
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Development of a rechargeable optical hydrogen peroxide sensor – sensor design and biological application†
Klaus
Koren
*a,
Peter Ø.
Jensen
b and
Michael
Kühl
*ac
aMarine Biological Section, Department of Biology, University of Copenhagen, 3000 Helsingør, Denmark. E-mail: klaus.koren@bio.ku.dk; mkuhl@bio.ku.dk
bDepartment of Clinical Microbiology, Rigshospitalet Copenhagen, 2100 Copenhagen, Denmark
cPlant Functional Biology and Climate Change Cluster, University of Technology Sydney, Ultimo, New South Wales 2007, Australia
First published on 12th May 2016
Abstract
Hydrogen peroxide (H2O2) is an important member of the reactive oxygen species (ROS) family. Among ROS, H2O2 is considered the most long-lived and can accumulate inside and outside of cells, where it is involved in both vital (signaling) and deadly (toxic) reactions depending on its concentration. Quantifying H2O2 within biological samples is challenging and often not possible. Here we present a quasi-reversible fiber-optic sensor capable of measuring H2O2 concentrations ranging from 1–100 μM within different biological samples. Based on a Prussian blue/white redox cycle and a simple sensor recharging and readout strategy, H2O2 can be measured with high spatial (∼500 μm) and temporal (∼30 s) resolution. The sensor has a broad applicability both in complex environmental and biomedical systems, as demonstrated by (i) H2O2 concentration profile measurements in natural photosynthetic biofilms under light stress reaching H2O2 concentrations as high as 15 μM, and (ii) the quantification of the transient increase of the extracellular concentration of H2O2 during stimulation of neutrophils.
1. Introduction
Hydrogen peroxide (H2O2) has two important roles in living organisms;1 on the one hand H2O2 is associated with cell death, dysfunction and ageing,2,3 on the other hand H2O2 is also involved in vital processes like cell signaling4 and combatting microbial intruders.5 As a reactive oxygen species (ROS), H2O2 is involved in a variety of redox processes within organisms.6 In contrast to less stable and more reactive ROS such as superoxide anion or hydroxyl radical, H2O2 exhibits a longer lifetime and a higher steady state concentration and can thus accumulate within a cell and even diffuse out.7Maintaining ROS levels below certain threshold concentrations is essential for cellular survival,2 but despite of the development of new fluorescent8–10 and genetically encoded probes,11 quantification of ROS concentrations remains challenging and is often not conducted in biomedical studies due to a lack of suitable methods.12,13 While several probes (with varying specificity) enable visualization of the localization and relative differences of certain ROS,14,15 the lack of quantitative data is recognized as a main bottle neck in understanding redox processes within cells or tissues.16
A possible alternative to fluorescent probes could be the use of mini- or microsensors. Optical17 as well as electrochemical18 microsensors have been used in a variety of biological systems and revealed quantitative information on small molecules like O2,19 H+(pH),20 H2S,21 N2O22 or NO.23 Microsensors with tip diameters well below 100 μm are now commercially available and facilitate scientist to measure a variety of analytes at high spatio-temporal resolution. As those sensors are still bigger or at best in the same size range as individual cells, the most promising member of the ROS family to measure with microsensors is H2O2 as it is most likely to be found outside cells. Several electrochemical H2O2 sensor concepts exist24,25 including some strategies towards intracellular measurements using nanoelectrodes.26–28 Researchers within the field are very active in terms of modifying the used electrodes in order to reduce the overpotential and improve electrode kinetics.29 One of the most important materials used to modify electrodes is Prussian blue (PB),30,31i.e., ferric hexacyanoferrate. By modifying electrodes with PB, the sensitivity and selectivity of electrochemical H2O2 sensors can be drastically improved.31 PB is often considered an “artificial peroxidase” due to its rather specific catalytic activity towards H2O2.32 In terms of (reversible) optical H2O2 sensors only a few sensing principles33 have been reported, but they failed to be applicable in biological systems due to lack of specificity.
Recently, the potential of exploiting the redox reaction induced change in the optical properties of PB has been realized.34,35 It was shown that upon exposure to a reducing agent, PB can be reduced to colorless Prussian white (PW) than can be oxidized back to PB by H2O2. Regeneration of the sensor was possible, but the kinetics were rather slow and the reduction and oxidation steps were separated in space compromising real-time sensing.34
In this study, we present a new quasi-reversible luminescence-based optical H2O2 sensor using PB as sensing element with fast kinetics and high stability. Recharging can be accomplished in close proximity to the sample using a “recharger gel” (containing ascorbic acid donating electrons to PB oxidized by H2O2). By using a differential readout scheme, we measured the rate of oxidation which is directly linked to the H2O2 concentration. We describe the sensor design and fabrication along with its measuring characteristics and demonstrate its application for quantitative H2O2 measurements in environmental and biomedical science.
