M. A. Borg, M. de Kraker, E. Scicluna, N. van de Sande-Bruinsma, E. Tiemersma, J. Monen and H. Grundmann, J. Antimicrob. Chemother., 2007, 60, 1310–1315 CrossRef CAS PubMed.
- C. C. Boehme, et al. , Lancet, 2011, 377, 1495–1505 CrossRef.
- A. Pantoja, C. Fitzpatrick, A. Vassall, K. Weyer and K. Floyd, Eur. Respir. J., 2013, 42, 708–720 CrossRef PubMed.
- UNITAID, Tuberculosis: Diagnostic Technology Landscape, World Health Organization, 2012 Search PubMed.
- R. Mariella, Biomed. Microdevices, 2008, 10, 777–784 CrossRef PubMed.
- A. Niemz, T. M. Ferguson and D. S. Boyle, Trends Biotechnol., 2011, 29, 240–250 CrossRef CAS PubMed.
- C. M. Kusuma, J. F. Kokai-kun and S. A. Sam, Society, 2005, 49, 3256–3263 CAS.
- D. Voet, J. G. Voet and C. W. Pratt, Fundamentals of Biochemistry, John Wiley & Sons, Inc, 3rd edn, 2008 Search PubMed.
- O. Salazar and J. a. Asenjo, Biotechnol. Lett., 2007, 29, 985–994 CrossRef CAS PubMed.
- T. Masaki and K. Nakamura, Agric. Biol. Chem., 1978, 42, 1443–1445 CrossRef CAS.
- S. M. Paule, D. M. Hacek, B. Kufner, K. Truchon, R. B. Thomson, K. L. Kaul, A. Robicsek and L. R. Peterson, J. Clin. Microbiol., 2007, 45, 2993–2998 CrossRef CAS PubMed.
- N. Kobayashi, H. Wu, K. Kojima, K. Taniguchi, S. Urasawa, N. Uehara, Y. Omizu, Y. Kishi, A. Yagihashi and I. Kurokawa, Epidemiol. Infect., 2009, 113, 259 CrossRef.
- P. LaBarre, K. R. Hawkins, J. Gerlach, J. Wilmoth, A. Beddoe, J. Singleton, D. Boyle and B. Weigl, PLoS One, 2011, 6, e19738 CAS.
- J. Singleton, D. Guelig, J. Buser, R. Burton, O. Edeh, K. Hawkins, B. Weigl and P. LaBarre, in IEEE Global Humanitarian Technology Conference (GHTC 2014), IEEE, 2014, pp. 721–725 Search PubMed.
- L. Lillis, D. Lehman, M. C. Singhal, J. Cantera, J. Singleton, P. Labarre, A. Toyama, O. Piepenburg, M. Parker, R. Wood, J. Overbaugh and D. S. Boyle, PLoS One, 2014, 9, e108189 Search PubMed.
- B. A. Rohrman and R. R. Richards-Kortum, Lab Chip, 2012, 12, 3082–3088 RSC.
- J. C. Linnes, A. Fan, N. M. Rodriguez, B. Lemieux, H. Kong and C. M. Klapperich, RSC Adv., 2014, 4, 42245–42251 RSC.
- N. M. Rodriguez, J. C. Linnes, A. Fan, C. K. Ellenson, N. R. Pollock and C. M. Klapperich, Anal. Chem., 2015, 87, 7872–7879 CrossRef CAS PubMed.
- E. Fu, T. Liang, P. Spicar-Mihalic, J. Houghtaling, S. Ramachandran and P. Yager, Anal. Chem., 2012, 84, 4574–4579 CrossRef CAS PubMed.
- J. Singleton, C. Zentner, J. Buser, P. Yager, P. LaBarre and B. H. Weigl, Proc. SPIE, 2013, 8615, 86150R CrossRef PubMed.
- J. R. Buser, S. Diesburg, J. Singleton, D. Guelig, J. D. Bishop, C. Zentner, R. Burton, P. LaBarre, P. Yager and B. H. Weigl, Lab Chip, 2015, 15, 4423–4432 RSC.
- J. R. Choi, J. Hu, R. Tang, Y. Gong, S. Feng, H. Ren, T. Wen, X. Li, W. A. B. Wan Abas, B. Pingguan-Murphy and F. Xu, Lab Chip, 2015, 16, 611–621 RSC.
- N. Panpradist, B. J. Toley, X. Zhang, S. Byrnes, J. R. Buser, J. A. Englund and B. R. Lutz, PLoS One, 2014, 9, e105786 Search PubMed.
- S. Dharmaraja, L. Lafleur, S. Byrnes, P. Kauffman, J. Buser, B. Toley, E. Fu, P. Yager and B. Lutz, Proc. SPIE, 2013, 8615, 86150X CrossRef.
- B. J. Toley, B. McKenzie, T. Liang, J. R. Buser, P. Yager and E. Fu, Anal. Chem., 2013, 85, 11545–11552 CrossRef CAS PubMed.
- B. J. Toley, J. A. Wang, M. Gupta, J. R. Buser, L. K. Lafleur, B. R. Lutz, E. Fu and P. Yager, Lab Chip, 2015, 15, 1432–1444 RSC.
- J. R. Buser, A. Wollen, E. K. Heiniger, S. Byrnes, P. C. Kauffman, P. D. Ladd and P. Yager, Lab Chip, 2015, 15, 1994–1997 RSC.
- S. A. Byrnes, J. D. Bishop, L. Lafleur, J. R. Buser, B. Lutz and P. Yager, Lab Chip, 2015, 15, 2647–2659 RSC.
- K. G. Shah, D. Guelig, S. Diesburg, J. Buser, R. Burton, P. LaBarre, R. Richards-Kortum and B. Weigl, PLoS One, 2015, 10, e0139449 Search PubMed.
- C. Liu, M. G. Mauk, R. Hart, X. Qiu and H. H. Bau, Lab Chip, 2011, 11, 2686–2692 RSC.
- MREInfo.com – Home, http://www.mreinfo.com, accessed September 2015.
- J. Kuypers, N. Wright and R. Morrow, J. Clin. Virol., 2004, 31, 123–129 CrossRef CAS PubMed.
- V. M. Cowton, D. R. McGivern and R. Fearns, J. Gen. Virol., 2006, 87, 1805–1821 CrossRef CAS PubMed.
- N. Casali and A. Preston, E. coli plasmid vectors: Methods and applications, Springer Science and Business Media, 2003, vol. 235 Search PubMed.
- J. Schüpbach and J. Böni, J. Virol. Methods, 1993, 43, 247–256 CrossRef.
- J. Kim, M. Johnson, P. Hill and B. K. Gale, Integr. Biol., 2009, 1, 574–586 RSC.
Footnote |
| † BHW now at Intellectual Ventures Laboratory, Bellevue, WA, United States of America. |
| This journal is © The Royal Society of Chemistry 2016 |