
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrated on-chip mass spectrometry reaction monitoring in microfluidic devices containing porous polymer monolithic columns
C.
Dietze
a,
S.
Schulze
a,
S.
Ohla
a,
K.
Gilmore
 b,
P. H.
Seeberger
*b and
D.
Belder
*a
b,
P. H.
Seeberger
*b and
D.
Belder
*a
aInstitut für Analytische Chemie, Universität Leipzig, Linnéstr. 3, D-04103 Leipzig, Germany. E-mail: belder@uni-leipzig.de
bBiomolecular Systems Department, Max-Planck-Institute for Colloids and Interfaces, Am Mühlenberg 1, D-14476 Potsdam, Germany. E-mail: peter.seeberger@mpikg.mpg.de
First published on 29th June 2016
Abstract
Chip-based microfluidics enable the seamless integration of different functions into single devices. Here, we present microfluidic chips containing porous polymer monolithic columns as a means to facilitate chemical transformations as well as both downstream chromatographic separation and mass spectrometric analysis. Rapid liquid phase lithography prototyping creates the multifunctional device economically.
Introduction
The integration of miniaturized chemical reactors1 and analytical techniques2,3 into single microfluidic devices4,5 is attractive for process development. Continuous flow micro-systems can combine microflow reactors with a separation column. Meanwhile, the seamless integration of synthesis and analytics accelerates the study of chemical processes for very fast reactions and at dimensions hardly reachable with conventional technologies. To date, flow reactors have been typically combined with electrophoresis for downstream separation and fluorescence detection.4,6 The immense potential of integrated devices to study stereoselective chemical conversions at the micro- and nanolitre scale was recently demonstrated in the context of whole cell enantioselective biocatalysis.7 While the integration of electrophoretic separation into microfluidic channel networks is straightforward, electrophoresis has rather limited applications as the separation of typical uncharged small organic molecules poses a challenge. As such, in order to explore the full potential of integrated chips by studying a broad range of organic reactions, a seamless connection or integration of liquid chromatography or LC/MS is desirable. These analytical techniques are the work horses of traditional chemical laboratories to analyse crude reaction mixtures.Liquid chromatography on-chip is technically much more challenging when compared to chip electrophoresis. While electrophoresis can be performed in an open channel at ambient pressure, liquid chromatography relies on packed separation columns and pressure driven flow, which complicates on-chip flow steering and world-to-chip interfacing. Although there is tremendous progress in chip-based HPLC.8 Chip electrochromatography (ChEC) is an interesting alternative in this context as it allows for chromatographic separations in unpressurized systems. As pumping is performed with electroosmotic flow (EOF) the obstacles of interfacing the chip device to HPLC tubings and high pressure pumps can be overcome.9,10
Recently, we have shown that prototyping of functional electrochromatography chips, which include an integrated electrospray emitter for MS detection, can be realized without sophisticated microsystem technology equipment.11 Such devices can be constructed from simple glass microscope slides by inserting a structured photopolymer layer. The technical demands for the liquid phase lithography process are low and the necessary equipment is available in most chemical laboratories. Hence, the approach is ideally suited to prototype functional integrated micro systems to study continuous flow processes.
Experimental
Chemicals
All chemicals were used as received. Polyethyleneglycol diacrylate (PEG-DA) (MW ∼ 258), PEG-DA (MW ∼ 575), 3-(trichlorosilyl)propyl methacrylate (TPM); trichloromethane, butyl acrylate, 1,3-butanediol diacrylate, 3-(trimethoxysilyl)propyl methacrylate, 2-acryl-amido-2-methyl-1-propanesulfonic acid (AMPS), 2,2-dimethoxy-2-phenylacetophenone (DMPA), ammonium acetate, thiourea, 2-bromoacetophenone, 1-heptanal and trypsin from bovine pancreas (12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 443 BAEE units per mg protein) were purchased from Sigma-Aldrich (Steinheim, Germany). Elastosil E43 was obtained from Wacker Chemie (Munich, Germany). Acetonitrile (Rotisolv HPLC gradient grade) and ethanol were acquired from Carl Roth (Karlsruhe, Germany). Buffer solutions were prepared using ultrapure water.
443 BAEE units per mg protein) were purchased from Sigma-Aldrich (Steinheim, Germany). Elastosil E43 was obtained from Wacker Chemie (Munich, Germany). Acetonitrile (Rotisolv HPLC gradient grade) and ethanol were acquired from Carl Roth (Karlsruhe, Germany). Buffer solutions were prepared using ultrapure water.