2. Experimental section
Chromium(III)-Activated Yttrium Aluminum Borate (Cr-YAB) was synthesized and characterized as reported elsewhere.36 Polyurethane hydrogel (Hydromed D4) was obtained from AdvanSource Biomaterials (http://www.advbiomaterials.com). Multimode low OH fibers with a core diameter of 400 μm, 30 μm cladding and equipped with ST connectors were purchased from Laser Components Nordic. The protective plastic coating was removed using standard stripping tools from RS Components. Micromanipulator with a motorized z-axis, optical O2 microsensors (50 μm tip diameter) and the fiber optical phase fluorimeter FirestingII were obtained from Pyro Science (Aachen, Germany, pyro-science.com).2.1. Sensor preparation
2.2 mg of Prussian blue (PB) and 140 mg of Cr-YAB were dispersed in 1 g of a 10% w/w D4 in ethanol/water (9/1) and the flat-cut end of an optical fiber was dip-coated with this dispersion. In detail, a small drop of the dispersion was placed on the tip of a flat spatula. The flat-cut fiber tip was slowly dipped into the drop until the entire tip was covered with the dispersion (as judged by naked eye or with a magnification glass). After slow retraction of the fiber from the dispersion, the sensor was dried at ambient air for half an hour and was ready to use thereafter.2.2. Recharging gel
To recharge/regenerate the sensor a 1% agarose gel containing 0.05 M of ascorbic acid in PBS (phosphate-buffered saline) adjusted to pH 7.4 was used. The ascorbic acid was dissolved in PBS, the buffer adjusted and subsequently the agar was added and dissolved at elevated temperatures. Around 3 ml of the hot solution was filled in 15 ml centrifugation tubes (vwr.com). After hardening the recharging gels could be stored in the fridge for several weeks.2.3. Measurement setup
The coated optical fiber was fixed in a Pasteur pipet using a rubber stopper and the pipet was mounted on a motorized micromanipulator controlled by the manufacturer's profiling software (Profix, Pyroscience GmbH, Germany). This allowed for rapid and reproducible vertical positioning of the sensor tip. A tube with the recharging gel was fixed below the sensor, the bottom of the tube was cut open, and the sensor was lowered through the gel in the tube. Calibration solutions or samples were placed underneath the tube opening and it was ensured that the recharging gel was in contact with the sample (no air between); see Fig. 2 and S10.† In general the following settings were used: the sensor was charged in the gel for 17 seconds and was lowered with a speed of 2 mm s−1 into the sample, where it was left for 5 seconds and then retracted into the gel. Measuring signals were recorded using the logger software of the fiber-optic phase fluorimeter (FirestingII, PyroScience GmbH, Germany). The modulation frequency of the meter was set to 470 Hz, the LED intensity and amplification were set to maximum levels, and the measurement time 1 data point each 0.75 s. The experimental setup is shown in Fig. 2. If not stated otherwise the measurements were performed at room temperature (22 °C) and checked with a digital thermometer. Every sensor was calibrated prior to and after the measurement.2.4. Biofilm measurements
Sediment samples covered with a well-developed brownish-green photosynthetic biofilm of diatoms and cyanobacteria were collected at a brackish seawater site at Løgstør Bredning, Limfjorden near the city of Aggersund, Denmark (57°00′ 02.15 N, 9°17′ 12.89 E). The samples were mounted in a flow chamber and flushed with a constant flow of aerated seawater from a seawater reservoir. The temperature of the seawater was kept constant with the help of an aquarium heater and was checked constantly with an electric thermometer (testo 110; http://www.testo.de). The biofilm was illuminated with a fiber-optic tungsten halogen lamp equipped with a collimating lens (KL-2500, Schott GmbH, Germany). The scalar irradiance at the biofilm surface was determined with an underwater quantum irradiance meter (ULM-500, WALZ, walz.com). The position of the microsensor measuring tips relative to the biofilm-surface were determined using an USB microscope (Dino-lite digital microscope, dino-light.eu). O2 concentration profiles were measured with a commercial fiber-optic O2 microsensor with 50 μm tip diameter (Pyro Science) that were calibrated according to the manufacturer.2.5. PMN measurements
