
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electrocatalytic monitoring of peptidic proton-wires†
V.
Dorčák
a,
M.
Kabeláč
b,
O.
Kroutil
c,
K.
Bednářová
a and
J.
Vacek
*d
aInstitute of Biophysics of the CAS, v.v.i., Kralovopolska 135, 612 65 Brno, Czech Republic
bDepartment of Chemistry, Faculty of Science, University of South Bohemia, Branisovska 31, 370 05 Ceske Budejovice, Czech Republic
cInstitute of Physics and Biophysics, Faculty of Science, University of South Bohemia, Branisovska 1760, 370 05 Ceske Budejovice, Czech Republic
dDepartment of Medical Chemistry and Biochemistry, Faculty of Medicine and Dentistry, Palacky University, Hnevotinska 3, 775 15 Olomouc, Czech Republic. E-mail: jan.vacek@upol.cz; Fax: +420 585632302; Tel: +420 585632303
First published on 15th June 2016
Abstract
The transfer of protons or proton donor/acceptor abilities is an important phenomenon in many biomolecular systems. One example is the recently proposed peptidic proton-wires (H-wires), but the ability of these His-containing peptides to transfer protons has only been studied at the theoretical level so far. Here, for the first time the proton transfer ability of peptidic H-wires is examined experimentally in an adsorbed state using an approach based on a label-free electrocatalytic reaction. The experimental findings are complemented by theoretical calculations at the ab initio level in a vacuum and in an implicit solvent. Experimental and theoretical results indicated Ala3(His–Ala2)6 to be a high proton-affinity peptidic H-wire model. The methodology presented here could be used for the further investigation of the proton-exchange chemistry of other biologically or technologically important macromolecules.
The exchange or transfer of protons is important for many biochemical processes including the respiratory chain, photosynthesis and enzyme catalysis. In proteins, proton transfer and proton binding affinities determine the charge and structure of their molecules, and thus have a large impact on their functioning and interactions with other molecules.1
A highly important phenomenon is the regulation of the function of some proteins by proton translocation, e.g. using the so-called proton-wires (H-wires) formed by water molecules captured in the channels localized within protein structures.2–4 They can be found in green fluorescent proteins,5 microbial rhodopsins,6 gramicidin,7 carbonic anhydrase,5,8 or ribosomal complexes.9 The mechanism of proton transport in such channels is based on proton diffusion through the hydrogen bond network of water molecules (the Grotthuss mechanism) followed by proton exchange with amino acid (aa) residues; very often His.5
In addition to the water H-wires in proteins, helical peptidic H-wires have also been proposed recently.10 The ability of these 21-mer His-rich peptides to accept a proton and transfer it via the His residues distributed along the peptide backbone was proposed only at a theoretical level. To the best of our knowledge, such proton binding and translocation events were investigated using only the tools of computational chemistry.10 In general, there is a lack of experimental approaches for the study of proton transfer or proton intramolecular movement in peptidic structures.
