
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceApplication of dynamic headspace and gas chromatography coupled to mass spectrometry (DHS-GC-MS) for the determination of oxygenated volatile organic compounds in refinery effluents†
Grzegorz
Boczkaj
*a,
Patrycja
Makoś
a and
Andrzej
Przyjazny
b
aDepartment of Chemical and Process Engineering, Chemical Faculty, Gdansk University of Technology, 11/12 G. Narutowicza St., 80-233 Gdansk, Poland. E-mail: grzegorz.boczkaj@gmail.com; Fax: +48 58 347 26 94; Tel: +48 697970303
bKettering University, 1700 University Avenue, Flint, MI 48504, USA
First published on 23rd March 2016
Abstract
The paper presents a new procedure for the determination of oxygenated volatile organic compounds (O-VOCs) in postoxidative effluents from the production of petroleum asphalt using dynamic headspace coupled to gas chromatography-mass spectrometry in the selected ion monitoring (SIM) mode (DHS-GC-MS). Among the GC capillary columns tested, a polar SLB-IL111 column with the ionic liquid stationary phase was found to be superior due to its high selectivity for n-alkanes and individual oxygenated volatile organic compounds. The low detection limit, good reproducibility and a wide linear range allows determination of O-VOCs at low concentration levels and applicability of the procedure to routine analyses of O-VOCs in industrial effluents with a very complex composition. The developed procedure was used for the analysis of real samples – raw effluents from the production of bitumens and effluents treated chemically through oxidation. Thirteen compounds at concentrations ranging from 0.01 μg mL−1 to 118.61 μg mL−1 were identified in the effluents. In addition, nine more compounds, mostly alcohols, aldehydes and ketones, were identified using the SCAN mode. The paper demonstrates the need for monitoring O-VOCs in processes of chemical treatment of effluents. Due to the pathways of oxidation of organic pollutants present in effluents, O-VOCs become secondary pollutants. A substantial increase in the concentration of some groups of compounds, i.e. phenol and its derivatives and aliphatic and cyclic alcohols, was found in oxidized effluents. The presence of these compounds has a negative effect on the activated sludge used in refinery wastewater treatment plants.
1 Introduction
Volatile organic compounds (VOCs) and oxygenated volatile organic compounds (O-VOCs) are considered to be priority pollutants of the atmosphere and aquatic environment. They are believed to have carcinogenic and mutagenic properties1,2 and a high degree of ecotoxicity and to play a significant role in the formation of secondary air pollutants – tropospheric ozone.3,4 In the European Union countries natural sources constitute only 20% of VOC emissions,5 the rest being of anthropogenic nature, such as emissions by the petrochemical industry, including the production of bitumens. During the production of bitumens, VOCs are formed at every stage of processing of crude oil, such as vacuum distillation or bitumen production. They can also be released from the final bitumen products and from the postoxidative effluents. More stringent environmental regulations imposed on refineries have resulted in a growing number of analytical procedures allowing a detailed identification of individual groups of compounds emitted to the atmosphere. These groups include mainly polycyclic aromatic hydrocarbons (PAHs),6 volatile organosulfur compounds (VSCs),7 volatile organonitrogen compounds (VNCs), aromatic and aliphatic hydrocarbons and oxygenated volatile organic compounds (O-VOCs).8–15Oxygenated volatile organic compounds, including alcohols, aldehydes, ketones, phenols, esters, ethers, carboxylic acids and their derivatives, are highly malodorous and toxic. Their identification in aqueous samples with a highly complex matrix poses numerous problems even at the sample collection step due to their hydrophobic nature, high reactivity and volatility of lower members of O-VOCs as well as their trace concentrations.16,17 At present, the most common analytical method allowing the determination of O-VOCs at concentration levels down to μg L−1 or even ng L−1 is gas chromatography. However, in order to obtain such low detection limits it is necessary to use an appropriate analyte isolation and/or enrichment technique. To this end, procedures combining gas chromatography with universal detection (flame ionization detection – FID or mass spectrometry – MS) and various extraction methods, i.e. solid-phase extraction (SPE),18 solid-phase microextraction (SPME),19 headspace solid-phase microextraction (HS-SPME),20,21 static headspace (SHS),22 simultaneous steam distillation–extraction (SDE)23 or single-drop microextraction (SDME),24 are used. Alternatively, selective GC detectors, such as the electron capture detector (ECD) or photoionization detector (PID),25–27 can be employed in combination with a derivatization step. The use of high-performance liquid chromatography (HPLC) enables identification of a wide variety of compounds at concentrations down to a μg L−1 level, while capillary electrophoresis and ion or ion-exchange chromatography are limited to identification of phenols and carboxylic acids.28–33 Electrochemical and spectroscopic techniques are also of interest; however, these methods permit only the determination of the total O-VOC content in the analyzed samples.34–39 Profiling of the emission of total VOCs as well as the determination of their boiling range can be accomplished by gas chromatography with flame ionization detection (GC-FID).7,40,41
Currently, due to environmental concerns, for the determination of the VOC content in water and wastewater samples, solvent-free sample preparation techniques are mainly used, which include static and dynamic headspace analysis techniques. For the determination of compounds from a group of VOCs, the fully automated DHS technique allows the determination of analytes at concentrations much lower compared to the static method.42 The same dependency was observed as compared to the SPME technique, for the low molecular weight compounds.43 The DHS coupled to GC-MS method is recommended by the US EPA for the determination of toxic VOCs in wastewater, however among the list of compounds, only 3 compounds belong to the group of O-VOCs, the others are mainly halogenated organic compounds, as well as aromatic hydrocarbons.44 Other examples in the literature, which describes the use of DHS-GC-MS techniques in the analysis of industrial wastewater, are related to the same groups of compounds.45–48
This paper describes a procedure for the determination of oxygenated volatile organic compounds using dynamic headspace coupled to gas chromatography-mass spectrometry (DHS-GC-MS) as a tool for routine control of the O-VOC content in refinery effluents.