Experimental details on the PMN measurements can be found in the ESI.†2.6. Data analysis
All data was analyzed using OriginPro 9 (Microcal, USA). As the sensor is based on a kinetic measuring principle, the change of signal over time correlates to the analyte concentration (see Fig. 3 and S12†). The intensity traces generated by the measurement device were imported and the first derivative was taken in order to obtain ΔS/Δt traces. As the change from PW to PB results in a decrease of the signal over time, −ΔS/Δt were extracted from the differential plot and correlated to the known H2O2 concentrations in case of calibrations. For the real measurements, measured −ΔS/Δt values were then converted into H2O2 concentrations with the help of the previously generated calibration curve that showed a linear correlation between −ΔS/Δt and [H2O2].3. Results
3.1. Sensor principle and design
The optical H2O2 sensor employs the pigment Prussian blue (PB) as sensing element undergoing a H2O2-dependent redox cycle. Prussian blue, i.e., ferric hexacyanoferrate, is one of the first discovered synthetic pigments37 and can be easily reduced to its uncolored form Prussian white (PW),38 that reacts rather specifically with H2O2 restoring the oxidized and deep blue-colored form (PB) and thus coupling the color change to H2O2 concentration (Fig. 1A). To enable a fast redox-cycling, i.e., reconditioning of PB to PW after H2O2 exposure, we constructed a recharger gel, containing ascorbic acid as a reductant that could be placed in close proximity to the sample.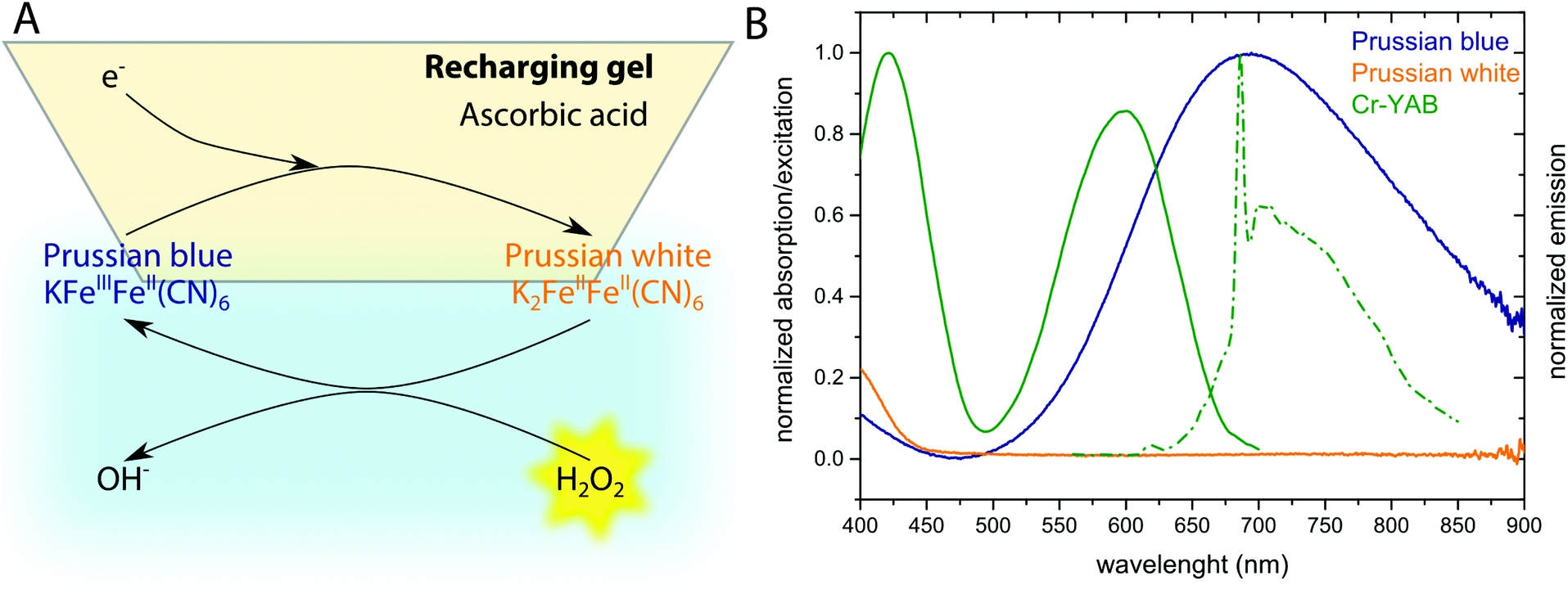 | ||
| Fig. 1 Prussian blue (PB) based optical H2O2 sensor concept. (A) Redox chemistry involved in the presented sensor. PB gets reduced when in the recharging gel, and reduced PW can be oxidized by H2O2 when the sensor is immersed into the sample. (B) Spectral properties of the indicator (PB/PW) and the coupled emitter Cr-YAB. Orange and blue curves show absorption spectra of PW and PB, respectively. Cr-YAB excitation (solid line) and luminescence emission spectra (dashed line) are shown in green. | ||
In order to produce a miniaturized optical fiber-based sensor, the H2O2 – induced color change of PB was coupled to a change in luminescence from chromium(III)-Activated Yttrium Aluminum Borate (Cr-YAB), a highly (photo)stable luminescent crystal.36 The excitation and emission spectra of Cr-YAB overlap with the broad absorption spectrum of PB (Fig. 1B), where oxidized PB will reduce the emission of Cr-YAB significantly due to the inner-filter effect, while colorless PW will not cause signal reduction. The spectral characteristics of the combined PB/Cr-YAB indicator immobilized on the tip of an optical fiber are compatible with a commercial fiber-optic meter for luminescence amplitude and lifetime measurements (FireStingII, Pyro Science GmbH, Germany). This instrument uses a 625 nm LED as excitation source and collects luminescence above 700 nm (RG9 glass filter) and enables flexible use with different types of optical fibers connected via a standard ST connector. An additional benefit of the emitter is that Cr-YAB is not susceptible to photobleaching and only shows cross-sensitivity to temperature (as every sensor) while being insensitive to other analytes like O2 or pH.36