Here we focused on peptidic H-wires composed of His (H) and Ala (A) residues alone as follows: A3[HA2]6 (HA2), A2[HA3]4HA2 (HA3), and A2[HA4]3HA3 (HA4).10 Their proton transfer abilities were examined experimentally for the first time via an approach based on the electrochemical investigation of their activity in the catalytic hydrogen evolution reaction (CHER) at a Hg-electrode. An essential part of the electrode process is a catalytic cycle (reactions (1) & (2), Scheme 1A) in which the transfer of protons from the acid constituent of a solution (BH, H+ donor) onto the electrode surface (H+ acceptor) is mediated by the His-containing catalyst adsorbed onto the electrode surface. The surface-bound hydrogen atoms then combine into more stable molecules of gaseous hydrogen, H2 (reaction (3), Scheme 1A). The consumption of electrons, accompanying the transfer of protons, is reflected in the so-called peak H (Scheme 1B) at highly negative potentials as a typical analytical output when constant-current chronopotentiometric stripping (CPS) is used.11
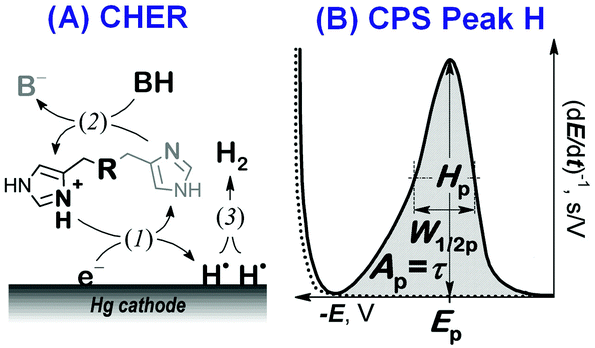 | ||
| Scheme 1 (A) The catalytic hydrogen evolution reaction (CHER) of a His-containing catalyst. Catalyst deprotonation (1) reprotonation/regeneration (2), and formation of molecular/gaseous hydrogen (3) are highlighted. BH: the acid component of the supporting electrolyte. (B) CPS peak H due to the CHER resulting from derivation of the measured E–t curve (raw data) to facilitate its evaluation. | ||
Usually, both an increase in the CPS peak H area Ap (corresponding to the transition time τ) and a shift in its potential Ep towards less negative values suggest a more effective mediation of the proton transfer. In CHER, only aa residues with functional groups bearing exchangeable protons can be involved, i.e. only imidazolium groups of His residues in peptidic H-wires can participate in the transfer of protons reflected by the CPS peak H.12–14 We applied the above-described methodology to study the proton transfer ability of H-wires HA2, HA3 and HA4.
At first, the H-wires were analyzed on the basis of the CPS peak H at a hanging mercury drop electrode (HMDE) in three different media. Initially, the peptides were accumulated on the electrode surface at an applied (accumulation) potential (EA) of +0.1 V from unstirred solutions at a concentration of 1 μM. After 60 s of accumulation time (tA), the electrode with adsorbed H-wire molecules was polarized by a cathodic (stripping) current (Istr) of −5 μA to reach highly negative potentials of around −2.0 V, where peak H was recorded. Other details of the electrochemical measurements are provided in section 1 of the ESI.†