2 Materials and methods
2.1 Materials
Please refer to ESI – Section S.1.†2.2 Real samples
Samples of raw postoxidative effluents from the production of petroleum bitumen 20/30 from the vacuum residue of Rebco crude oil, having a strongly basic pH (ca. 10.5), were collected behind the plate separator which separated the condensed organic phase from the aqueous phase. Characteristics of the effluents and conditions of their formation can be found in previous papers.7,11,15 The aqueous phase of the effluents was analyzed using DHS-GC-MS. As an example of the usage of the developed procedure, the following effluent samples from preliminary investigations of chemical degradation of pollutants by various oxidation techniques were analyzed: effluent I − sono-cavitation (induced by sonication) + UV + ozonation + addition of hydrogen peroxide (H2O2), effluent II − ozonation. A SONOREX Technik AQ reactor (Bandelin, Germany) was used in the studies. The reactor allows oxidizing compounds using a sono-cavitation phenomenon which is induced by ultrasounds. The reactor was equipped with a UV lamp. Additional oxidants – ozone and hydrogen peroxide were injected at the inlet along with the introduced effluent. Detailed characteristics of the procedures and discussion of their effectiveness will be presented in a separate paper.2.3 Apparatus
Please refer to ESI – Section S.2.†2.4 Procedure
Chromatographic analysis: two fused silica capillaries were introduced through the septum – one of them reaching the bottom of the vial fed hydrogen purging the solution while the other one transporting the gas with the analytes to the sorbent trap was placed 0.5 cm below the septum (Fig. S1†). The purging process was carried out for 5 min (20 mL min−1) at room temperature (20 °C) (trap temperature 30 °C), followed by desorption for 4 min at 250 °C. The gas flow during desorption was turned on when the trap reached 245 °C. The desorbed analytes were passed through a fused silica transfer line heated to 200 °C directly to the gas chromatograph. DHS-GC-MS analysis conditions: capillary column SLB-IL 111, carrier gas: hydrogen at 1 mL min−1, injection port temperature: 250 °C, ion source temperature (EI, 70 eV) 200 °C, GC-MS transfer line temperature 310 °C; temperature program: 40 °C (5 min) – ramped at 5 °C min−1 – 220 °C (10 min), SCAN mode from a mass-to-charge ratio of 34 to 220 m/z and SIM mode for the m/z values selected for individual compounds.
3 Results and discussion
3.1 Selection of capillary column
In order to optimize the chromatographic separation of 36 O-VOCs, the standard solution was chromatographed using three capillary columns with various polarities. The retention times for individual compounds along with their peak numbers and selectivity factors with respect to the preceding compound and n-nonane are compiled in Tables S1–S3.† This hydrocarbon was selected due to the fact that for the most polar stationary phase (IL-111) lower n-alkanes were eluted at the dead time. A detailed analysis of the obtained results with regard to the selectivity of the studied phases and separation mechanisms is presented in Section S.4.1 (ESI†).The most polar column SLB-IL 111 with the stationary phase being an ionic liquid (1,5-di(2,3-dimethylimidazolium)pentane bis(trifluoromethylsulfonyl)imide) (Fig. 1) had the highest selectivity towards the standard mixture. Coelution was observed only for a few compounds, such as 3-pentanol and 2-pentanone as well as 2,4-dimethyl-3-pentanone and 3-heptanone.
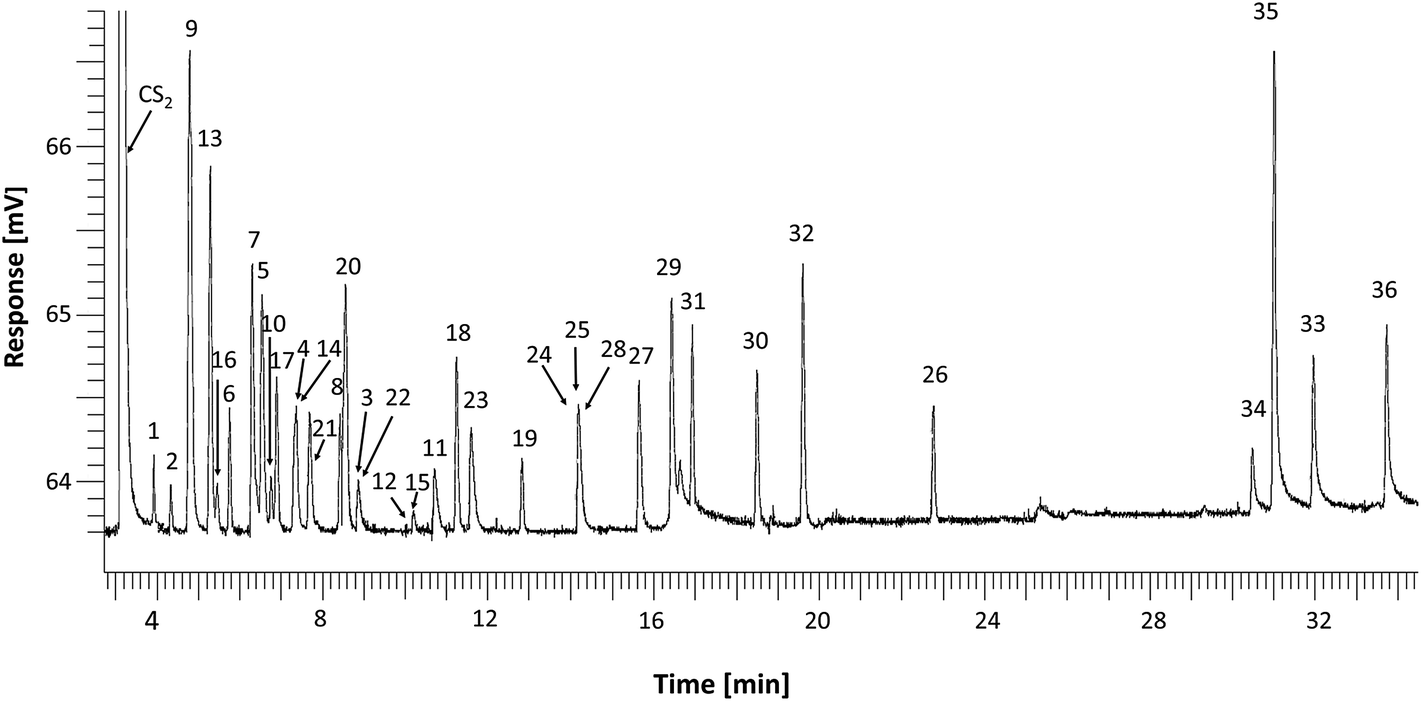 | ||
| Fig. 1 Chromatogram of a mixture of standards of alcohols, ketones, aldehydes, phenols, esters and ethers separated on a SLB-IL 111 column. | ||
On the basis of the results obtained, the SLB-IL111 column with the ionic liquid stationary phase was selected for further work, since it ensures the highest selectivity towards O-VOCs, as demonstrated by the selectivity factors relative to the preceding compound (Fig. S4†). The few coelutions should not interfere with the procedures making use of GC-MS due to substantial differences in mass spectra of the separated compounds and the occurrence of numerous specific fragmentation ions. Furthermore, previous investigations7,11,15 revealed that the samples of postoxidative effluents contain n-alkanes, which under the DHS conditions are released in the n-C5 to n-C9 range while small amounts of n-C10 to n-C13were also present. Under the selected separation conditions n-alkanes up to n-C8 are eluted at the dead time; thus, the use of IL-111 column largely reduces the matrix effect.