Chip fabrication and experimental procedures
Glass-polymer-MS-chips (outer dimensions 76 mm × 26 mm) were prepared via liquid phase lithography based on a method described earlier.11 The following is a brief summary of the fabrication process and the experimental procedures. The chips are based on glass slides (Carl Roth, Karlsruhe, Germany) which were all cleaned and treated with a 5 mmol L−1 solution of TPM in n-heptane and trichloromethane (4:1, v/v) for 2 min. The bottom glass slide is prepared with spacers (tape from HellermannTyton, Tornesch, Germany) and the glued fused silica capillary (CS Chromatographie Service GmbH, Langerwehe, Germany, outer diameter (OD): 164 μm; inner diameter (ID): 100 μm) as ESI-emitter. Microfluidic access holes in the top glass slide were created by powderblasting (Sandstrahler Point II, Barth, Königsbach-Stein, Germany). After the preparation, 250 μL monomer mixture (1% (w/w) DMPA in PEG-DA (MW ∼ 258)) was dispensed between a bottom and a top glass slide. The respective photomask (foil offset print, 3600 DPI, DTP-System-Studio, Leipzig, Germany) with the desired layout was placed on top of the glass slide and the glass-polymer sandwich chip was illuminated with an exposer unit (SÜSS MicroTec AG, Munich, Germany) equipped with a mercury lamp (13 mW cm−2 at 365 nm) for 1.3 s. The uncured prepolymer was removed and the resulting microfluidic channel network flushed with ethanol. To perform chip electrochromatography the chip was equipped with an ion-conductive hydrogel wall in-between the separation channel and the makeup flow channel. The hydrogel wall (60% PEG-DA (MW ∼ 575) with 1% DMPA in ultrapure water) was implemented by laser-based (355 nm) polymerization. Finally, a monolithic column was integrated in the reaction channel as well as in the injection cross and separation channel following the procedure published by Ngola et al.10 The polymerization mixture consists of 685 μL butyl acrylate, 297 μL 1,3-butanediol diacrylate, 3 μL 3-(trimethoxysilyl) propyl methacrylate, 5 mg of DMPA and AMPS dissolved in 2010 μL casting solvent (60% acetonitrile, 20% ethanol, 20% 5 mM phosphate buffer pH 6.8 (v/v/v)). After degassing, the polymerization mixture was filled in all attached glass reservoirs on the chip. The chip was illuminated with UV light (UV exposure unit 1S, Gie-Tec GmbH, Munich, Germany) at a distance of 10 cm for 10 min after a resting time of 10 min. The chip was stored and flushed with same buffer as for the analytical separation (80% acetonitrile, 20% 5 mM ammonium acetate buffer pH 8 (v/v)).
To perform mass spectrometry detection the commercial ESI-source was removed. Finished glass-polymer chips were placed in front of the mass spectrometer (LC/MS 2010 EV, Shimadzu, Duisburg, Germany) on a home-made x,y,z-positioning stage. Electrical contact was realized with a four channel high voltage power supply (model HCV 40M-10000, FuG Elektronik GmbH, Rosenheim, Germany) and a custom-built poly(methylmethacrylate) plate with integrated platinum electrodes to connect the microfluidic channels. The conditioned chip was filled with separation buffer and the reactant solutions were filled in the corresponding inlet vials. The reactants were flushed electrokinetically through the reaction structure and continuously focused in the injection cross. A small amount of the reaction mixture is then injected in the separation channel by a voltage-controlled pinched injection program. The individual electrical potentials are given below.
Enzyme reaction and thiazole synthesis
Focusing parameters: R 2.00 kV (1.00 kV, 0.50 kV); SO −0.41 kV; BI (2) −0.31 kV; makeup flow 1.40 kV.Separation parameters: R 3.50 kV; SO 3.50 kV; BI (2) 5.50 kV; makeup flow 2.00 kV.
Mannich reaction
Additionally a potential was applied at BI (1) to dilute the reaction solution with eluent.Focusing parameters: R 2.00 kV; BI (1) 0.20 kV; SO −0.63 kV; BI (2) −0.31 kV; makeup flow 1.40 kV.
Separation parameters: R 3.50 kV; BI (1) 3.50 kV; SO 3.50 kV; BI (2) 5.50 kV; makeup flow 2.00 kV.
The used channel layout is schematically illustrated in Fig. 1.
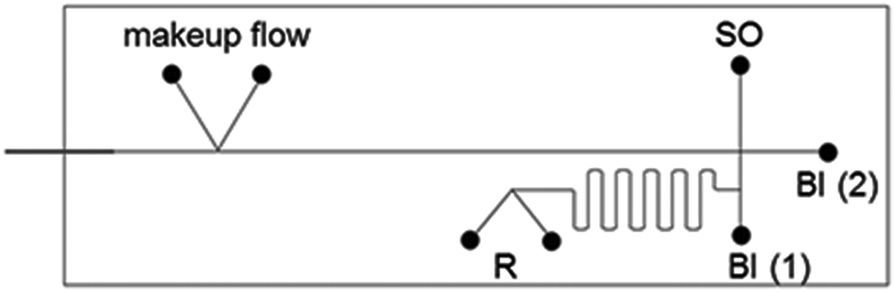 | ||
| Fig. 1 Schematic representation of the chip layout for reaction and separation on-chip. SO-sample inlet; BI-buffer inlet, R-reactants inlet. | ||
The data were recorded by LabSolution (Shimadzu), transferred and analyzed by Clarity Station (DataApex).