Well-developed and much higher peaks were obtained under buffered conditions, while more than 1000-fold smaller peaks were produced in unbuffered solution (Fig. 1). Buffered medium, providing an excess of slightly acidic H+ donors, is thus necessary for immediate reprotonation/regeneration of the catalyst in its adsorbed state from the bulk to close/form the catalytic cycle in CHER13 (Scheme 1A).
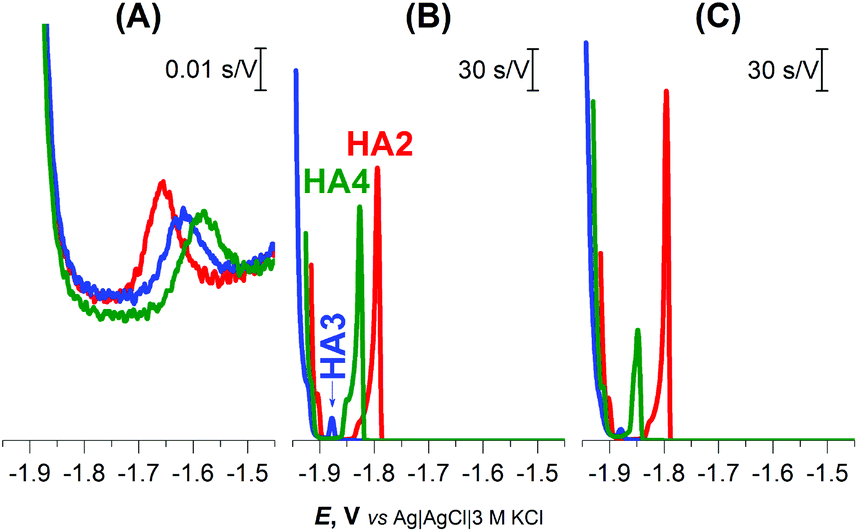 | ||
| Fig. 1 CPS peaks H of 1 μM HA2, HA3, and HA4 at HMDE in (A) 0.1 M NaCl, pH 5.5, (B) 0.1 M Na-phosphate buffer, pH 7 and (C) McIlvaine buffer of pH 7, an Istr of −5 μA. | ||
In the buffered media, the biggest peak H at the least negative potential (Ep of −1.8 V) was produced by peptide HA2. The peptides HA3 and HA4 gave smaller responses at significantly more negative potentials (Fig. 1B and C). This finding indicates that HA2 is an effective proton transporter, which is in agreement with the previously reported computational results.10 However, in contrast to our results, the theoretical investigation showed HA3 to be a more effective proton transporter than HA4. To interpret the theoretical vs. experimental differences in HA3 and HA4 proton transfer efficacy, it should be taken into account that proton transfer for H-wires in the form of an adsorbed layer at the Hg-surface is detected by CPS. In this way, protons can be transported via various mechanisms, including intermolecular and cooperative transfer effects, in comparison to the idealistic theoretical setup based on proton intramolecular translocation as reported in the literature.10
As a result, in subsequent experimental work we focused primarily on investigating HA2 together with the H6 peptide, which served as a positive control. H6 was chosen as the control because it contains six His residues, like HA2. The CPS peaks H of both peptides, including the original E–t curves, are shown in Fig. 2.
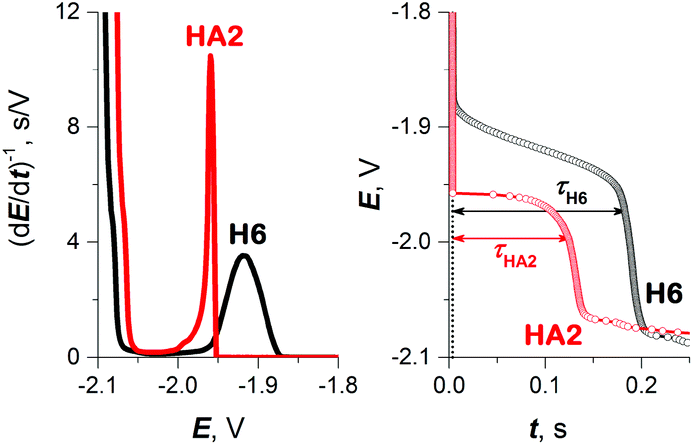 | ||
| Fig. 2 CPS peaks H (left) and their corresponding E–t curves (right) of 10 μM HA2 and H6 at HMDE in 50 mM Na-phosphate buffer of pH 7, an Istr of −50 μA. | ||