3.2 Optimization of the DHS-GC-MS procedure
Dynamic headspace (DHS), also called the purge-and-trap (P&T) technique, was selected due to the complex matrix of effluent samples (containing to a large extent nonvolatile components, a strongly basic pH and a high concentration of sulfides) and the presence of a wide variety of volatile chemical compounds. The sorbent trap was packed with Tenax, a porous polymer recommended by the US Environmental Protection Agency for trapping compounds with a medium to high boiling point, and allowing the determination of volatile organic compounds at a low concentration level (μg mL−1 and ng mL−1).49 The DHS parameters were selected based on US EPA recommendations from standard methods 524.2, 5030a and 8260b as well as our experience in the determination of analytes in high-load industrial effluents.50–52 At the same time, a modified P&T system with a simplified design was used to purge analytes. Due to the difficulty with cleaning standard P&T containers (wall memory effect), they were replaced with disposable 12 mL vials and purging and trapping of the analytes was performed using two fused silica capillary columns (Fig. S1†). Such an approach was successfully used in the investigation of emission of VOCs from hot bitumens by DHS-GC-MS.7 The purging conditions used did not aim at 100% recovery of the analytes due to a large difference in concentrations between raw and treated effluents. Instead they aimed at obtaining satisfactory sensitivity and linear range of the procedure.Optimization of the PT-GC-MS procedure began with the preparation of standard solutions. The analytes were first dissolved in acetone and then diluted with water in a vial. The mixture containing 5 standards was used to optimize injection of the analytes desorbed from the DHS system. The solutions were injected in three modes: split (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), splitless (1.0; 1.2) and splitless (3.8; 4.0). In the splitless modes, opening of the purge valve in the injection port takes place after 1 min and 3.8 min, respectively, and the split valve is opened after 1.2 and 4.0 min, respectively. Based on the signal-to-noise ratio (S/N) (Table S4†), split mode was found to be the optimum injection mode yielding symmetrical, sharp peaks and a lowered noise level. This is due to the fact that in split mode the analytes are removed from the trap using a much higher flow rate of the purge gas, which results in a reduction of precolumn band broadening.
:1), splitless (1.0; 1.2) and splitless (3.8; 4.0). In the splitless modes, opening of the purge valve in the injection port takes place after 1 min and 3.8 min, respectively, and the split valve is opened after 1.2 and 4.0 min, respectively. Based on the signal-to-noise ratio (S/N) (Table S4†), split mode was found to be the optimum injection mode yielding symmetrical, sharp peaks and a lowered noise level. This is due to the fact that in split mode the analytes are removed from the trap using a much higher flow rate of the purge gas, which results in a reduction of precolumn band broadening.
The caustic wastewater from the bitumen production produced during the oxidation of vacuum residue, in many instances, contains a condensed and partly emulsified oil in the aqueous phase.7,11,15 The oil phase removal takes place in the plate separator and additionally using demulsification. The presence of residues of the oil phase is not a problem for a refinery wastewater treatment plant, because the type of activated sludge used in this plant is adapted to the degradation of wastewater containing a high load of hydrocarbons. However, particularly important is the presence of oxygenated volatile organic compounds that are highly toxic to the refinery active sludge. The selection of conditions for the developed method of O-VOC analysis was, therefore, a consequence of the need for the determination of the O-VOCs in a matrix with a high content of hydrocarbons. High selectivity is achieved by using a highly polar ionic liquid stationary phase, which selectively retains the compounds from the group of O-VOCs, while allowing elution with almost no retention of a large spectrum of saturated hydrocarbons. The second “dimension” of selectivity is achieved through the use of a mass spectrometer in selected ion monitoring (SIM) mode.
The experimental LOD values for all the investigated compounds ranged from 0.005 μg mL−1 to 20 μg mL−1. The LOD values for each compound were determined on the basis of signal-to-noise ratio values for the characteristic ion m/zint using (SIM) mode. LOQ values were calculated from eqn (2) (S.3.3).† At the assumed signal-to-noise ratio (6 S/N) for all the analytes, the peak areas of the analytes were reproducible. The lowest detection limits were found for 2,3-dihydropyran, anisole, benzyl alcohol, o-cresol, m-cresol and phenol. On the other hand, the highest LOD values, equal to 20 μg mL−1, were obtained for acetaldehyde due to the high volatility of the analyte. In addition, relative standard deviation (RSD) values were determined for each analyte from three independent analyses for 100 μg mL−1 and found to be below 5%, and also for the LOQ value of individual compounds, whose RSD values were below 5.61%, which confirms the good repeatability of the developed procedure. Considerable differences in LOD values result from the differences in analyte volatility, polarity, water solubility and extent of ionization as well as differences in mass spectra and the selected m/z values at which the determinations are carried out. The LOD and LOQ values demonstrate that the procedure can be used for the determination of low levels of O-VOCs, which makes it suitable for the determination of oxygenated volatile organic compounds in postoxidative effluents.