The optical fiber tip with the immobilized PB/Cr-YAB indicator can be moved through the recharger gel with the help of a motorized micromanipulator. This enables fast measurements, as the sensor can be dipped in the sample for a few seconds and recharged directly thereafter by retracting it into the gel (Fig. 2). One measurement and recharging cycle takes ∼30 seconds, which is fast enough to follow many relevant biological processes involving H2O2. In terms of spatial resolution the size of the fiber tip (in this case 400 μm) and the diffusibility of H2O2 have to be considered. The diffusion coefficient of H2O2 in water, D(H2O2) at room temperature is ∼1.4 × 10−5 cm2 s−1.39 Within a t = 5 s measuring period, peroxide can thus diffuse a distance of ∼(2Dt)1/2 = 118 μm. As the sensor tip is larger than diffusive distance of H2O2 we conclude that a spatial resolution of <500 μm can be achieved.
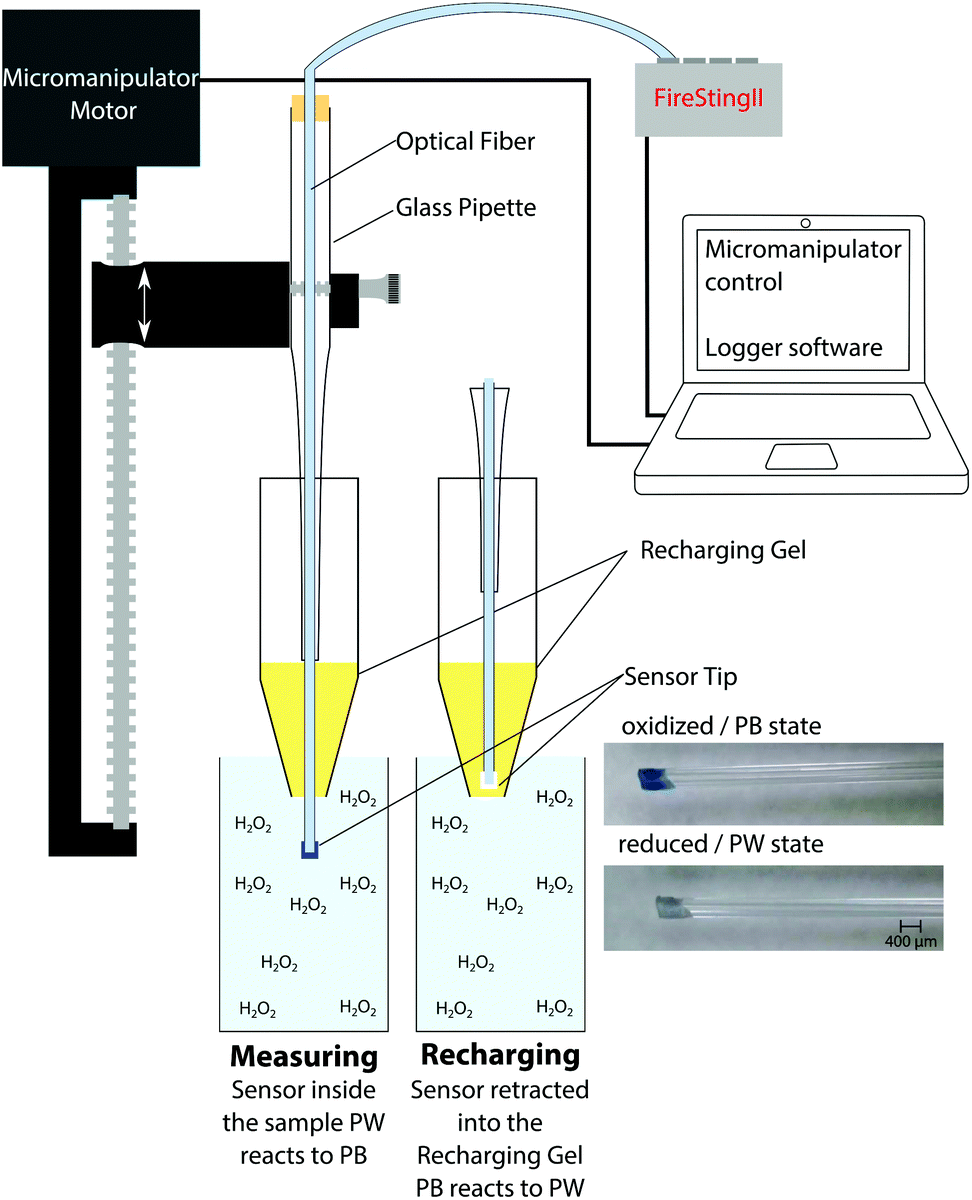 | ||
| Fig. 2 Schematic drawing of the H2O2 sensor measurement setup. The optical fiber sensor is fixed on a motorized micromanipulator that moves the sensor tip in and out of the recharging gel and the sample. The photographs show the fiber tip with immobilized PB/Cr-YAB in its oxidized and reduced state. | ||
As the reaction of PW with H2O2 is responsible for the change in signal intensity, a differential readout mode was chosen. By using the first derivative of the luminescence signal intensity, a more robust sensor readout was obtained. Even in cases when the recharging was not fully completed, the measured decline in signal intensity upon H2O2 exposure could be quantitatively correlated to the analyte concentration. By using differential signal readout, the sensor is less affected by optical interferences such as ambient light as long as light intensity does not change dramatically within the measurement cycle. The readout principle is shown in Fig. 3, where an example of the luminescence signal dynamics, its first derivative, and the obtained calibration curve for different H2O2 concentrations are shown. From the calibration curve of this sensor a detection limit of 0.4 μM and a limit of quantification of 1.3 μM was determined. This makes the reported sensor suitable for measurements in the range between 1 and 100 μM. We stress that the novel H2O2 sensor is not equilibrium based, in contrast to e.g. optical O2 sensors. The new sensor is based on the H2O2-dependent kinetics of the optical indicator redox status. This means that it is not necessary to await equilibrium and enables fast data acquisition; <30 s between measurement points. The kinetics based sensor concept also has implications for sensor stability and sensitivity.
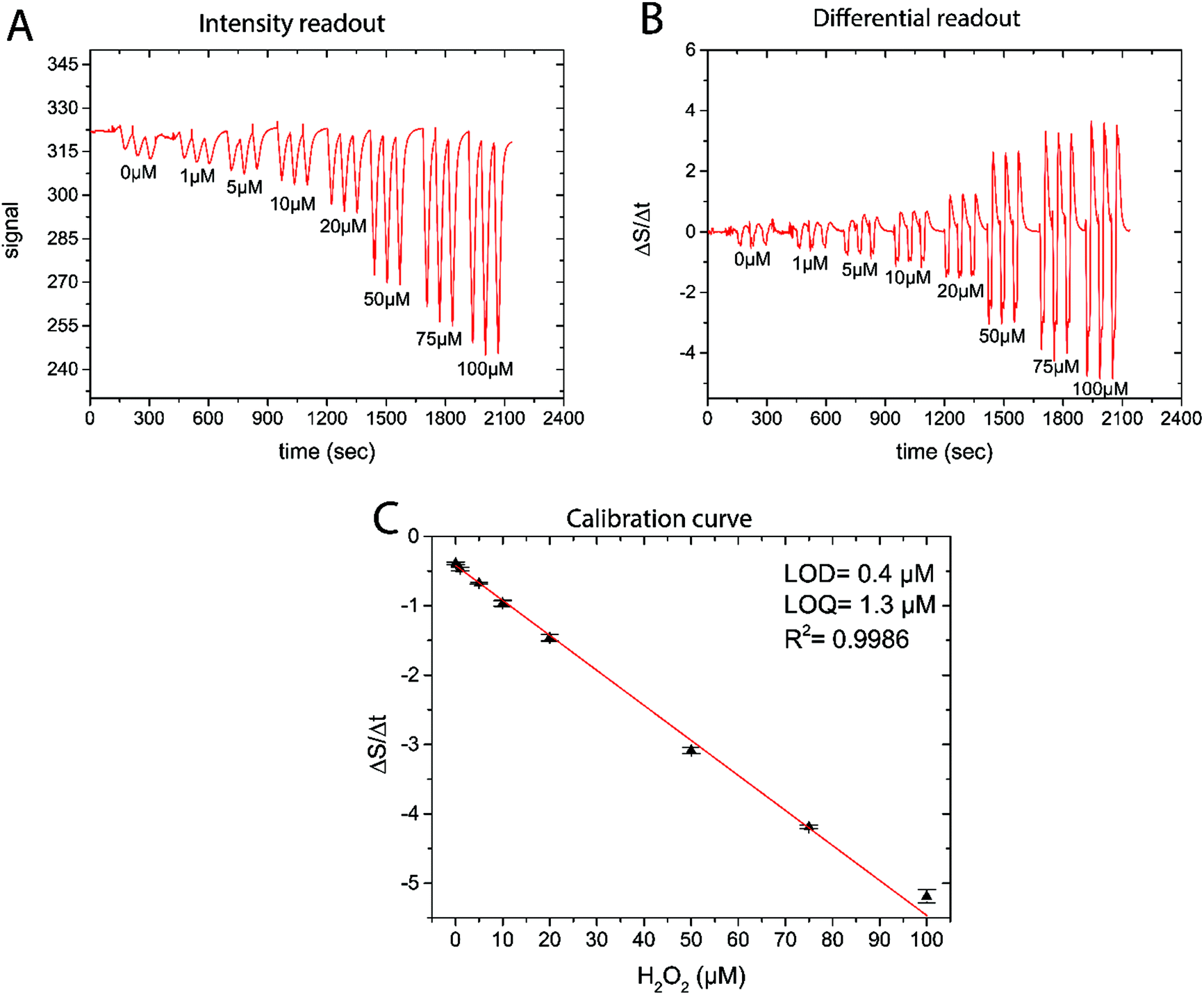 | ||
| Fig. 3 (A) Raw sensor luminescence intensity trace during calibration in eight different H2O2 containing solutions (pH = 7.4). (B) Differentiated sensor signal trace showing a higher reproducibility than the original raw signal data. (C) Calibration curve obtained from the differential readout (mean ± SD; n = 3). | ||
3.2. Cross-sensitivities and limitations
As the H2O2-catalyzed conversion of PW to PB is responsible for the change in signal, all factors either stabilizing or destabilizing PW lead to cross-sensitivities. Obviously, temperature is affecting the sensitivity and therefore has to be kept as constant as possible. Additionally, we found that increased O2 levels lead to an increase in sensor sensitivity. When calibrations were conducted in O2 free and O2 saturated water the sensitivity increased by ∼18% with rising oxygenation (see ESI Fig. S6†). Also the effect of pH on the sensor performance has to be taken into account. Increased pH leads to a decreased sensitivity (see ESI Fig. S5†). The calibration obtained at pH 9 exhibited a slope ∼20% less steep than a calibration curve obtained at pH 4. In contrast, calibration curves measured at pH 4 and 7.4 only showed a 7% difference in sensitivity. This can be explained by PW being