To obtain more reliable data for comparing the proton transfer in HA2 and H6, we measured their CPS responses under conditions suggesting full electrode coverage, i.e. at 10 μM or higher concentration (Fig. S1 in ESI†). Thus, the surface concentration of the catalyst was kept constant in the subsequent experiments, while the effect of pH on the CPS responses of HA2 and H6 was investigated. With decreasing pH-value, both the extent of peptide protonation and the proton donor concentration in the solution increased. As a result, an increase in the peak H area (Ap) and height (Hp) of both peptides was observed with decreasing pH from 7.5 to 6.3, and between pH 6.3 and 5.6 it changed only a little (Fig. 3). The pH-value of this break (6.3) is very close to the pKa value of the His imidazolium group, i.e. ∼6.1.15
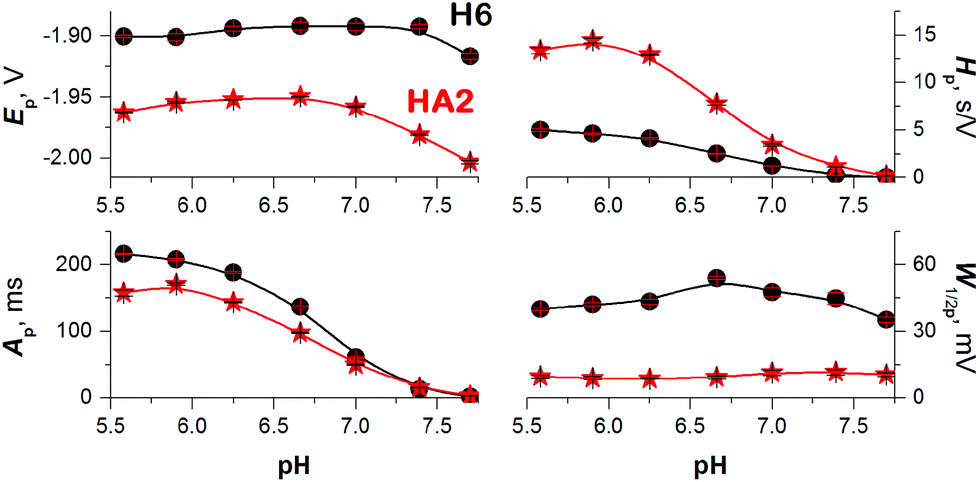 | ||
| Fig. 3 Dependences of 10 μM HA2 (asterisk) and H6 (circle) peak H potential (Ep), area (Ap), height (Hp), and half-width (W1/2p) on the pH-value of 50 mM Na-phosphate buffer at an Istr of −50 μA. Data are represented as means ± SD of four measurements. Error bars smaller than the plotted symbols are not visible. | ||
It was recently shown using several model proteins that Istr is a driving parameter affecting their interfacial behavior and structural integrity during a CPS scan. Thus, we increased cathodic Istr intensity to avoid (i) reorientation of the catalyst molecules during the electrode process,16 and (ii) disruption of the catalyst structural features upon exposure to highly negative electrode potentials.17
We can conclude that at −Istr higher than 95 μA, both of the above effects were eliminated (Fig. 4 and S2 in ESI†). Between 35 and 95 μA, an S-shaped transition can be observed with HA2. This transition most likely reflects interfacial structural changes, i.e. distortion in HA2's structure, which is in agreement with previously reported studies,16–19 especially the study where bovine serum albumin was investigated under practically the same experimental conditions.17 At −Istr lower than 35 μA, the prolonged exposure of HA2 molecules in the adsorbed layer to negative potentials could lead to complete structure destabilization, i.e. unfolding. The unfolding is due to electrostatic and hydrophobic interactions with the highly negatively charged electrode surface. This most likely results in the disruption of hydrogen bonds, the predominant stabilizing element of HA2's structure. The unfolded structures give higher electrochemical responses (expressed as Istrτ1/2 in Fig. 4) than the folded ones, because of the higher accessibility of the electrocatalytically active sites of HA2 to the electrode surface. In this way unfolded HA2 is involved in CHER more easily than the compact folded HA2 configuration. Similar examples and interpretations of electric field-induced destabilization of biomacromolecular structures (polypeptides and nucleic acids) can be found in the ref. 11 and 20.