For each of the analytes a 7-point calibration curve was determined, which was then used for the determination of O-VOCs in effluent samples. Calibration parameters along with the linear range are compiled in Table 1. For 2,3-dihydropyran and tetrahydropyran the upper limit of linearity is 20 μg mL−1 which can make analysis of real samples more difficult. For most of the compounds the upper limits of linearity, equal to 100 μg mL−1, are satisfactory. A statistical data treatment revealed that, for some of the compounds including benzyl alcohol and 2-methyl-2-butanol, the ranges of linearity selected on the basis of the coefficient of determination had errors. For those compounds the R2 values were respectively 0.9998 and 0.9904. In spite of satisfactory correlation coefficient values, the results obtained using the second method, which is described in Section S.3.3† and also using standard residual analysis,53 suggest that the concentrations of these compounds do not show linearity for the assumed lower limit of linearity (0.5 μg mL−1). For both compounds, the real range of linearity is 10–100 μg mL−1.
| No. | Compound | t R [min] | m/zid | m/zint |
|
LOD [μg mL−1] | LOQ [μg mL−1] | RSD(100 ppm) [%] | RSD(LOQ) [%] | Slope | Intercept | Correlation coefficient (R2) | Linear range [μg mL−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Acetaldehyde | 1.29 | 43 | 44 | 0.572 ± 0.011 | 20 | 40 | 4.98 | 5.01 | 492.89 | 494.61 | 0.9998 | 40–100 |
| 2 | Propionaldehyde | 1.38 | 58 | 59 | 4.912 ± 0.282 | 0.5 | 1 | 2.13 | 4.86 | 1506.8 | 1082.4 | 0.9999 | 1–100 |
| 3 | Tetrahydrofuran | 1.54 | 72 | 71 | 1.134 ± 0.041 | 0.5 | 0.1 | 1.69 | 1.97 | 83417 |
2095.2 | 0.9989 | 0.5–100 |
| 4 | 2,3-Dihydropyran | 1.54 | 83 | 84 | 0.390 ± 0.005 | 0.005 | 0.01 | 2.93 | 2.84 | 4166.6 | 2872.5 | 0.9944 | 0.5–20 |
| 5 | Tetrahydropyran | 1.69 | 85 | 86 | 1.112 ± 0.022 | 0.05 | 0.1 | 3.02 | 2.98 | 88993 |
1697.6 | 0.9996 | 0.5–20 |
| 6 | Ethyl acetate | 1.69 | 61 | 88 | 2.193 ± 0.063 | 0.05 | 0.1 | 3.29 | 3.99 | 95237 |
3681.8 | 0.9966 | 1–100 |
| 7 | Methyl acrylate | 1.85 | 85 | 68 | 5.321 ± 0.342 | 0.05 | 0.1 | 3.78 | 4.14 | 87289 |
42592 |
0.9964 | 10–100 |
| 8 | 2-Butanol | 2.06 | 74 | 71 | 1.151 ± 0.034 | 0.05 | 0.1 | 0.97 | 3.97 | 4296 | 1277.8 | 0.9971 | 0.5–100 |
| 9 | 2-Methyl-2-butanol | 2.13 | 73 | 59 | 0.571 ± 0.011 | 0.5 | 1 | 3.17 | 4.36 | 1831.2 | 3494.6 | 0.9998 | 1–100 |
| 10 | Ethyl propionate | 2.13 | 102 | 74 | 1.230 ± 0.020 | 0.5 | 1 | 3.65 | 3.97 | 19328 |
1284.7 | 0.9915 | 10–100 |
| 11 | 2-Butanone | 2.22 | 72 | 43 | 0.254 ± 0.011 | 0.5 | 1 | 3.14 | 4.38 | 11392 |
858.31 | 0.9997 | 1–100 |
| 12 | Ethyl acrylate | 2.29 | 85 | 82 | 0.963 ± 0.052 | 0.05 | 0.1 | 4.14 | 4.26 | 89733 |
38846 |
0.9995 | 10–100 |
| 13 | 3-Methyl-2-butanone | 2.47 | 86 | 71 | 3.524 ± 0.131 | 0.5 | 1 | 4.25 | 4.64 | 10167 |
877.15 | 0.9994 | 1–100 |
| 14 | 2-Methyl-1-propanol | 2.47 | 74 | 55 | 1.421 ± 0.022 | 0.05 | 0.1 | 1.87 | 2.22 | 9103.2 | 1436.7 | 0.9904 | 10–100 |
| 15 | Isobutyl acetate | 2.65 | 73 | 86 | 3.272 ± 0.170 | 0.5 | 1 | 3.89 | 4.11 | 81066 |
21096 |
0.9969 | 1–100 |
| 16 | 1-Propanol | 2.65 | 59 | 60 | 1.651 ± 0.044 | 0.5 | 1 | 3.16 | 4.23 | 1597.2 | 3492.1 | 0.9930 | 1–100 |
| 17 | Paraldehyde | 2.65 | 117 | 89 | 0.483 ± 0.004 | 0.5 | 1 | 4.68 | 5.18 | 1115 | 1092 | 0.9996 | 20–100 |
| 18 | 3-Pentanol | 3.05 | 86 | 73 | 0.960 ± 0.011 | 0.5 | 1 | 4.89 | 5.04 | 2302.9 | 1002.9 | 0.9990 | 10–100 |
| 19 | 2-Pentanone | 3.05 | 86 | 71 | 2.123 ± 0.022 | 0.05 | 0.1 | 2.88 | 3.76 | 47987 |
1215.8 | 0.9999 | 1–100 |
| 20 | 1-Butanol | 3.22 | 71 | 73 | 1.944 ± 0.033 | 0.5 | 1 | 2.65 | 3.89 | 1811.3 | 1534 | 0.9987 | 10–100 |