Results and discussion
In order to realize the intended microfluidic device seamlessly integrating a chemical micro-flow reactor with downstream electrochromatography-MS, we developed a microfluidic layout which is schematically shown in Fig. 2. The chip system's microfluidic channel layout includes two inlets for the starting materials. This channel leads into a cross section that interconnects the synthetic and analytic functions of the microfluidic chip. Portions of the reaction mixture can be directed to the chromatographic channel (5 cm long) containing a polymer monolithic stationary phase with reversed-phase separation properties. The separation column leads into an integrated fused-silica capillary that functions as an electrospray emitter. The necessary electrical contact for EOF generation and the ESI-process at the column end is realized by a K-shaped channel structure with an integrated ion-conductive membrane.11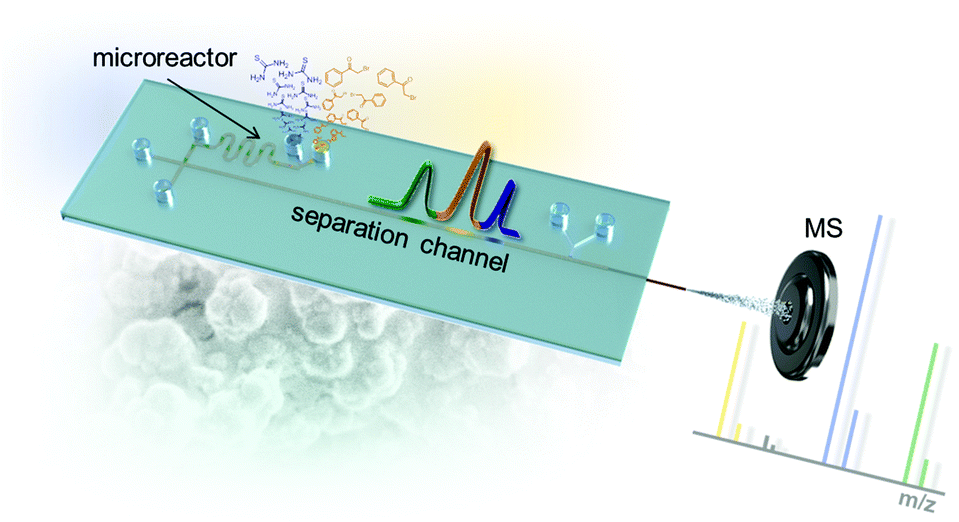 | ||
| Fig. 2 Microfluidic layout of the chip with integrated serpentine microreactor structure, electrochromatographic separation column and electrospray ionization tip leading into a mass spectrometer. | ||
While we previously developed methods to prototype single functional chips containing either a electrochromatography functionality or a simple open channel flow reactor,11 the integration of both elements turned out to be more challenging than expected. A major difficulty for the intended seamless integration was the necessary directed and reproducible flow steering including sample injection from the reaction channel into the separation channel filled with the polymeric monolith. The porous polymer monolithic column is generated inside the microfluidic channels by radical photo polymerization of an acrylate-based monomer solution.10
In principle, the porous monolith is only required as chromatographic stationary phase in the separation channel. But in practice the combination of a common open reaction channel with a polymer filled separation channel posed a challenge. Beside manufacturing issues, the controlled and reproducible flow steering via electroosmotic flow turned out to be troublesome. These issues could be overcome if the polymer monolith was generated in all channels including the meandering reaction channel. A respective photograph of such a chip is shown later in Fig. 5a. In such chip devices precise control and steering of the electroosmotic fluid flows within the entire microfluidic network was possible. The presence of the monolith in the reaction channel also promotes reagent mixing.12,13 After successful development of a reliable method to prototype the chips their functionality was tested in chemical model reactions.
A simple enzymatic trypsin cleavage of the peptide Cbz-L-Arg-MCA, yielding Cbz-protected arginine and a coumarin derivative as products, served as a first model reaction to test and optimize the chip device. Enzyme and substrate were pipetted into the individual reactant inlets and were electroosmotically pumped through the reactor by applying electrical potentials via electrodes inserted in all reservoirs.