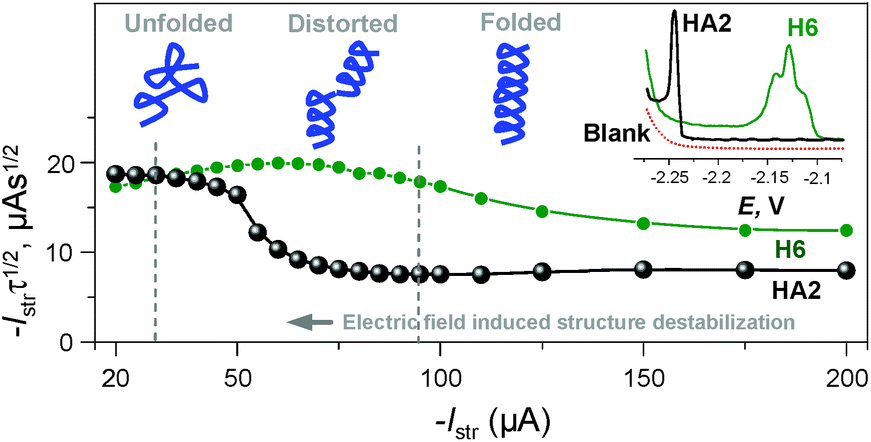 | ||
| Fig. 4 Dependence of the Istrτ1/2 values of HA2 and H6 on an Istr at a 50 mM concentration of Na-phosphate buffer solution of pH 6. Inset: peak H of HA2 and H6 at an Istr of −150 μA. For other details, see Fig. S2 in the ESI.† The depictions of peptide structures here are only schematic and not to scale. | ||
The typical S-shaped transition was not observed with H6, since it has the structure of a random coil (see below Fig. 5B and 6). The typical CPS responses of HA2 and H6 acquired at an Istr of −150 μA are shown in the inset of Fig. 4.
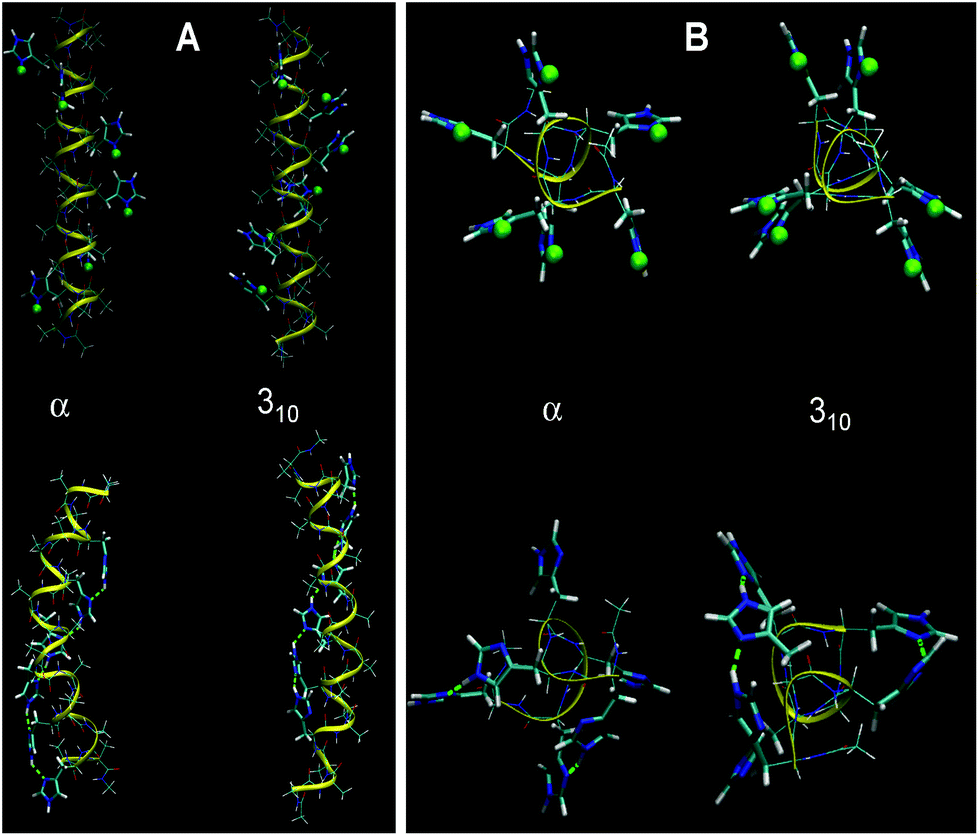 | ||
| Fig. 5 Optimized α- and 310-structures of HA2 (A) and H6 random coil (B) in a fully protonated (top) and unprotonated (below) state using water as a solvent. Protons bound to His residues are highlighted as green spheres. The structures calculated in a vacuum are shown in Fig. S3 in the ESI.† | ||
 | ||
| Fig. 6 CD spectra of 10 μM HA2 and H6 in 50 mM Na-phosphate buffer at (A) pH 6, (B) pH 7 and (C) pH 8. The depictions of peptide structures are only schematic and not to scale. | ||
On the basis of our experimental data, the proton transfer mechanism of HA2 is different from that of H6. It is evident that both peptides have high proton transfer ability. HA3 and HA4 did not give a CPS response under the experimental conditions used (Fig. 4, inset). This result confirmed that HA2 is a more effective proton transporter than HA3 and HA4, not only under the initial experimental conditions (Fig. 1B and C) but also under the optimized conditions excluding the inevitable effects attributed to the specific interfacial behavior and structural distortion of adsorbed peptidic H-wires at the negatively charged electrode surface.