| 21 | 3-Methyl-1-butanol | 4.52 | 70 | 55 | 0.714 ± 0.012 | 0.5 | 1 | 3.01 | 3.46 | 1159.8 | 1235.4 | 0.9982 | 1–100 |
| 22 | 2-Hexanone | 4.94 | 100 | 85 | 1.253 ± 0.013 | 0.05 | 0.1 | 2.32 | 2.97 | 13808 |
24207 |
0.9909 | 0.5–100 |
| 23 | 1-Pentanol | 5.43 | 70 | 55 | 0.781 ± 0.011 | 0.5 | 1 | 2.13 | 3.35 | 3364.1 | 1539.4 | 0.9997 | 10–100 |
| 24 | 3-Heptanone | 6.54 | 85 | 72 | 1.511 ± 0.022 | 0.05 | 0.1 | 2.65 | 4.67 | 12315 |
22281 |
0.9961 | 0.5–100 |
| 25 | 2,4-Dimethyl-3-pentanone | 6.56 | 85 | 114 | 2.584 ± 0.090 | 0.5 | 1 | 3.28 | 2.87 | 10981 |
1088.1 | 0.9999 | 1–100 |
| 26 | 1-Hexanol | 8.07 | 84 | 57 | 0.731 ± 0.021 | 0.5 | 1 | 3.16 | 3.47 | 5356.8 | 9334.9 | 0.9998 | 1–100 |
| 27 | Cyclopentanone | 8.71 | 57 | 84 | 1.45 ± 0.06 | 0.5 | 1 | 2.78 | 2.18 | 12953 |
13239 |
0.9938 | 1–100 |
| 28 | Cyclohexanol | 9.94 | 82 | 57 | 0.461 ± 0.010 | 0.5 | 1 | 2.55 | 4.12 | 1096.2 | 1357.1 | 0.9999 | 1–100 |
| 29 | Anisole | 10.32 | 108 | 78 | 1.381 ± 0.042 | 0.005 | 0.01 | 1.87 | 1.48 | 74374 |
29073 |
0.9993 | 10–100 |
| 30 | Cyclohexanone | 11.35 | 55 | 98 | 2.101 ± 0.073 | 0.05 | 0.1 | 5.00 | 4.22 | 14868 |
23805 |
0.9905 | 0.5–100 |
| 31 | 3-Methylcyclohexanone | 12.49 | 112 | 55 | 0.314 ± 0.004 | 0.05 | 0.1 | 4.54 | 3.98 | 9575.7 | 6786.3 | 0.9953 | 1–100 |
| 32 | Furfural | 15.23 | 95 | 96 | 0.974 ± 0.013 | 0.05 | 0.1 | 2.75 | 3.26 | 4142.9 | 300.42 | 0.9987 | 1–100 |
| 33 | Benzyl alcohol | 22.4 | 108 | 107 | 1.292 ± 0.032 | 0.005 | 0.01 | 4.41 | 5.61 | 1481.9 | 1295.6 | 0.9998 | 10–100 |
| 34 | o-Cresol | 22.98 | 108 | 107 | 1.021 ± 0.022 | 0.005 | 0.01 | 3.40 | 4.01 | 2974.4 | 5176.1 | 0.9957 | 0.5–100 |
| 35 | Phenol | 23.55 | 94 | 66 | 2.741 ± 0.113 | 0.005 | 0.01 | 4.64 | 5.08 | 2231.2 | 6439.7 | 0.9956 | 0.5–100 |
| 36 | m-Cresol | 25.28 | 108 | 107 | 1.054 ± 0.025 | 0.005 | 0.01 | 3.43 | 4.25 | 2059.5 | 3380.3 | 0.9952 | 0.5–100 |
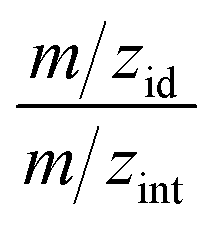
The DHS technique owes its popularity to the significant increase of the method sensitivity.54,55 Enrichment, based on trapping the removed analytes from the samples, in a stream of inert gas, which is fed to the sample in a highly dispersed form, allows in many cases the use of relatively simple apparatus at the final stage of determination. The degree of recovery, which is characterized by the DHS technique, generally ranges between 91 and 105%. In this paper, the recovery of the analytes was not studied, due to the fact that “typical” conditions of DHS, standard apparatus and sorption traps which meet the requirements of most EPA procedures regarding DHS techniques were used. The main objective of this study was to develop a method enabling the routine determination of analytes in a wide range of industrial wastewater with complex matrices. A confirmation of the linear relationship, in the desired concentration range, between the response of the analytical system with the MS detector in SIM mode and the analyte concentration in a sample of wastewater was considered as sufficient confirmation of the method ability to generate accurate and reproducible results of determination. In this case, the degree of repeatability of recovery by gas extraction, which is confirmed by low RSD values for the studied concentration ranges of calibration mixtures, is a confirmation of the quality of results at an acceptable level for routine analysis of industrial wastewater. At the same time, the conditions used do not have to provide one hundred percent recovery of the analytes by purging the sample, but should only allow obtaining satisfactory sensitivity at the shortest possible time of gas extraction.