At the channel cross section, portions of the bypassing reaction media can be directed into the adjacent separation channel by voltage switching according to a pinched injection scheme.14 The injected sample plug is then electroosmotically driven along the column and separated chromatographically. After reaching the integrated fused-silica capillary at the end of the separation channel, which serves as electrospray emitter, the eluting compounds are ionized and detected in positive ion mode in a mass spectrometer. A representative electrochromatogram obtained after on-chip tryptic cleavage of Cbz-L-Arg-MCA and subsequent electrochromatographic separation with ESI-MS detection is shown in Fig. 3.
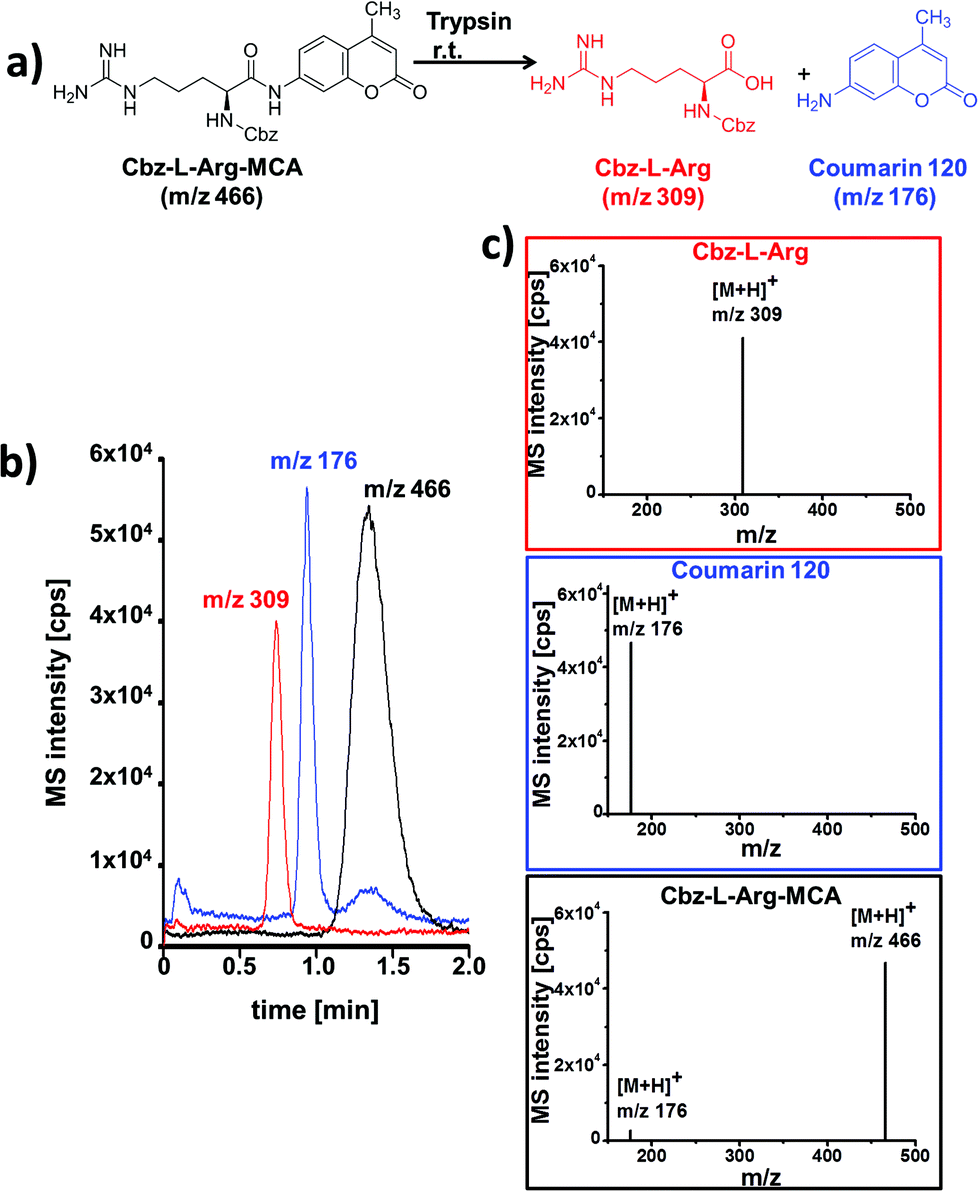 | ||
| Fig. 3 (a) Tryptic cleavage of Cbz-L-Arg-MCA; (b) electrochromatogram obtained after on-chip tryptic cleavage of Cbz-L-Arg-MCA and ChEC-based separation with ESI-MS detection; (c) mass spectra for each separated peak (Cbz-L-Arg red framed; coumarin 120 blue framed; Cbz-L-Arg-MCA black framed) with background subtraction. Data presented as extracted ion chromatograms; initial concentrations: 21 μM trypsin; 1 mM Cbz-L-Arg-MCA.; separation and reaction medium: MeCN/5 mM NH4OAc pH 8 (80/20 vol%); column length: 5 cm; eff. separation field strength: 758 V cm−1; ESI-MS detection: SIM pos. mode, 2 Hz. | ||
The displayed data correspond to a residence time in the reaction structure of 2.5 minutes, which is defined by the length of this channel and the applied electrical potential. Baseline separation of all reaction components was achieved in less than two minutes. The rather broad signal of the starting material Cbz-L-Arg-MCA can be explained by overloading of the column. Reliable peak assignment was possible by MS-detection, as documented by the extracted mass spectra shown in Fig. 3c. In comparison with a simple coupling of a flow reactor with MS, the additional chromatographic functionality enables to reliably distinguish enzyme induced product formation from fragmentation of the starting material in the MS. This is evident from the MS-spectrum of the eluting starting material, as well as of the mass trace at m/z 176 which indicates ESI-MS induced fragmentation of Cbz-L-Arg-MCA. This was confirmed by further control experiments without catalyst.