An important aspect in the further interpretation of both the experimental and theoretical data presented here is the knowledge of the structure of both peptides. Accordingly, we performed a calculation of the helical structures with all His residues protonated in a vacuum and in an implicit water environment. The 310-helix and α-helix configurations were taken into account, as reported previously.10 With unprotonated polypeptides, the α-helical structure is more stable than the 310-one (by 19 kJ mol−1 in a vacuum and 49 kJ mol−1 in water). This is probably connected to the strong ability of Ala residues to form the α-helix. Optimized conformations of HA2 and H6 at fully protonated or unprotonated states in an aqueous environment are shown in Fig. 5. The calculated structures indicate that the state of protonation has a smaller impact on the structure for the HA2 peptide than for the H6 random coil peptide.
The proton affinity was calculated by computing the energy difference between the optimized unprotonated molecule and the fully protonated molecule (Table 1). The protonation energies are comparable for the different conformations. However, the HA2 peptide has a slightly higher proton affinity than H6 in water (by 30 kJ mol−1), in a vacuum the difference is substantial (170 kJ mol−1). Details of the calculated structures for HA2 and H6 can be found in section 2 in the ESI.†
| Proton affinity | |
|---|---|
| Vacuum | |
| H6-310 | 937.0 |
| H6-α | 940.9 |
| HA2-310 | 1112.5 |
| HA2-α | 1099.7 |
| Water | |
| H6-310 | 1682.7 |
| H6-α | 1694.1 |
| HA2-310 | 1718.6 |
| HA2-α | 1714.3 |
In the next part of the experimental work, the structures of HA2 and H6 were investigated by circular dichroism (CD) spectroscopy under the same experimental conditions as for CPS measurement; for details see section 3 in the ESI.† HA2 exhibited a strong negative band near 200 nm, revealing that ordered structures are present (Fig. 6).21 The CD spectrum of H6 exhibited only a low molar ellipticity in the examined pH-range, indicating a random coil organization with no dominating organized structure. It is evident that HA2 has a compact structure compared to the H6, with no periodicity in its structure.
In conclusion, further application of the electrocatalytic assay presented in this communication could provide new knowledge in the field of protonation equilibria and proton transfer in peptides. The experimental data are supported by ab initio calculations and CD spectroscopy of peptidic H-wires at different pH levels. Both our experimental data and calculated proton affinities indicate that HA2 is an effective molecule that can be used as a model for both experimental and theoretical investigations in the future. The proposed methodology is not only able to recognize protonated vs. unprotonated states (static parameter) but also to assess the proton transfer ability (dynamic parameter) in peptidic H-wires via CPS peak H characteristics, as specified in Scheme 1B. In terms of the mechanistic elucidation focused on the proton transfer process, the goal of further experimental work will be the self-assembled organization of peptidic H-wires onto the electrode surface,22,23 which could enable us to also investigate a mechanism of proton movement in more detail.
This work was supported by the Czech Science Foundation (14-08032S, 13-08651S and P205/12/0466), by the MEYS of the Czech Republic/COST Action BM1203 (project no. LD14033) and by the CAS (RVO 68081707). The access to computing and storage facilities owned by parties and projects contributing to the National Grid Infrastructure MetaCentrum, provided under the programme “Projects of Large Infrastructure for Research, Development, and Innovations” (LM2010005) is highly appreciated.