less stable at increased pH. It was observed that only the PW form is prone to cause sensor instability and changes in sensitivity. The PB state was stable even at pH 9 for over 17 hours without showing any indicator leakage or decomposition that would lead to a signal increase (see ESI Fig. S1†). Obviously, the PB/PW redox couple is strongly affected by reducing agents. As ascorbic acid is used within the recharging gel, the sensor will be strongly responding to ascorbic acid. This ultimately limits the applicability in systems with high ascorbic acid concentrations. On the contrary, hydrogen sulfide (H2S) and glutathione (GSH) did not affect the sensor signal (data not shown). The sensor response to other types of ROS was evaluated (see Fig. S7†) and no cross-sensitivity towards any of the tested ROS was found. An apparent response to superoxide could be attributed to H2O2 produced from dismutation. A technical aspect regarding cross-sensitivity is that ascorbic acid may diffuse out of the gel or H2O2 into the gel. We tried to reduce this effect by using a minimal contact area between the gel and the sample (diameter <5 mm). Obviously by using a 3 dimensional (xyz) micromanipulator recharging could be achieved more remote from the sample and would overcome this limitation. Further information on the sensor performance can be found in the ESI.†3.3. Sensor stability
As every sensor is unique due to the fiber tip coating process, calibration before use is essential. It was observed that sensor sensitivity to H2O2 could vary quite dramatically; between −0.05 and −0.005 ΔS/Δt μM−1. Therefore, a short (at least 4 point) calibration was obtained prior to every measurement series. Calibrations were obtained by diluting a fresh H2O2 solution in the respective medium (buffer). All calibrations showed high linearity, and recalibration after the measurement yielded identical calibration curves; recalibration was done to test for sensor drift throughout the measurement. The repeatability of the sensor reading was tested and showed a RSD of ≤5% (see Fig. S4†). The same sensor could be used for multiple subsequent measurements over several days. The main limitation in terms of sensor stability was the mechanical stability of the sensor coating. Typically before any other degradation was observed, the sensor chemistry detached from the optical fiber and a more stable immobilization of the sensor chemistry is under investigation.3.4. Biological H2O2 measurements
We tested the new optical H2O2 sensor in several biological applications spanning from measurements in defined enzyme systems, over concentration profiling in a complex natural biofilm to quantification of H2O2 concentration dynamics in activated neutrophils.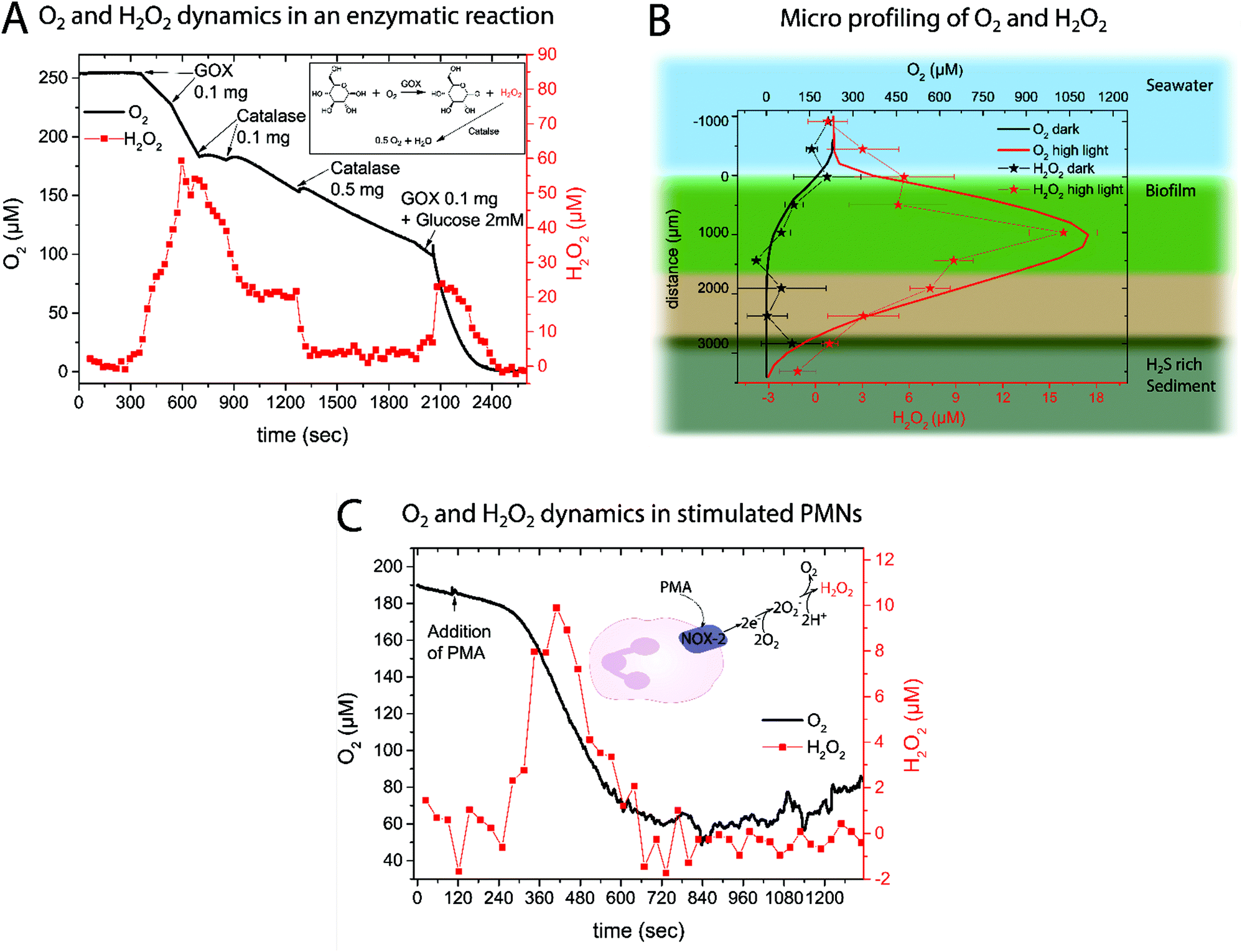 | ||
| Fig. 4 (A) Dynamics of O2 and H2O2 concentrations measured simultaneously with an O2 micro-optode and the new H2O2 sensor in a buffered glucose solution (1 mM) upon addition of glucose oxidase (GOX) and catalase at the marked time points. The insert shows the chemical reactions involved. (B) Micro profiling of O2 (solid line) and H2O2 (stars) concentrations in a natural photosynthetic biofilm under high irradiance (2000 μM photons m−2 s−1; red lines and symbols) and in the dark (black lines and symbols). The H2O2 profile shows the average of 3 measurements ± SD; for O2 only one of the three measured and highly similar profiles is plotted. (C) Respiratory burst in activated neutrophils as shown by O2 and H2O2 concentration dynamics in a PMN suspension (5 × 106 cells per mL in Krebs–Ringer buffer at 37 °C) upon addition of an external stimuli (PMA) as indicated by an arrow. The insert shows a simplified version of the processes leading to H2O2 buildup. | ||