After establishing the functionality of the chip device, the synthesis of 2-amino-4-phenylthiazole was used as a model for a typical organic transformation.15,16 The starting materials, thiourea and 2-bromoacetophenone, were electroosmotically pumped through the meandering channel and portions of the reaction mixture were analyzed by the adjacent chromatography. The residence time in the flow reactor can be prolonged or shortened by decreasing or increasing the electrical field strength across the micro-channel. A representative electrochromatogram obtained after on-chip thiazole synthesis for 5 min and subsequent ChEC-based separation with ESI-MS detection is shown in Fig. 4.
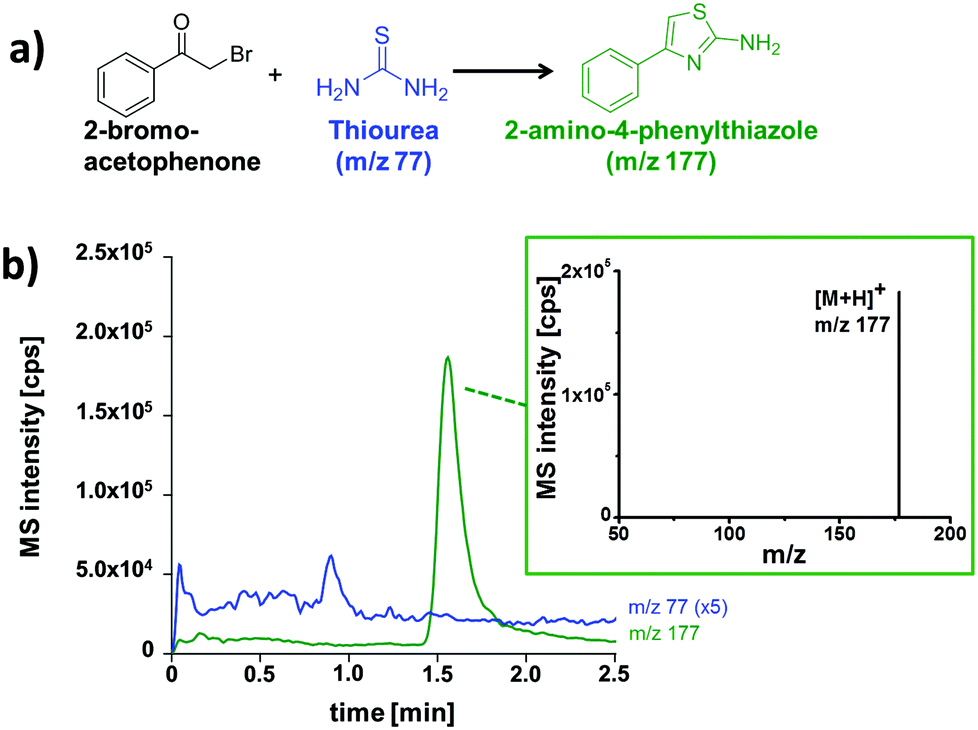 | ||
| Fig. 4 (a) Thiazole synthesis; (b) electrochromatogram obtained after on-chip thiazole synthesis for 5 min and ChEC-based separation with ESI-MS detection and mass spectrum of the product with background subtraction. Data presented as extracted ion chromatograms; initial concentrations: 1 mM 2-bromoacetophenone, 2.5 mM thiourea; separation and reaction medium: MeCN/5 mM NH4OAc pH 8 (80/20 vol%); column length: 5 cm; eff. separation field strength: 758 V cm−1; ESI-MS detection: SIM pos. mode, 2 Hz. | ||
As visible from the recorded ion chromatograms, the reaction mixture can be well separated by electrochromatography within two minutes. The compound 2-bromoacetophenone can, however, not be detected due to poor ionization by the ESI-process. In a further set of experiments the reaction progress in relationship to the residence time in the microreactor structure was also investigated (Table 1). Longer residence times in the reactor channel (2.5 min vs. 7 min) resulted in significantly more thiazole product (m/z 177) accompanied by a decrease of the recorded signal of the starting material (m/z 77).