3.3 Determination of O-VOCs in postoxidative effluents
In the first stage of investigations, O-VOCs were identified in samples of raw effluents using DHS-GC-MS in selected ion monitoring (SIM) mode. The retention data determined during the optimization of separation conditions and the mass spectra of reference substances were used to construct the sequence of detection conditions in the selective ion monitoring mode. For the range of retention time values of each reference substance, the settings of the mass spectrometer have been selected, for ions with characteristic mass-to-charge ratio values for each compound (m/zint – value at which the peak was integrated and m/zid – value used to confirm identification based on the ratio of intensities for m/zint and m/zid). For the confirmation of identification, the tolerance intervals ±0.2% were assumed for the retention time, and ±10% for the ratio of intensities for m/zint and m/zid. The values of the ratio of ion intensities for standard compounds are compiled in Table 1. In this way “double” identification was ensured, based on the determination of the presence of analytes using the retention time and the confirmation of identification with the m/zint to m/zid ratio.This resulted in identification of 13 O-VOCs in samples of raw effluents, for which the analytical procedure was optimized. On the basis of peak areas and calibration curves, the concentrations of individual components were determined in real samples (Table 2). Detailed analysis of the results is given in Section S.4.2.† A particularly large increase was observed for phenol and o-cresol as well as cyclohexanol. In the discussed case where oxidation aided by sonic cavitation is a preliminary stage of treatment of the effluent prior to sending the process effluent to the refinery treatment plant, such an increase in alcohol content is highly undesirable. The activated sludge used during biological treatment of refinery effluents has completely different characteristics compared to the activated sludge used in the treatment of municipal wastewater. Strains of microorganisms present in the activated sludge used for the treatment of refinery effluents are highly intolerant to alcohols, including phenols. The increase in alcohol content in the effluents can result in a substantial decrease in the effectiveness of biological treatment of the effluents despite a significant reduction in the total load of pollutants. The increase in the concentration of phenols is a problem because of the strict limits of their content in the effluent discharged into waterways, which is 0.1 mg L−1.56
| No. | Name | Concentration [ppm] | ||
|---|---|---|---|---|
| Raw effluent | Effluent I (sono-cavitation + UV + ozonation + H2O2) | Effluent II (ozonation) | ||
| 1 | 2,3-Dihydropyran | 2.00 ± 0.06 | 0.18 ± 0.02 | — |
| 2 | 2-Butanol | 14.75 ± 0.24 | 3.27 ± 0.04 | — |
| 3 | Ethyl acrylate | 0.11 ± 0.06 | — | — |
| 4 | 2-Pentanone | 18.70 ± 0.17 | 27.60 ± 0.32 | 0.18 ± 0.01 |
| 5 | 2-Hexanone | 8.89 ± 0.24 | 9.80 ± 0.12 | — |
| 6 | 1-Hexanol | 7.84 ± 0.07 | 18.31 ± 0.47 | — |
| 7 | Cyclohexanol | 19.28 ± 0.59 | 48.62 ± 0.29 | — |
| 8 | Cyclohexanone | 3.29 ± 0.04 | 6.90 ± 0.04 | — |
| 9 | 3-Methylcyclohexanone | 0.181 ± 0.004 | 0.75 ± 0.09 | — |
| 10 | Furfural | 39.16 ± 0.69 | 5.45 ± 0.15 | 0.49 ± 0.04 |
| 11 | o-Cresol | — | 37.19 ± 0.53 | — |
| 12 | Phenol | — | 118.61 ± 0.97 | — |
| 13 | m-Cresol | — | 0.01 ± 0.01 | — |
By performing the analysis in the SCAN mode, additional O-VOCs, mostly aldehydes, ketones and alcohols, were identified. Using their characteristic ions, peak areas and tentative concentrations of these analytes were determined (Table S5†). A SCAN chromatogram of O-VOCs from raw postoxidative effluent and treated by ozonation is depicted in Fig. 2. The concentrations of the identified compounds are compiled in Table S5.† The O-VOCs additionally identified in the SCAN mode were determined quantitatively based on the averaged response factors calculated separately for each group of O-VOCs. The response factors are listed below in Table S5.†
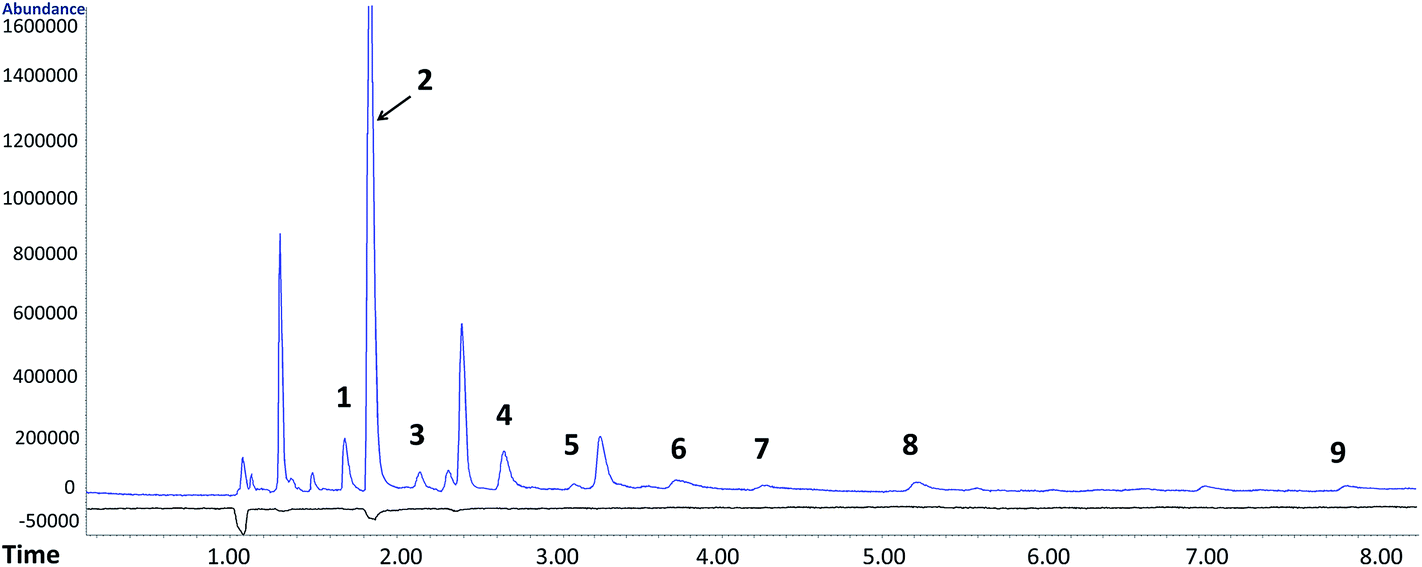 | ||
| Fig. 2 An overlay of SCAN chromatograms of raw postoxidative effluent (blue) and ozonated effluent (black). Identified compounds: (1) 2-propanol, (2) acetone, (3) pentanal, (4) 3-methylbutanal, (5) 2-penten-1-ol, (6) hexanal, (7) octanal, (8) 1,2-cyclopentanediol, and (9) 3-hydroxybutanal. | ||
Detailed analysis of the pathways of conversion and the effectiveness of various processes of effluent treatment aided by sono-cavitation will be the subject of a future paper. In this paper samples were used only as an example of application of the developed method. The opposite effect of compared methods of sewage treatment revealed the importance of the control of O-VOC changes during the process.