Notes and references
- V. R. I. Kaila, M. Wikstrom and G. Hummer, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6988–6993 CrossRef CAS PubMed.
- E. Meyer, Protein Sci., 1992, 1, 1543–1562 CrossRef CAS PubMed.
- A. Migliore, N. F. Polizzi, M. J. Therien and D. N. Beratan, Chem. Rev., 2014, 114, 3381–3465 CrossRef CAS PubMed.
- G. M. Ullmann and E. Bombarda, Biol. Chem., 2013, 394, 611–619 CrossRef CAS PubMed.
- A. Shinobu and N. Agmon, J. Phys. Chem. A, 2009, 113, 7253–7266 CrossRef CAS PubMed.
- K. Gerwert, E. Freier and S. Wolf, Biochim. Biophys. Acta, Bioenerg., 2014, 1837, 606–613 CrossRef CAS PubMed.
- R. Pomes and B. Roux, Biophys. J., 1996, 71, 19–39 CrossRef CAS PubMed.
- Q. Cui and M. Karplus, J. Phys. Chem. B, 2003, 107, 1071–1078 CrossRef CAS.
- Y. S. Polikanov, T. A. Steitz and C. A. Innis, Nat. Struct. Mol. Biol., 2014, 21, 787–793 Search PubMed.
- G. E. Lopez, I. Colon-Diaz, A. Cruz, S. Ghosh, S. B. Nicholls, U. Viswanathan, J. A. Hardy and S. M. Auerbach, J. Phys. Chem. A, 2012, 116, 1283–1288 CrossRef CAS PubMed.
- E. Palecek, J. Tkac, M. Bartosik, T. Bertok, V. Ostatna and J. Palecek, Chem. Rev., 2015, 115, 2045–2108 CrossRef CAS PubMed.
- V. Dorcak, V. Ostatna and E. Palecek, Electrochem. Commun., 2013, 31, 80–83 CrossRef CAS.
- V. Dorcak, V. Vargova, V. Ostatna and E. Palecek, Electroanalysis, 2015, 27, 910–916 CrossRef.
- V. Vargova, M. Zivanovic, V. Dorcak, E. Palecek and V. Ostatna, Electroanalysis, 2013, 25, 2130–2135 CrossRef CAS.
- CRC Handbook of Chemistry and Physics, CRC Press, Boca Raton, Florida, 66th edn, 1985 Search PubMed.
- V. Dorcak, M. Bartosik, V. Ostatna, E. Palecek and M. Heyrovsky, Electroanalysis, 2009, 21, 662–665 CrossRef.
- H. Cernocka, V. Ostatna and E. Palecek, Electrochim. Acta, 2015, 174, 356–360 CrossRef CAS.
- V. Ostatna, H. Cernocka and E. Palecek, J. Am. Chem. Soc., 2010, 132, 9408–9413 CrossRef PubMed.
- E. Palecek, V. Ostatna, H. Cernocka, A. C. Joerger and A. R. Fersht, J. Am. Chem. Soc., 2011, 133, 7190–7196 CrossRef CAS PubMed.
- E. Palecek and M. Bartosik, Chem. Rev., 2012, 112, 3427–3481 CrossRef CAS PubMed.
- N. J. Greenfield, Nat. Protoc., 2006, 1, 2876–2890 CrossRef CAS PubMed.
- J. J. Gooding and N. Darwish, Chem. Rec., 2012, 12, 92–105 CrossRef CAS PubMed.
- B. J. Pepe-Mooney and R. Fairman, Curr. Opin. Struct. Biol., 2009, 19, 483–494 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Section 1, details of electrochemical experiments, section 2, in silico methods, section 3, CD spectroscopy analyses. See DOI: 10.1039/c6an00869k |
| This journal is © The Royal Society of Chemistry 2016 |