| Reaction time [min] | Monitored compound | [M + H]+ | Peak area |
|---|---|---|---|
| m/z | [cps min] | ||
| 2.5 | Thiazole product | 177 | 12.760 |
| Thiourea | 77 | 2.500 | |
| 5.0 | Thiazole product | 177 | 32.530 |
| Thiourea | 77 | 2.060 | |
| 7.0 | Thiazole product | 177 | 124.040 |
| Thiourea | 77 | 1.630 |
To extend the solvent scope beyond aqueous media, non-aqueous reaction mixtures were tested. For this proof of concept study an organocatalytic Mannich reaction served as model system (Fig. 5b).17 As for aqueous media, it was possible to pump the reactants and the catalyst dissolved in pure acetonitrile electroosmotically through the meandering reaction channel. In order to extend the reaction time, we worked in a stop-flow mode, by switching off the electrical potentials for ten minutes. Thereafter, a portion of the reaction mixture was directed towards the separation channel by a pinched injection protocol. To ensure a reliable injection process, the non-aqueous reaction solution was mixed with aqueous LC eluent prior entering the channel cross section. As this dilutes the reaction mixture this results in reduced signal intensities in the downstream electrochromatography-MS analysis. An annotated photograph of the chip illustrating this procedure and a typical electrochromatogram are depicted in Fig. 5. Two distinct peaks were baseline separated in less than two minutes. According to their mass-to-charge ratio these signals correspond to the Mannich product (m/z 322) and the residual imine starting material (m/z 208). A closer look at the mass spectra reveals that the product is partly fragmented during the electrospray process, as described previously8 and verified in control experiments.
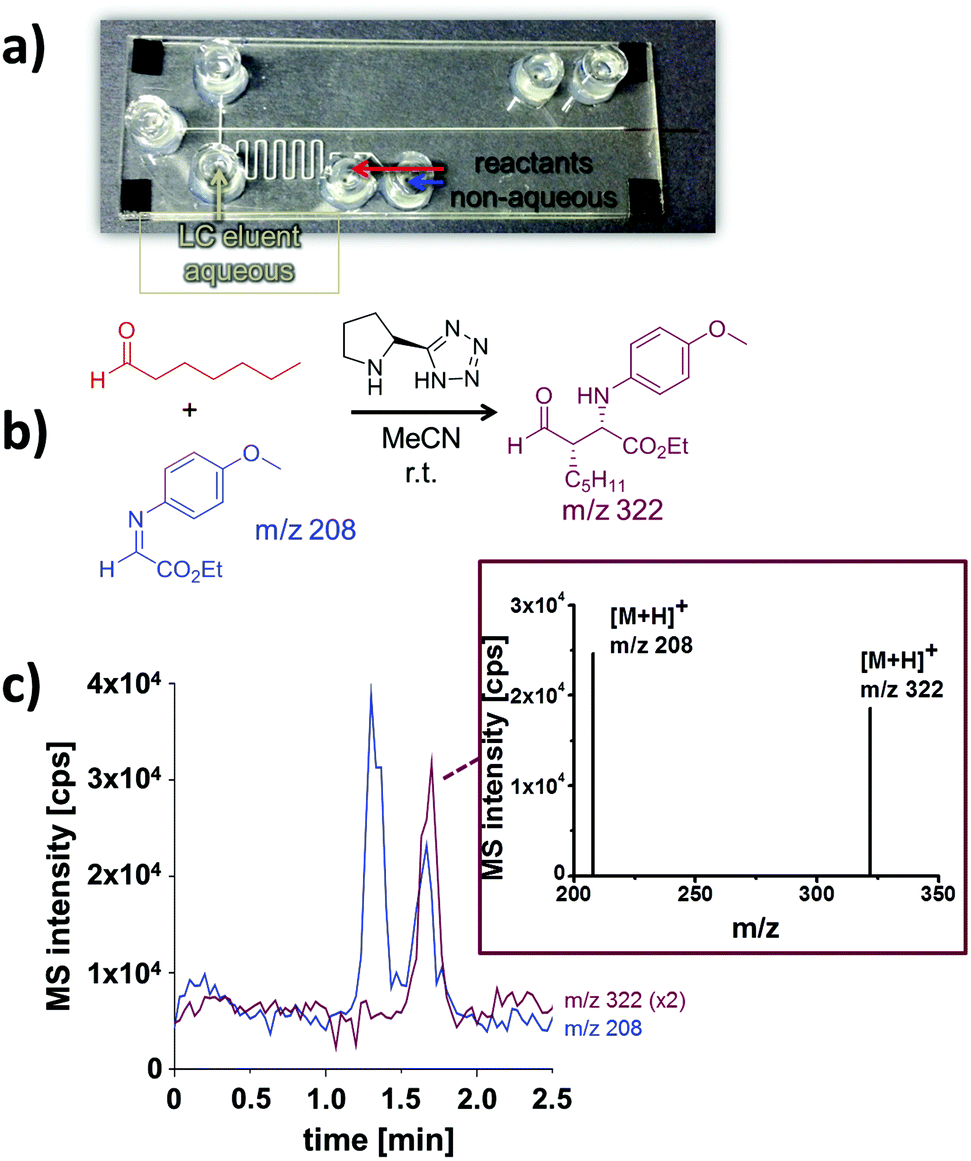 | ||
| Fig. 5 Investigation of a Mannich reaction under non-aqueous conditions by integrated electrochromatography with ESI-MS detection. (a) Glass-polymer chip with integrated monolithic column; (b) Mannich reaction; (c) electrochromatogram obtained after on-chip Mannich reaction and mass spectrum of the product. Data presented as extracted ion chromatograms; initial concentrations: 1 mM α-iminoglyoxylate; 9.6 mM 1-heptanal; 0.4 mM organocatalyst (5-(pyrrolidin-2-yl)tetrazole); reaction medium: MeCN; separation medium: MeCN/5 mM NH4OAc pH 8 (80/20 vol%); column length: 5 cm; eff. separation field strength: 758 V cm−1; ESI-MS detection: SIM pos. mode, 2 Hz. | ||
Conclusion