To compare the O-VOC changes with “total” parameters used to control the wastewater treatment processes, the BOD and COD test results, as well as Microtox test results (EC20 and EC50 parameters) are summarized in Table S6.† The BOD and COD values were reduced after using both methods of wastewater treatment. Also EC20 and EC50 revealed the reduction of the wastewater toxicity, after using treatment processes (an increase in EC20 and EC50 parameters indicates a decrease in the toxicity). The highest treatment efficiency was obtained during the application of the method of treatment using H2O2. The opposite results were obtained in the case of O-VOC content test results. The cause of this is effective oxidation of groups of compounds with higher molecular weights and their partial conversion into volatile organic compounds.
4 Conclusions
The paper demonstrates the effectiveness of the dynamic headspace-gas chromatography-mass spectrometry (DHS-GC-MS) procedure for the determination of low levels of oxygenated volatile organic compounds (O-VOCs) in refinery effluents.Among the capillary columns investigated, a very polar column, SLB-IL 111, with an ionic liquid as the stationary phase was found to be superior for the separation of O-VOCs, as it has a high selectivity towards n-alkanes and oxygenated volatile organic compounds. The few observed coelutions do not interfere with the GC-MS analyses due to considerable differences in the mass spectra of coeluting compounds.
The main calibration parameters obtained for the 36 standards, i.e. LOD ranging from 0.005 μg mL−1 to 0.5 μg mL−1 and LOQ along with a wide linear range allow the determination of O-VOCs at low concentration levels. Furthermore, good repeatability confirms the usefulness of the optimized procedure and its applicability to routine analyses of O-VOCs in complex matrices.
The significance of the developed procedure is illustrated in Fig. S4,† which shows the percent change in O-VOC content and chemical oxygen demand (COD) following various methods of chemical treatment of raw effluents. The results of the investigations reveal that O-VOCs are an important class of chemical compounds that need to be monitored during the treatment of effluents due to the fact that in many cases they are secondary pollutants formed through oxidation of compounds initially present in the effluent. The effectiveness of the process expressed by total parameters, i.e. COD, demonstrates the expected reduction in content. The reduction depends on the strength of the oxidizing agent and the dose used during treatment. However, the investigations described in this paper reveal that excessively strong oxidation results in an increase in O-VOC content.
Acknowledgements
The authors would like to thank Lotos Asfalt, Ltd (Lotos Group, Inc.) for their cooperation in this project. The authors gratefully acknowledge the financial support from the National Science Center, Warsaw, Poland – decision no. DEC-2013/09/D/ST8/03973.References
- International Agency for Research on Cancer, Monographs on the Evaluation of the Carcinogenic Risk of Chemicals to Man, WHO, Geneva, 1999, vol. 71 Search PubMed.
- Environmental Protection Agency, Health Effects Notebook for Hazardous Air Pollutants, 2013, http://www.epa.gov/ttnatw01/hlthef/hapindex.html, accessed: November 2015 Search PubMed.
- B. Dimitriades, J. Air Waste Manage. Assoc., 1999, 49, 831–838 CAS.
- M. E. Jenkin and G. D. Hayman, Atmos. Environ., 1999, 33, 1275–1293 CrossRef CAS.
- European Environmental Agency, EMEP/CORINAIR, Emission Inventory Guidebook, Technical report No 30, Copenhagen, Denmark, 2005 Search PubMed.
- E. Gilgenast, G. Boczkaj, A. Przyjazny and M. Kamiński, Anal. Bioanal. Chem., 2011, 401, 1059–1069 CrossRef CAS PubMed.
- G. Boczkaj, A. Przyjazny and M. Kamiński, Chemosphere, 2014, 104, 23–30 CrossRef PubMed.
- M. J. Ahrens and C. V. Depree, Chemosphere, 2010, 81, 1526–1535 CrossRef CAS PubMed.
- H. C. A. Brandt and P. C. D. de Groot, Water Res., 2001, 35, 4200–4207 CrossRef CAS PubMed.
- I. Burstyn, H. Kromhout and P. Boffetta, Am. Ind. Hyg. Assoc. J., 2000, 61, 715–726 CAS.
- G. Boczkaj, M. Kamiński and A. Przyjazny, Ind. Eng. Chem. Res., 2010, 49, 12654–12664 CrossRef CAS.
- F. M. Davie, P. F. Nolan and T. W. S. Hoban, J. Loss Prev. Process Ind., 1993, 6, 139–143 CrossRef.
- F. Deygout, Environ. Prog. Sustainable Energy, 2011, 30, 102–112 CrossRef CAS.
- W. Wei, S. Cheng, G. Li, G. Wang and H. Wang, Atmos. Environ., 2014, 89, 358–366 CrossRef CAS.
- G. Boczkaj, A. Przyjazny and M. Kamiński, Ind. Eng. Chem. Res., 2014, 53, 1503–1514 CrossRef CAS.
- R. Beale, P. S. Liss, J. L. Dixon and P. D. Nightingale, Anal. Chim. Acta, 2011, 706, 128–134 CrossRef CAS PubMed.
- K. Grodowska and A. Parczewski, Acta Poloniae Pharmaceutica–Drug Research, 2010, 67, 3–12 CAS.
- M. L. Davi and F. Gnudi, Water Res., 1999, 33, 3213–3219 CrossRef CAS.
- T. Górecki, P. Martos and J. Pawliszyn, Anal. Chem., 1998, 70, 19–27 CrossRef PubMed.
- A. Kabir, C. Hamlet and A. Malik, J. Chromatogr. A, 2004, 1047, 1–13 CrossRef CAS PubMed.
- F. Zhou, X. Li and Z. Zeng, Anal. Chim. Acta, 2005, 538, 63–70 CrossRef CAS.
- M. Serrano, M. Gallego and M. Silva, J. Chromatogr. A, 2013, 1307, 158–165 CrossRef CAS PubMed.
- P. Bartak, P. Frnkova and L. Cap, J. Chromatogr. A, 2000, 867, 281–287 CrossRef CAS PubMed.
- C. Deng, N. Yao, N. Li and X. Zhang, J. Sep. Sci., 2005, 28, 2301–2305 CrossRef CAS PubMed.
- R.-S. Jian, Y.-S. Huang, S.-L. Lai, L.-Y. Sung and C.-J. Lu, Microchem. J., 2013, 108, 161–167 CrossRef CAS.
- J. Nawrocki, A. Dabrowska and A. Borcz, Water Res., 2002, 36, 4893–4901 CrossRef CAS PubMed.
- S.-W. Tsai and C.-M. Chang, J. Chromatogr. A, 2003, 1015, 143–150 CrossRef CAS PubMed.
- X. Liu, Y. Ji, Y. Zhang, H. Zhang and M. Liu, J. Chromatogr. A, 2007, 1165, 10–17 CrossRef CAS PubMed.
- G. Boczkaj, M. Jaszczołt, A. Przyjazny and M. Kamiński, Anal. Bioanal. Chem., 2013, 405, 6095–6103 CrossRef CAS PubMed.
- A. K. K. V. Pillai, K. Gautam, A. Jain and K. K. Verma, Anal. Chim. Acta, 2009, 632, 208–215 CrossRef CAS PubMed.
- J. A. Morales, H. I. de Medina, M. G. de Nava, H. Velhquez and M. Santana, J. Chromatogr. A, 1994, 671, 193–196 CrossRef CAS.
- K. Tsukagoshi, T. Kameda, M. Yamamoto and R. Nakajima, J. Chromatogr. A, 2002, 978, 213–220 CrossRef CAS PubMed.
- S. Morales and R. Cela, J. Chromatogr. A, 2000, 896, 95–104 CrossRef CAS PubMed.
- K. V. Ozdokur, L. Pelit, H. Ertas, S. Timur and F. N. Ertas, Talanta, 2012, 98, 34–39 CrossRef PubMed.
- I. E. Mulazımoglu, A. D. Mulazımoglu and E. Yılmaz, Desalination, 2011, 268, 227–232 CrossRef.
- W. Cao, X. Mu, J. Yang, W. Shi and Y. Zheng, Spectrochim. Acta, Part A, 2007, 66, 58–62 CrossRef PubMed.
- W. Liu, W. Cao, W. Liu, K. Du and P. Gong, Spectrochim. Acta, Part A, 2012, 85, 283–287 CrossRef CAS PubMed.
- J. Arana, E. T. Rendón, J. M. D. Rodrígue, J. A. H. Melián, O. G. J. Díaz and P. Peña, Chemosphere, 2001, 44, 1017–1023 CrossRef CAS PubMed.
- J. Wei, Y. Song, X. Tu, L. Zhao and E. Zhi, Chem. Eng. J., 2013, 218, 319–326 CrossRef CAS.
- G. Boczkaj and M. Kamiński, Anal. Bioanal. Chem., 2013, 405, 8377–8382 CrossRef CAS PubMed.
- G. Boczkaj, A. Przyjazny and M. Kamiński, Anal. Bioanal. Chem., 2011, 399, 3253–3260 CrossRef CAS PubMed.
- R. A. Ketola, V. T. Virkki, M. Ojala, V. Komppa and T. Kotiaho, Talanta, 1997, 44, 373–382 CrossRef CAS PubMed.
- M. Povolo and G. Contarini, J. Chromatogr. A, 2003, 985, 117–125 CrossRef CAS PubMed.
- U.S. Environmental Protection Agency, Method 1624c, Volatile Organic Compounds by Isotope Dilution GCMS, http://water.epa.gov/scitech/methods/cwa/bioindicators/upload/2007_11_27_methods_method_1624c.pdf, accessed: November 2015.
- X.-A. Ning, J.-Y. Wang, R.-J. Li, W.-B. Wen, C.-M. Chen, Y.-J. Wang, Z.-Y. Yang and J.-Y. Liu, Chemosphere, 2015, 136, 50–55 CrossRef CAS PubMed.
- A. S. Costa, L. P. C. Romão, B. R. Araújo, S. C. O. Lucas, S. T. A. Maciel, A. J. Wisniewski and M. R. Alexandre, Bioresour. Technol., 2012, 105, 31–39 CrossRef CAS PubMed.
- H. S. Dórea, J. R. L. Bispo, K. A. S. Aragão, B. B. Cunha, S. Navickiene, J. P. H. Alves, L. P. C. Romão and C. A. B. Garcia, Microchem. J., 2007, 85, 234–238 CrossRef.
- A. D. Nikolaou, S. K. Golfinopoulos, M. N. Kostopoulou, G. A. Kolokythas and T. D. Lekkas, Water Res., 2002, 36, 2883–2890 CrossRef CAS PubMed.
- U.S. Environmental Protection Agency, 1999, Method TO-17, http://www.epa.gov/ttn/amtic/files/ambient/airtox/to-17r.pdf, accessed: November 2015.
- U.S. Environmental Protection Agency, 1995, Method 524.2, https://www.epa.gov/sites/production/files/2015-06/documents/epa-524.2.pdf, accessed: March 2016.
- U.S. Environmental Protection Agency, 1992, Method 5030a, http://www3.epa.gov/wastes/hazard/testmethods/sw846/pdfs/Method%205030A, %20Revision%201%20-%201992.pdf, accessed: March 2016.
- U.S. Environmental Protection Agency, 1996, Method 8260B, http://www.caslab.com/EPA-Methods/PDF/8260b.pdf, accessed: March 2016.
- L. Huber, Validation and Qualification in Analytical Laboratories, Interpharm Press, Buffalo Grove, 1998 Search PubMed.
- H.-H. Zhang, J.-L. Li, G.-P. Yang, Y.-C. Song and N. Jin, Chin. J. Anal. Chem., 2015, 43, 333–337 CAS.
- E. Martınez, S. Lacorte, I. Llobet, P. Viana and D. Barcelo, J. Chromatogr. A, 2002, 959, 181–190 CrossRef.
- Regulation of the Minister of the Environment of 24 July 2006 on conditions to be met when discharging waste to waters or to the ground, and on substances with particularly harmful effects on the water environment, Journal of Laws No. 137 item 984 and of 2009 No. 27 item 169.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ay03043a |
| This journal is © The Royal Society of Chemistry 2016 |