Obviously the cross-sensitivity towards O2 and pH has to be considered when analyzing the data. It is well known that due to photosynthesis not only the O2 levels but also the pH increases.40 Luckily those effects were found to counteract on the sensor sensitivity and therefore minimize the cross-effects in the light; nevertheless leading to uncertainty. In the dark and also in the bulk water no H2O2 was measured, albeit some offset in the zero response of the new H2O2 sensor was observed with depth in the biofilm38 (see Discussion).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 45 and accelerated O2 consumption.46 Simultaneous measurements of O2 and H2O2 concentration dynamics in isolated neutrophils activated with PMA confirmed the expected changes (Fig. 4C). About 2 minutes after PMN stimulation by PMA, an increased O2 consumption and H2O2 production was observed. The extracellular H2O2 concentration peaked at ∼10 μM after 7 min. When extracellular O2 levels started to stabilize, extracellular H2O2 disappeared, presumably due to activity of catalase and myeloperoxidase.47 A replicate of the experimental data in Fig. 4C can be found in the ESI (Fig. 8†).
45 and accelerated O2 consumption.46 Simultaneous measurements of O2 and H2O2 concentration dynamics in isolated neutrophils activated with PMA confirmed the expected changes (Fig. 4C). About 2 minutes after PMN stimulation by PMA, an increased O2 consumption and H2O2 production was observed. The extracellular H2O2 concentration peaked at ∼10 μM after 7 min. When extracellular O2 levels started to stabilize, extracellular H2O2 disappeared, presumably due to activity of catalase and myeloperoxidase.47 A replicate of the experimental data in Fig. 4C can be found in the ESI (Fig. 8†).
4. Discussion
Hydrogen peroxide (H2O2) is a key analyte in environmental and biomedical research. Quantification of this reactive analyte enables the study of the current redox status of cells and tissues. Hitherto, optical H2O2 detection has focused on the use of cell permeable dyes selective to H2O2. Despite their high spatial resolution, the application of such indicators mainly yields qualitative information on the presence/absence of H2O2 and is difficult, if not impossible, to extract quantitative measures of H2O2 concentration from such probes.Here we present a fiber-optic H2O2 sensor for H2O2 concentration measurements with a spatial resolution of ∼500 μm and a time resolution of ∼30 seconds. The system is based on the PB induced reduction of the emission of a secondary emitter. In order to provide the highest possible photostability, an inorganic emitter, namely Cr-YAB was chosen. Although this crystalline material was previously used for temperature sensing, it has to be noted that mainly the luminescence decay time is sensitive to temperature, while the intensity is much less affected.36
A crucial step of applying the new H2O2 sensor for biological measurements is the possibility of rapid regenerating. This was achieved by producing a recharger gel and using automated approach and retract protocols of a computer controlled micromanipulator. It was possible to position the recharger gel in close proximity to the sample; e.g. 5 mm above the biofilm or in the calibration solution (see Fig. 2 and ESI†). This is a major advancement in comparison to irreversible H2O2 probes. While irreversible probes are superior for localizing H2O2 especially within cells or tissues,8 this sensor enables H2O2 quantification over a long time. Computer controlled positioning into the sample and retraction of the sensor into the recharging gel facilitates easy handling.
Monitoring an enzyme reaction using the proposed sensor showed that it is possible to follow the evolution of H2O2 over time. With intervals between measurement points of around 30 seconds, the sensor system is able to follow H2O2 dynamics during relatively fast reactions. The obtained H2O2 concentration traces are in very good agreement with the simultaneously measured O2 dynamics.
To the best of our knowledge this is the first quantification of H2O2 in a natural biofilm with an optical H2O2 sensor. Previously, scanning electrochemical analysis was used to measure H2O2 concentration in a photosynthetic biofilm48 or amperometric microsensors were used to study the effect of externally added H2O2 (>50 mM)49,50 on a lab-grown biofilm. In the presented data it is remarkable that the H2O2 profile follows the O2 profile indicating that increased photosynthetic O2 production leads to an increase of ROS. This is in good agreement with previous observations51,52 suggesting that H2O2 production is associated with photosynthesis under high light stress.53 ROS are important potential byproducts in the photosynthesis pathway and can be produced at Photosystem I54 and II51via a variety of reactions. Our measurements thus clearly demonstrate that H2O2 production is tightly linked to the local O2 concentration and that H2O2 can accumulate in biofilms as a reaction to light stress.55
We note that the standard deviation of the measured H2O2 concentration profiles is rather high and that the observed cross-sensitivities to pH and O2 are to be taken into account as photosynthetic biofilms show pronounced O2 and pH dynamics.40 A quantification of O2 and pH interference on H2O2 sensor calibration can be found in the ESI.† The fact that the measured H2O2 levels in the dark apparently became negative might be due to the reductive environment that builds up when O2 is gone leading to stabilization of PW. The cross-sensitivity to pH and reductants that will stabilize the PW form are the current limitations of the sensor. Nevertheless, the measured profile gives valuable information on the buildup of H2O2 and the new H2O2 sensor is an important new tool for studies investigating how environmental conditions affect in situ ROS production in natural aquatic systems.
As an example of biomedical application the respiratory burst of neutrophils was investigated. The H2O2 is produced by neutrophils as a response to bacterial infections5 by converting O2 to first superoxide (O2−) and subsequently H2O2 (see insert in Fig. 4C). This process is facilitated using the plasma membrane-associated NADPH oxidase that gets activated upon contact with bacteria or due to stimulation (e.g. by PMA). So far H2O2 generation in this highly important immune response has been monitored using electrochemical systems.45,47 Here we showed that the rechargeable optical H2O2 sensor is capable of measuring H2O2 generation in this bio-medically relevant system. The obtained O2 and H2O2 time traces follow each other and lead to the conclusion that the consumed O2 is rapidly converted into superoxide and subsequently H2O2. The obtained results are in good agreement with previous findings45 regarding the extracellular H2O2 concentration.
5. Conclusion
We developed a versatile rechargeable fiber-optic H2O2 sensor for fast, spatially resolved and quantitative measurement of extracellular H2O2 concentration in complex media. While electrochemical H2O2 quantification has seen a lot of development over the years,29 this paper presents the first fiber-optic H2O2 sensor enabling quantitative concentration measurements in environmental and biomedical applications. As H2O2 often reacts irreversibly with optical indicators,9 the possibility of recharging the sensor makes this sensing principle very promising for a wide range of applications. By bringing the recharging process in close proximity to the sample and by applying differential readout, a fast and robust measurement scheme was developed that is applicable in a wide range of complex environmental and even some biomedical samples. Future development should try to decrease current cross-sensitivities, lower the detection limit and go towards further miniaturization to also enable less invasive measurement in tissues.Acknowledgements
Sofie L. Jakobsen is thanked for technical assistance and for collecting the biofilm sample. Prof. Ingo Klimant and Dr Sergey M. Borisov from the Graz University of Technology are thanked for fruitful discussions throughout the development of this sensor and for kindly providing the Cr-YAB used in this study. Dr Roland Thar, CEO of Pyro Science, is thanked for his help and advice. Dr Mette Kolpen, Rigshospitalet Copenhagen, is thanked for helping isolate the neutrophil cells. This study was supported by a grant from the Villum Foundation, a Sapere-Aude Advanced grant from the Danish Research Council for Independent Research, and a grant by the Danish Research Council for Independent Research | Technical and Production Sciences.References
- D. R. Gough and T. G. Cotter, Cell Death Dis., 2011, 2, e213 CrossRef CAS PubMed.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS PubMed.
- B. Halliwell and J. Gutteridge, Free Radicals in Biology and Medicine, Oxdord Univerity Press, Oxford, 2007 Search PubMed.
- M. P. Murphy, A. Holmgren, N.-G. Larsson, B. Halliwell, C. J. Chang, B. Kalyanaraman, S. G. Rhee, P. J. Thornalley, L. Partridge, D. Gems, T. Nyström, V. Belousov, P. T. Schumacker and C. C. Winterbourn, Cell Metab., 2011, 13, 361–366 CrossRef CAS PubMed.
- W. M. Nauseef, Immunol. Rev., 2007, 219, 88–102 CrossRef CAS PubMed.
- C. C. Winterbourn, Methods Enzymol., 2013, 528, 3–25 CAS.
- C. C. Winterbourn, Nat. Chem. Biol., 2008, 4, 278–286 CrossRef CAS PubMed.
- J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973–984 CrossRef CAS PubMed.
- A. R. Lippert, G. C. Van de Bittner and C. J. Chang, Acc. Chem. Res., 2011, 44, 793–804 CrossRef CAS PubMed.
- X. Chen, X. Tian, I. Shin and J. Yoon, Chem. Soc. Rev., 2011, 40, 4783–4804 RSC.
- V. V. Belousov, A. F. Fradkov, K. A. Lukyanov, D. B. Staroverov, K. S. Shakhbazov, A. V. Terskikh and S. Lukyanov, Nat. Methods, 2006, 3, 281–286 CrossRef CAS PubMed.
- M. B. Grisham, Comp. Biochem. Physiol., Part A: Mol. Integr. Physiol., 2013, 165, 429–438 CrossRef CAS PubMed.
- C. C. Winterbourn, Biochim. Biophys. Acta, 2014, 1840, 730–738 CrossRef CAS PubMed.
- B. C. Dickinson, C. Huynh and C. J. Chang, J. Am. Chem. Soc., 2010, 132, 5906–5915 CrossRef CAS PubMed.
- J. F. Woolley, J. Stanicka and T. G. Cotter, Trends Biochem. Sci., 2013, 38, 556–565 CrossRef CAS PubMed.
- M. M. Tarpey and I. Fridovich, Circ. Res., 2001, 89, 224–236 CrossRef CAS PubMed.
- M. Kühl, Methods Enzymol., 2005, 397, 166–199 Search PubMed.
- N. P. Revsbech, Methods Enzymol., 2005, 397, 147–166 CAS.
- I. Klimant, V. Meyer and M. Kühl, Limnol. Oceanogr., 1995, 40, 1159–1165 CrossRef CAS.
- M. Kühl, Y. Cohen, T. Dalsgaard, B. B. Jørgensen and N. P. Revsbech, Mar. Ecol.: Prog. Ser., 1995, 117, 159–172 CrossRef.
- P. Jeroschewski, C. Steuckart and M. Kühl, Anal. Chem., 1996, 68, 4351–4357 CrossRef CAS.
- M. Kolpen, M. Kühl, T. Bjarnsholt, C. Moser, C. R. Hansen, L. Liengaard, A. Kharazmi, T. Pressler, N. Høiby and P. Ø. Jensen, PLoS One, 2014, 9, e84353 Search PubMed.
- T. Malinski and Z. Taha, Nature, 1992, 358, 676–678 CrossRef CAS PubMed.
- C. Calas-Blanchard, G. Catanante and T. Noguer, Electroanalysis, 2014, 26, 1277–1286 CrossRef CAS.
- N. V. Kulagina and A. C. Michael, Anal. Chem., 2003, 75, 4875–4881 CrossRef CAS PubMed.
- D. W. M. Arrigan, Analyst, 2004, 129, 1157–1165 RSC.
- M. Delvaux, A. Walcarius and S. Demoustier-Champagne, Anal. Chim. Acta, 2004, 525, 221–230 CrossRef CAS.
- Y. Wang, J.-M. Noël, J. Velmurugan, W. Nogala, M. V. Mirkin, C. Lu, M. Guille Collignon, F. Lemaître and C. Amatore, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11534–11539 CrossRef CAS PubMed.
- W. Chen, S. Cai, Q.-Q. Ren, W. Wen and Y.-D. Zhao, Analyst, 2012, 137, 49–58 RSC.
- F. Ricci and G. Palleschi, Biosens. Bioelectron., 2005, 21, 389–407 CrossRef CAS PubMed.
- A. A. Karyakin, Electroanalysis, 2001, 13, 813–819 CrossRef CAS.
- A. A. Karyakin and E. E. Karyakina, Sens. Actuators, B, 1999, 57, 268–273 CrossRef CAS.
- O. S. Wolfbeis, A. Dürkop, M. Wu and Z. Lin, Angew. Chem., Int. Ed., 2002, 41, 4495–4498 CrossRef CAS.
- J. F. Botero-Cadavid, A. G. Brolo, P. Wild and N. Djilali, Sens. Actuators, B, 2013, 185, 166–173 CrossRef CAS.
- H. A. Khorami, J. F. Botero-Cadavid, P. Wild and N. Djilali, Electrochim. Acta, 2014, 115, 416–424 CrossRef CAS.
- S. M. Borisov, K. Gatterer, B. Bitschnau and I. Klimant, J. Phys. Chem. C, 2010, 114, 9118–9124 CAS.
- J. Brown, Philos. Trans. R. Soc. London, 1724, 33, 17–24 CrossRef.
- R. Koncki, T. Lenarczuk and S. Głąb, Anal. Chim. Acta, 2000, 424, 27–35 CrossRef CAS.
- S. A. M. van Stroe-Biezen, F. M. Everaerts, L. J. J. Janssen and R. A. Tacken, Anal. Chim. Acta, 1993, 273, 553–560 CrossRef CAS.
- M. Nielsen, L. H. Larsen, L. D. M. Ottosen and N. P. Revsbech, Sens. Actuators, B, 2015, 215, 1–8 CrossRef CAS.
- C. W. Baldridge and R. W. Gerard, Am. J. Physiol., 1932, 103, 235–236 Search PubMed.
- B. M. Babior, R. S. Kipnes and J. T. Curnutte, J. Clin. Invest., 1973, 52, 741–744 CrossRef CAS PubMed.
- G. Y. N. Iyer, M. F. Islam and J. H. Quastel, Nature, 1961, 192, 535–541 CrossRef CAS.
- L. R. DeChatelet, P. S. Shirley and R. B. Johnston, Blood, 1976, 47, 545–554 CAS.
- S. T. Test and S. J. Weisst, Biol. Chem., 1984, 259, 399–405 CAS.
- J. E. Repine, J. G. White, C. C. Clawson and B. M. Holmes, J. Clin. Invest., 1974, 54, 83–90 CrossRef CAS PubMed.
- A. J. Kettle and C. C. Winterbourn, Biochemistry, 2001, 40, 10204–10212 CrossRef CAS PubMed.
- X. Liu, M. M. Ramsey, X. Chen, D. Koley, M. Whiteley and A. J. Bard, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2668–2673 CrossRef CAS PubMed.
- X. Lu, F. Roe, A. Jesaitis, Z. Lewandowski and X. Liu, Biotechnol. Bioeng., 1998, 59, 156–162 CrossRef CAS PubMed.
- P. S. Stewart, F. Roe, J. Rayner, J. G. Elkins, Z. Lewandowski, U. A. Ochsner and D. J. Hassett, Appl. Environ. Microbiol., 2000, 66, 836–838 CrossRef CAS PubMed.
- P. Pospísil, Biochim. Biophys. Acta, 2009, 1787, 1151–1160 CrossRef PubMed.
- M. P. Lesser, Annu. Rev. Physiol., 2006, 68, 253–278 CrossRef CAS PubMed.
- A. Latifi, M. Ruiz and C. C. Zhang, FEMS Microbiol. Rev., 2009, 33, 258–278 CrossRef CAS PubMed.
- K. Apel and H. Hirt, Annu. Rev. Plant Biol., 2004, 55, 373–399 CrossRef CAS PubMed.
- J. Collen, M. J. Del Rio, G. Garcia-Reina and M. Pedersen, Planta, 1995, 196, 225–230 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00864j |
| This journal is © The Royal Society of Chemistry 2016 |