In summary, we have introduced a novel approach for the seamless combination of chemical reactions, electrochromatographic separation and electrospray ionization in one single rapid prototyped microfluidic device. With this device, microflow reactions can be followed by downstream integrated LC-MS in quasi real time. We successfully applied the approach to various microflow reactions ranging from simple enzymatic conversions to organocatalytic Mannich transformations. As the fast prototyping process does not rely on complex microsystem technology equipment, such devices can be generated in most chemical laboratories. This enabling technology helps to gain a better insight into chemical transformations at the microscale.18References
- (a) D. T. McQuade and P. H. Seeberger, J. Org. Chem., 2013, 78, 6384–6389 CrossRef CAS PubMed; (b) K. Geyer, J. D. C. Codée and P. H. Seeberger, Chem. – Eur. J., 2006, 12, 8434–8442 CrossRef CAS PubMed; (c) A. J. DeMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed; (d) R. L. Hartman, J. P. McMullen and K. F. Jensen, Angew. Chem., Int. Ed., 2011, 50, 7502–7519 CrossRef CAS PubMed; (e) J. P. McMullen and K. F. Jensen, Anal. Chem., 2010, 3, 19–42 CAS.
- (a) S. Thurmann, A. Dittmar and D. Belder, J. Chromatogr., A, 2014, 1340, 33–39 CrossRef PubMed; (b) S. Thurmann, C. Lotter, J. J. Heiland, B. Chankvetadze and D. Belder, Anal. Chem., 2015, 87, 5568–5576 CrossRef CAS PubMed; (c) J. S. Moore and K. F. Jensen, Angew. Chem., Int. Ed., 2014, 53, 470–473 CrossRef CAS PubMed; (d) P. Jáč and G. K. E. Scriba, J. Sep. Sci., 2013, 36, 52–74 CrossRef PubMed.
- (a) D. Janasek, J. Franzke and A. Manz, Nature, 2006, 442, 374–380 CrossRef CAS PubMed; (b) D. Mark, S. Haeberle, G. Roth, F. von Stetten and R. Zengerle, Chem. Soc. Rev., 2010, 39, 1153–1182 RSC; (c) M. L. Kovarik, D. M. Ornoff, A. T. Melvin, N. C. Dobes, Y. Wang, A. J. Dickinson, P. C. Gach, P. K. Shah and N. L. Allbritton, Anal. Chem., 2013, 85, 451–472 CrossRef CAS PubMed.
- S. Ohla, R. Beyreiss, S. Fritzsche, P. Glaser, S. Nagl, K. Stockhausen, C. Schneider and D. Belder, Chem. – Eur. J., 2012, 18, 1240–1246 CrossRef CAS PubMed.
- (a) S. Jezierski, D. Belder and S. Nagl, Chem. Commun., 2013, 49, 11644–11646 RSC; (b) S. Fritzsche, S. Ohla, P. Glaser, D. S. Giera, M. Sickert, C. Schneider and D. Belder, Angew. Chem., Int. Ed., 2011, 50, 9467–9470 CrossRef CAS PubMed; (c) M. C. Mitchell, V. Spikmans and A. J. d. Mello, Analyst, 2001, 126, 24–27 RSC.
- (a) J. Wang, M. P. Chatrathi and B. Tian, Anal. Chem., 2000, 72, 5774–5778 CrossRef CAS PubMed; (b) B.-F. Liu, M. Ozaki, Y. Utsumi, T. Hattori and S. Terabe, Anal. Chem., 2003, 75, 36–41 CrossRef CAS PubMed; (c) N. P. Beard, J. B. Edel and A. J. DeMello, Electrophoresis, 2004, 25, 2363–2373 CrossRef CAS PubMed; (d) D. Belder, M. Ludwig, L.-W. Wang and M. T. Reetz, Angew. Chem., Int. Ed., 2006, 45, 2463–2466 CrossRef CAS PubMed.
- K. M. Krone, R. Warias, C. Ritter, A. Li, C. G. Acevedo-Rocha, M. T. Reetz and D. Belder, J. Am. Chem. Soc., 2016, 138, 2102–2105 CrossRef CAS PubMed.
- (a) S. Thurmann and D. Belder, Anal. Bioanal. Chem., 2014, 406, 6599–6606 CrossRef CAS PubMed; (b) S. Ehlert, L. Trojer, M. Vollmer, T. van de Goor and U. Tallarek, J. Mass. Spectrom., 2010, 45, 313–320 CrossRef CAS PubMed; (c) S. Thurmann, A. Dittmar and D. Belder, J. Chromatogr., A, 2014, 1340, 59–67 CrossRef CAS PubMed; (d) J. Xie, Y. Miao, J. Shih, Y.-C. Tai and T. D. Lee, Anal. Chem., 2005, 77, 6947–6953 CrossRef CAS PubMed; (e) C. Lotter, J. J. Heiland, S. Thurmann, L. Mauritz and D. Belder, Anal. Chem., 2016, 88, 2856–2863 CrossRef CAS PubMed; (f) P. A. Levkin, S. Eeltink, T. R. Stratton, R. Brennen, K. Robotti, H. Yin, K. Killeen, F. Svec and J. M. J. Fréchet, J. Chromatogr., A, 2008, 1, 55–61 CrossRef PubMed.
- (a) R. Shediac, S. M. Ngola, D. J. Throckmorton, D. S. Anex, T. J. Shepodd and A. K. Singh, J. Chromatogr., A, 2001, 925, 251–263 CrossRef CAS; (b) T. B. Stachowiak, F. Svec and J. M. Fréchet, J. Chromatogr., A, 2004, 1044, 97–111 CrossRef CAS; (c) C. Hackl, R. Beyreiss, D. Geissler, S. Jezierski and D. Belder, Anal. Chem., 2014, 86, 3773–3779 CrossRef CAS PubMed.
- S. M. Ngola, Y. Fintschenko, W.-Y. Choi and T. J. Shepodd, Anal. Chem., 2001, 73, 849–856 CrossRef CAS PubMed.
- C. Dietze, T. Scholl, S. Ohla, J. Appun, C. Schneider and D. Belder, Anal. Bioanal. Chem., 2015, 407, 8735–8743 CrossRef CAS PubMed.
- D. A. Mair, T. R. Schwei, T. S. Dinio, F. Svec and J. M. J. Fréchet, Lab Chip, 2009, 9, 877 RSC.
- T. Rohr, C. Yu, M. H. Davey, F. Svec and J. M. J. Fréchet, Electrophoresis, 2001, 22, 3959–3967 CrossRef CAS PubMed.
- S. Jacobson, R. Hergenroeder, A. W. Moore and J. M. Ramsey, Anal. Chem., 1994, 66, 1107–1113 CrossRef CAS.
- T. M. Potewar, S. A. Ingale and K. V. Srinivasan, Tetrahedron, 2008, 64, 5019–5022 CrossRef CAS.
- T.-A. Meier, R. J. Beulig, E. Klinge, M. Fuss, S. Ohla and D. Belder, Chem. Commun., 2015, 51, 8588–8591 RSC.
- (a) A. Odedra and P. H. Seeberger, Angew. Chem., Int. Ed., 2009, 48, 2699–2702 CrossRef CAS PubMed; (b) F. G. Finelli, L. S. M. Miranda and R. O. M. A. de Souza, Chem. Commun., 2015, 51, 3708–3722 RSC; (c) R. Martín-Rapún, S. Sayalero and M. A. Pericàs, Green Chem., 2013, 15, 3295 RSC.
- S. V. Ley, D. E. Fitzpatrick, R. J. Ingham and R. M. Myers, Angew. Chem., Int. Ed., 2015, 54, 3449–3464 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |