
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Quantification of selected volatile organic compounds in human urine by gas chromatography selective reagent ionization time of flight mass spectrometry (GC-SRI-TOF-MS) coupled with head-space solid-phase microextraction (HS-SPME)
Paweł
Mochalski
*a and
Karl
Unterkofler
ab
aBreath Research Institute of the University of Innsbruck, Rathausplatz 4, A-6850 Dornbirn, Austria. E-mail: pawel.mochalski@uibk.ac.at; Fax: +43-512-504-6724636; Tel: +43-512-504-24636
bVorarlberg University of Applied Sciences, Hochschulstr. 1, A-6850 Dornbirn, Austria
First published on 19th May 2016
Abstract
Selective reagent ionization time of flight mass spectrometry with NO+ as the reagent ion (SRI-TOF-MS(NO+)) in conjunction with gas chromatography (GC) and head-space solid-phase microextraction (HS-SPME) was used to determine selected volatile organic compounds in human urine. A total of 16 volatiles exhibiting high incidence rates were quantified in the urine of 19 healthy volunteers. Amongst them there were ten ketones (acetone, 2-butanone, 3-methyl-2-butanone, 2-pentanone, 3-methyl-2-pentanone, 4-methyl-2-pentanone, 2-hexanone, 3-hexanone, 2-heptanone, and 4-heptanone), three volatile sulphur compounds (dimethyl sulfide, allyl methyl sulfide, and methyl propyl sulfide), and three heterocyclic compounds (furan, 2-methylfuran, 3-methylfuran). The concentrations of the species under study varied between 0.55 nmol L−1 (0.05 nmol mmol−1creatinine) for allyl methyl sulfide and 11.6 μmol L−1 (1.54 μmol mmol−1creatinine) for acetone considering medians. Limits of detection (LODs) ranged from 0.08 nmol L−1 for allyl methyl sulfide to 1.0 nmol L−1 for acetone and furan (with RSDs ranging from 5 to 9%). The presented experimental setup assists both real-time and GC analyses of volatile organic compounds, which can be performed consecutively using the same analytical system. Such an approach supports the novel concept of hybrid volatolomics, an approach which combines VOC profiles obtained from two or more body fluids to improve and complement the chemical information on the physiological status of an individual.
1. Introduction
The analysis of volatile organic compounds (VOCs) released by the human organism emerges as a promising powerful tool for medical diagnosis and therapeutic monitoring, tracking of microbiota activity, or exposure to environmental pollutants and/or toxins.1–3 It targets markers in different human fluids and tissues including breath, urine, saliva, feces, blood, and sweat. Urine is a particularly interesting reservoir of potential disease markers and exhibits a number of advantages over other body fluids. It is readily and noninvasively collectable and may be sampled as often as it is desirable without discomfort for the patient. A wide range of different types of biomarkers from volatiles to non-volatiles can accumulate in urine and subsequently be excreted during urination providing the opportunity to detect changes in cells/tissues metabolism related to disease development including cancer. The major disadvantages of this fluid embrace the variation in dilution and the rate of urine production. Indeed, there is growing evidence provided by a number of recent studies suggesting that the disease state influences the pattern of volatile organic compounds in urine and that the chemical analysis of this pattern could be a valuable diagnostic tool. For instance, urine odor was shown to contain disease biomarkers, which allow detection of bladder cancer,4–6 colorectal cancer,7 kidney cancer,8 or prostate cancer.9–13The composition of urine in humans has received broad attention and a number of sophisticated analytical techniques have been employed to support the identification of volatiles in urine, or urine head-space. These embrace sensor arrays (e-noses),6,11,12 and their combination with gas chromatography,5 selected ion flow tube mass spectrometry (SIFT-MS),14,15 ion mobility spectrometry,7,16 nuclear magnetic resolution spectroscopy (NMR),17 or gas chromatography mass spectrometry (GC-MS).18–21 A compendium of 1849 VOCs forming the human volatolome (pool of all VOCs in human organism) provides 279 species identified in urine.22 Interestingly, the majority of reports yield only qualitative data, or semi-quantitative data, i.e. names of identified compounds, their occurrence in urine samples, or method-specific features of the signal (e.g., peak areas, signal relative strength). Quantitative data are relatively sparse and limited to single compounds, or selected classes of species.21,23–30
Selective reagent ionization time of flight mass spectrometry (SRI-TOF-MS)2,31,32 is a powerful analytical technique for detecting and quantifying VOCs in biological, medical, and environmental matrices. This is due to its versatility, excellent sensitivity, and real-time response. In particular, the last feature is intensively exploited in breath gas analysis to track short-time changes in VOCs levels, which may follow rapid physiological processes occurring in the body.33–36 The employment of a time-of-flight (TOF) mass filter in SRI-TOF-MS instruments provided a higher mass (m/z) resolving power (up to 5000 m/Δm), which supports the separation of isobaric ions; whereas, soft chemical ionization of volatiles by precursor (reagent) ions such as H3O+, NO+, or O2+ facilitates the interpretation of spectra.
Due to the above-mentioned lack of studies reporting the levels of VOCs in human urine, the primary goal of this work was to provide reliable reference concentrations for selected urinary VOCs. Altogether 16 volatiles abundant and omnipresent in urine were pre-selected on the basis of the earlier qualitative studies: 10 ketones, 3 volatile sulphur compounds and 3 heterocyclic compounds.18–20 For this purpose selective reagent ionization time of flight mass spectrometry in conjunction with gas chromatography (GC) and solid-phase microextraction (SPME) as the pre-concentration method was used. While the coupling of the SRI-TOF-MS instrument with a GC column compensated the low selectivity of SRI-TOF-MS and assisted the identification of urinary species, SPME supported the extraction of VOCs of interest from the liquid phase. SPME was proved to be an efficient technique for the extraction of urinary VOCs by numerous studies.18–20,24 Moreover, the SRI-TOF-MS instrument worked with NO+ as the reagent ion (SRI-TOF-MS(NO+)). A valuable feature of NO+ reactions with volatile organic compounds is the diversity of reaction mechanisms.37,38 These embrace charge transfer, hydride ion (H−) transfer, hydroxide ion (OH−) transfer, alkoxide ion (OR−) transfer, and NO+ ion-molecule association. Interestingly, different chemical classes of VOCs undergo characteristic reaction processes with NO+, which is analytically very useful and in some cases helps identify structural isomers.
2. Experimental
2.1. Materials and calibration mixtures
Liquid multi-compound calibration mixtures were prepared from pure liquid substances. The reference substances with purities ranging from 98 to 99.8% were purchased from Sigma-Aldrich (Austria), Fluka (Switzerland), and SAFC (USA).Urine volatiles were calibrated using human urine samples. Calibration solutions were prepared in three steps. The primary solutions were produced in silanized glass vials (nominal volume 50 mL, real volume 58 mL, Macherey-Nagel, Germany) crimped with 1.3 mm butyl/PTFE septa (Macherey-Nagel, Germany) by adding 1–500 μL (depending on the desired urine concentration) of a pure liquid substance into 57 mL of distilled water (arium® pro, Sartorius Stedim Biotech GmbH, Germany), followed by intensive stirring for 15 minutes at room temperature. Next, the primary solution was additionally diluted by transferring 50 μL of a primary standard into 55 mL of water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1100). The final calibration solutions were prepared by transferring appropriate aliquots of a secondary solution into crimped vials containing urine samples. Calibration curves were obtained on the basis of 2-fold analyses of 5 distinct and independent concentration levels.
:1100). The final calibration solutions were prepared by transferring appropriate aliquots of a secondary solution into crimped vials containing urine samples. Calibration curves were obtained on the basis of 2-fold analyses of 5 distinct and independent concentration levels.
2.2. Human subjects and urine sampling
A cohort of 19 healthy volunteers (10 males, 9 females, age range 24–58 years, all non-smokers) was recruited. All subjects gave written informed consent to participate and completed a questionnaire describing their health status, as well as recent food intake. The sample collection was approved by the Ethics Commission of the Innsbruck Medical University. No special dietary regimes were applied.For VOCs analyses approximately 18 mL of morning urine were collected into 20 mL glass vials closed with 1.3 mm butyl/PTFE septa (both from Macherey-Nagel, Germany) immediately after urinating. In addition to this, 9 mL of urine were transferred into plastic urine-monovette vessels (Sarstedt, Germany) for the subsequent creatinine analysis. In parallel with urine sampling, one blank sample containing 18 mL of distilled water (arium® pro, Sartorius Stedim Biotech GmbH, Germany) was taken per volunteer using the same materials as in the case of urine sampling for VOCs’ analysis. This was done to identify possible contaminants stemming from sources other than urine. Blank samples were analyzed analogously as urine samples and the resulting concentration levels were subtracted from the respective values in the associated urine samples.
2.3. Urine sample preparation and HS-SPME procedure
Extraction of volatiles from urine samples was performed in headspace vials (20 mL, Gerstel, Germany) crimped with 1.3 mm butyl/PTFE septa and containing stirring bars. Vials were evacuated by means of a membrane pump and 0.25 mL of Dulbecco's phosphate-buffered saline (PAA Laboratories, Austria) and 1.25 mL of distilled water were added with the help of a glass syringe. The addition of the former aimed at the normalization of pH of processed samples. Next, a small amount (0.3–0.4 μL) of an anti-foam agent for purge and trap (Antifoam C, Restek, USA) was added to the vials to prevent the urine from foaming. Finally, pressure in the vials was balanced with high-purity nitrogen (6.0–99.9999%) and 0.5 mL of urine was transferred into it using a Hamilton syringe (Hamilton, Switzerland). Effectively, the urine sample was diluted at the ratio of 1:3. This was done to compensate the matrix effect between urine samples and water.30
Head space solid-phase microextraction (HS-SPME) was carried out automatically using a multipurpose sampler MPS (Gerstel, Germany). Prior to the extraction, the HS vials were incubated at 37 °C for 20 min in a temperature-controlled agitator and stirred intensively (750 rpm). Extraction was achieved by inserting a 75 μm Carboxen-PDMS SPME fiber (Supelco, Canada)18,19 into the vials and exposing it to the head-space gas for 50 minutes.19 This extraction time was found to be a reasonable trade-off between good detection limits and sampling duration. Subsequently, the fiber was introduced into the inlet of the gas chromatograph where the volatiles of interest were thermally desorbed at 290 °C in a splitless mode (1 min). The fiber was conditioned at 290 °C for 5 minutes prior to each analysis.
2.4. GC-SRI-TOF-MS analysis
Chromatographic separation was performed using an Agilent 7890A gas chromatograph (Agilent, USA). During desorption the inlet operated in the splitless mode (1 min), followed by a split mode at the ratio of 1 to 50. The urine volatiles were separated using an Rt-Q-BOND column (30 m × 0.25 mm, film thickness 8 μm, 100% divinylbenzene phase, Restek, USA) working in a constant flow of helium at 1 mL min−1. The column temperature program was as follows: 60 °C for 1 min, increase at a rate of 5 °C min−1 to 260 °C, constant temperature of 260 °C for 13 min. The effluent from the column was introduced into the drift tube of the SRI-TOF-MS instrument via a heated (150 °C) empty fused silica tube (1.5 m × 0.25 mm), which also served as a particle trap column. One end of the tube protruded 2–3 mm into the volume of the drift tube, whereas, the second was connected with the GC column using a quick-seal column connector (Markes International Ltd, UK). Such a connection facilitates quick change of the GC column without venting the mass spectrometer and protects the SRI-TOF-MS instruments from damage induced by possible particles shedding in PLOT columns. The standard transfer line of the SRI-TOF-MS instrument, used during real-time analyses, sampled dry and purified air at a steady flow rate of 20 mL min−1. This flow of a VOCs-free gas was necessary to reduce the background of VOCs in the MS and to maintain its appropriate analytical conditions.An Ionicon Analytik (Innsbruck, Austria) type 8000 SRI-TOF-MS instrument operating in the NO+ mode was used during the analyses.31 The settings of the ion source were chosen as follows: ion source current 5 mA, source voltage (Us) 20 V, source-out voltage (Uso) 70 V, and source valve opening 40%. With these settings the levels of the major impurity ions relative to that of the NO+ precursor ion were H3O+; 0.1–0.2%, O2+; 2–2.5%, and NO2+; 1.5–2%. The NO+/VOCs reactions occurred in the drift tube at a total pressure of 2.3 mbar and a gas temperature of 60 °C. The voltage along the drift section was set to 500 V leading to an E/N ratio of approximately 110 Td. The spectral scans of the TOF analyzer ranged from approximately m/z 2.7 to 500 and were acquired in a time of 500 ms by co-adding 12500 single 40 μs long TOF-MS extractions recorded at a sampling frequency of 1/Δt = 10 GHz. This sampling rate was found to be a reasonable trade-off between the satisfactory number of scans over the peaks needed for quantification and MS sensitivity. The actual mass resolution obtained from the detected peaks was approximately 4000 at m/z 100. The total duration of a single measurement was 54 minutes, which corresponds to the length of the column temperature program. The mass calibration was based on three impurity peaks always present in the spectra: H3O+ (19.0178), 15NO+ (30.9945), and NO2+ (45.9924).
The identification of compounds relied on the exact mass measurement of detected parent ions, the characteristic reaction mechanisms of VOCs with NO+, and the comparison of the retention times of peaks of interest with retention times obtained for standard mixtures prepared from pure compounds. Peak integration was based on the extracted time profiles of parent ions. The retention times of VOCs under scrutiny and parent ions used for their identification and quantification are presented in Table 1.
| VOC | CAS | Parent ion formula | Parent ion m/z [Th] | R t [min] | LOD [nmol L−1] | RSD [%] | R 2 | Linear range [nmol L−1] |
|---|---|---|---|---|---|---|---|---|
| Furan | 110-00-9 | C4H4O+ | 68.0265 | 15.38 | 1.0 | 7.4 | 0.986 | 3.0–31.3 |
| Acetone | 67-64-1 | C3H6O·NO+ | 88.0411 | 16.08 | 1.0 | 9.0 | 0.974 | 3.0–52000 |
| Dimethyl sulfide | 75-18-3 | C2H6S+ | 62.0199 | 16.60 | 0.4 | 8.0 | 0.980 | 1.2–24.0 |
| Furan, 2-methyl- | 534-22-5 | C5H6O+ | 82.0429 | 21.78 | 0.1 | 6.5 | 0.994 | 0.3–24.0 |
| Furan, 3-methyl- | 930-27-8 | C5H6O+ | 82.0429 | 22.18 | 0.1 | 7.6 | 0.992 | 0.3–20.0 |
| 2-Butanone | 78-93-3 | C4H8O·NO+ | 102.0567 | 22.22 | 0.3 | 8.4 | 0.996 | 0.9–637 |
| 2-Butanone, 3-methyl- | 563-80-4 | C5H10O·NO+ | 116.0741 | 26.37 | 0.23 | 8.0 | 0.994 | 0.69–23.5 |
| Allyl methyl sulfide | 10152-76-8 |
C4H8S+ | 88.0370 | 27.25 | 0.08 | 7.0 | 0.995 | 0.25–17.0 |
| 2-Pentanone | 107-87-9 | C5H10O·NO+ | 116.0722 | 27.33 | 0.17 | 5.0 | 0.997 | 0.52–720 |
| Methyl propyl sulfide | 3877-15-4 | C4H10S+ | 90.0515 | 28.03 | 0.12 | 7.0 | 0.991 | 0.37–18.7 |
| 2-Pentanone, 4-methyl- | 108-10-1 | C6H12O·NO+ | 130.0880 | 30.83 | 0.29 | 7.5 | 0.994 | 0.88–17.0 |
| 2-Pentanone, 3-methyl- | 565-61-7 | C6H12O·NO+ | 130.0900 | 31.08 | 0.31 | 8.0 | 0.991 | 0.9–14.0 |
| 3-Hexanone | 589-38-8 | C6H12O·NO+ | 130.0887 | 31.87 | 0.42 | 8.0 | 0.998 | 1.28–20.4 |
| 2-Hexanone | 591-78-6 | C6H12O·NO+ | 130.0892 | 32.22 | 0.52 | 8.5 | 0.998 | 1.55–22.6 |
| 4-Heptanone | 123-19-3 | C7H14O·NO+ | 144.1047 | 35.90 | 0.35 | 9.0 | 0.998 | 1.05–956 |
| 2-Heptanone | 110-43-0 | C7H14O·NO+ | 144.1075 | 36.53 | 0.31 | 9.2 | 0.993 | 0.92–18.10 |
2.5. Creatinine measurements
The creatinine concentrations in urine samples were determined via a routine colorimetric, alkaline picrate method (Jaffé-method) and the Advia 1800 Chemistry System (Siemens).3. Results and discussion
3.1. Validation parameters
Validation parameters for VOCs under study are presented in Table 1. Limits of detection (LODs) were calculated using the standard deviations of 6 consecutive blank signals and the algorithm presented by Huber.39 The LOD values ranged from 0.08 nmol L−1 for allyl methyl sulfide to 1.0 nmol L−1 for acetone and furan. The limit of quantification (LOQ) was defined as 3 × LOD. Relative standard deviations (RSDs) were calculated on the basis of consecutive analyses of five urine samples obtained from the same volunteer. The RSDs varied from 5.0% for 2-pentanone to 9.2% for 2-heptanone and were recognized as satisfactory for the goals of this study. The system response was found to be linear within the investigated concentration levels, with coefficients of variation falling within the range of 0.974–0.998. A part of an exemplary chromatogram from a HS-SPME-GC-SRI-TOF-MS analysis of human urine is shown in Fig. 1.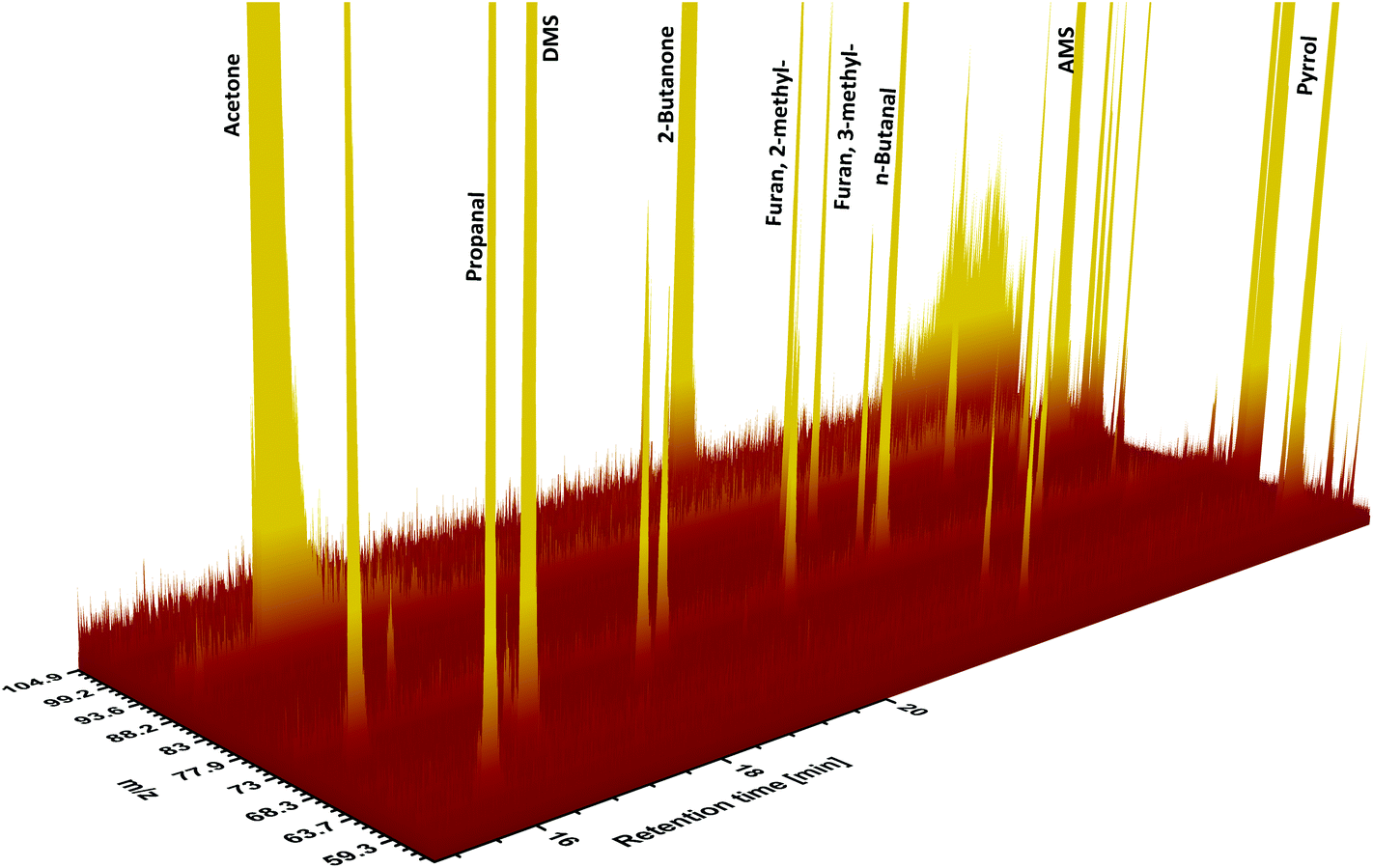 | ||
| Fig. 1 A part of an exemplary chromatogram from the HS-SPME-GC-SRI-TOF-MS analysis of urinary VOCs. | ||
Table 1 summarizes the parent ions of species under study in SRI-TOF-MS working with NO+ as the reagent ion. Ketones reacted with NO+via ion-molecule association forming NO+M adduct ions;40 whereas, the charge transfer generating the respective M+ ions was the basic ionization mechanism in the case of all furans and sulphur compounds of interest.41,42 The extracted time profiles of these molecular ions were used for the peak integration.
3.2. Quantification of selected urinary VOCs
Overall 16 compounds were quantified using HS-SPME-GC-SRI-TOF-MS in the NO+ mode. Amongst them there were ten ketones (acetone, 2-butanone, 3-methyl-2-butanone, 2-pentanone, 3-methyl-2-pentanone, 4-methyl-2-pentanone, 2-hexanone, 3-hexanone, 2-heptanone, and 4-heptanone), three volatile sulphur compounds (dimethyl sulfide, allyl methyl sulfide, and methyl propyl sulfide), and three heterocyclic compounds (furan, 2-methylfuran, and 3-methylfuran). Their associated detection and quantification incidences as well as the observed concentration ranges are given in Table 2 and depicted in Fig. 2. The presented concentration levels are diminished (if applicable) by the respective values obtained for the associated blank samples. Since urine exhibits a variation in dilution, the concentrations of species under scrutiny were normalized by dividing by the urinary creatinine (UCr) concentration obtained for the same urine sample and reported also as nanomol of target analyte per millimol of creatinine.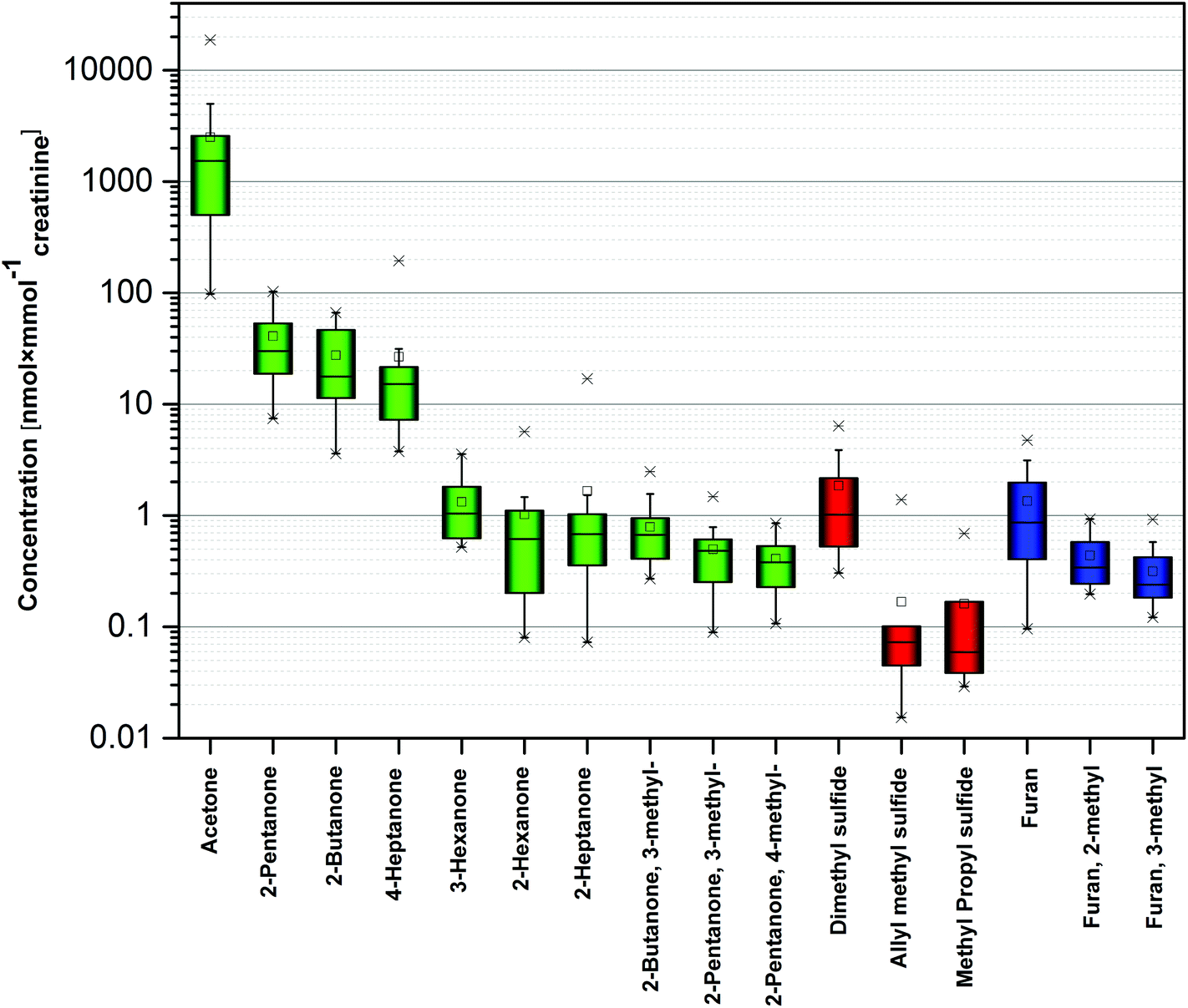 | ||
| Fig. 2 Concentration of selected VOCs in human urine. Different colors correspond to different chemical families. | ||
| VOC | Incidence nd(nq) | Range (median) [nmol L−1] | Range (median) [nmol mmol−1creatinine] | Literature data |
|---|---|---|---|---|
| a Refers to mean value. | ||||
| Furan | 19(19) | 1.06–28.8 (11.3) | 0.1–4.75 (0.87) | (a) LOQ (14.7) − 46 nmol L−1 (n = 100)27 |
| Acetone | 19(19) | 2300–107000 (11600) |
98–18700 (1540) |
(a) 800–17600 (3900) nmol mmol−1creatinine (n = 22)17 |
| (b) 22400 ± 41000 nmol L−1 (n = 66)28 |
||||
| (c) 34000–95000 (52000a) nmol L−1 (n = 20)25 |
||||
| Dimethyl sulfide | 19(19) | 2.46–84.7 (12.3) | 0.3–6.37 (1.02) | (a) <2.26 nmol L−1 (n = 6)26 |
| Furan, 2-methyl- | 19(19) | 1.74–6.88 (4.06) | 0.2–0.93 (0.34) | |
| Furan, 3-methyl- | 19(19) | 1.03–5.59 (2.97) | 0.12–0.92 (0.24) | |
| 2-Butanone | 19(19) | 32.3–729.2 (292) | 3.59–66.6 (17.7) | |
| 2-Butanone, 3-methyl- | 19(19) | 2.03–21.5 (6.92) | 0.27–2.48 (0.67) | |
| Allyl methyl sulfide | 19(17) | 0.24–9.18 (0.68) | 0.02–1.39 (0.07) | |
| 2-Pentanone | 19(19) | 104–987 (294) | 7.46–103 (30) | |
| Methyl propyl sulfide | 13(7) | 0.32–7.53 (0.76) | 0.03–0.69 (0.06) | |
| 2-Pentanone, 4-methyl- | 18(18) | 1.48–7.77 (4.28) | 0.11–0.86 (0.37) | |
| 2-Pentanone, 3-methyl- | 19(19) | 2.07–11 (4.23) | 0.09–1.48 (0.48) | |
| 3-Hexanone | 19(19) | 2.23–39 (11.93) | 0.53–3.56 (1.04) | |
| 2-Hexanone | 19(19) | 0.65–34.4 (6.07) | 0.08–5.67 (0.61) | |
| 4-Heptanone | 19(19) | 18.6–1280 (167) | 3.76–194 (15.2) | (a) 905–7400 (1600) nmol L−1 (n = 51)21 |
| 2-Heptanone | 19(19) | 0.61–111 (6.64) | 0.07–17 (0.68) | |
The observed concentrations ranged from 0.55 nmol L−1 (0.05 nmol mmol−1creatinine) for allyl methyl sulfide to 11.6 μmol L−1 (1.54 μmol mmol−1creatinine) for acetone considering medians. However, the levels of the majority of species did not exceed 100 nmol L−1 (10 nmol mmol−1creatinine). The highest median concentrations were noted for acetone (11.6 μmol L−1, or 1.54 μmol mmol−1creatinine), 2-pentanone (294 nmol L−1, or 30 nmol mmol−1creatinine), 2-butanone (292 nmol L−1, or 17.7 nmol mmol−1creatinine), and 4-heptanone 167 nmol L−1, or 15.2 nmol mmol−1creatinine). 14 species exhibited incidence rates of 100%. Moreover, 4-methyl-2-pentanone was found in all samples except one; whereas, methyl propyl sulfide was detected in only 13 samples.
Due to the aforementioned shortage of quantitative data on the urine volatolome, it is difficult to relate the results obtained within this study to the literature studies. Nevertheless, several urinary concentration values of species under scrutiny can be found in the literature. In occupational exposure settings Brega et al.25 compared the acetone levels in urine of healthy volunteers and workers exposed to acetone. The concentrations of this compound in urine of controls (n = 20) ranged from 34 to 95 μmol L−1. In the same context, Satoh et al.28 provided acetone levels for a slightly larger control group (n = 66), which were spread around 22.4–41 μmol L−1. More recently, Bouatra et al.17 reported urinary concentrations of acetone in healthy volunteers (n = 22), which fell within the range of 0.8–17.6 μmol mmol−1creatinine (median: 3.9 μmol mmol−1creatinine). The results obtained within this study agree very well with these data. Regarding other ketones, 4-heptanone levels ranging from 905 to 7400 nmol L−1 (median:1600) were observed by Wahl et al.21 in urine of 51 healthy subjects. The presence of 4-heptanone in the human volatolome is being attributed to the metabolism of di(2-ethylhexyl) phthalate (DEHP) and di-2-(ethylhexyl)adipate (DEHA)-plasticizers used in polyvinyl chloride products.43 More specifically, DEHP is a common constituent of medical devices, whereas, DEHA is present in food contact films.44 In humans DEHP and DEHA are rapidly metabolized to 2-ethylhexanol, which is next oxidized to 2-ethylhexanoic acid and finally to 2-heptanone and 4-heptanone.43 The discrepancy between the levels of 4-heptanone obtained within this work and by Wahl et al. can stem from the differences in the exposure of involved populations to the aforementioned plasticizers. Amongst other species furan was determined in urine of 100 healthy individuals by Jun et al.27 It was detected in 56 individuals; however, only 15 concentration values exceeded the LOQ of the method (14.7 nmol L−1). The observed concentrations ranged up to 46 nmol L−1 and agree with the values reported in this work. Single measurements of urinary dimethyl sulfide in humans (n = 6) were performed by Liu et al.26 DMS was detected in urine of 4 volunteers and its levels did not exceed 2.26 nmol L−1. A comparison of the urinary VOCs levels obtained within this study with data from the literature is presented in Table 2.
3.3. Potential sources of urinary VOCs under study in humans
Although the origin of urinary volatiles remains ambiguous, several sources could explain their presence. These include (i) metabolic production related to the physiological processes in the body (both normal and abnormal), (ii) activity of microflora in human organism, (iii) environmental exposure, and (iv) diet and its metabolites.Two potential pathways may be involved in the production of ketones in humans: (i) oxidation of secondary alcohols catalyzed by alcohol dehydrogenases (ADHs) and (ii) β-oxidation of branched-chain fatty acids. Although primary alcohols are the most preferred substrates for ADHs, they were also demonstrated to convert secondary alcohols into ketones.45–47 For example, 2-hexanone could be the product of 2-hexanol oxidation; whereas, 2-pentanone may stem from 2-pentanol.45 The respective secondary alcohols can derive in turn from dietary sources, or be the products of the alkanes’ metabolism.48–50 Regarding β-oxidation of branched-chain fatty acids, 2-ethylhexanoic acid was shown to be metabolized to 2-heptanone and 4-heptanone as discussed above.43 Perhaps other ketones also can be produced via this metabolic pathway. Acetone is the major ketone produced in the human organism with high abundances in breath,51–53 skin emanations,54,55 and urine.19,20 The sources of these volatiles include (i) endogenous decarboxylation of acetyl–CoA,52,56 (ii) 2-propanol metabolism,46 and (iii) diet. However, the latter two are of minor importance.
The occurrence of allyl methyl sulfide in human tissues is being attributed to garlic intake;57 whereas, methyl propyl sulfide was demonstrated to appear in human breath after onion consumption.58 Furthermore, dimethyl sulfide most likely stems from the metabolization of sulfur-containing amino acids methionine and cysteine in the transamination pathway.59 More specifically, it is formed via the methylation of methyl mercaptane by thiol S-methyltransferase in the liver.60 However, the bacterial production of this volatile cannot be neglected.59
Furan is a common constituent of heat-processed foods (canned, or jarred) and coffee.61 Thus a dietary source seems to contribute primarily to the pool of this specie in humans. Moreover, furan, 2-methylfuran and 3-methylfuran were found to be significantly related to smoking.62
4. Conclusions
The present study aimed at the determination of selected volatile organic compounds in human urine. 16 volatiles were quantified in the urine of 19 volunteers by selective reagent ionization time of flight mass spectrometry with NO+ as the reagent ion in conjunction with gas chromatography (GC) and head space solid-phase microextraction (HS-SPME-GC-SRI-TOF-MS(NO+)). The observed median concentrations ranged over several orders of magnitude from 0.55 nmol L−1 (0.05 nmol mmol−1creatinine) for allyl methyl sulfide to 11.6 μmol L−1 (1.54 μmol mmol−1creatinine) for acetone and agree reasonably well with the available literature values. These results are expected to partially fill the literature data gap with respect to quantitative data on urinary concentrations of volatile organic compounds in healthy individuals.The coupling of selective reagent ionization time of flight mass spectrometry with gas chromatography provides a number of advantages. First, it does not hinder the real-time measurements in SRI-TOF-MS. Consequently, real-time and GC analyses can be performed consecutively using the same analytical system, without additional modifications in the experimental set-up. This feature assists the novel concept of hybrid volatolomics, an approach which combines VOC profiles obtained for two or more body fluids to improve and complement the chemical information on the physiological status of an individual.63 Next, the soft chemical ionization simplifies the product ion distributions and, thereby, improves the baseline and reduces its drift induced by column bleed. This in turn supports integration and detection of chromatographically separated species.
Acknowledgements
P. M. and K. U. gratefully acknowledge support from the Austrian Science Fund (FWF) under Grant No. P24736-B23. This work has received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 644031. We also appreciate funding from the Austrian Agency for International Cooperation in Education and Research (OeAD-GmbH, project SPA 05/202-FEM_BREATH). We thank the government of Vorarlberg, Austria for its generous support.References
- A. Amann and D. Smith, Breath analysis for clinical diagnosis and therapeutic monitoring, World Scientific, New Jersey, 2005 Search PubMed.
- Volatile Biomarkers: Non-invasive Diagnosis in Physiology and Medicine, ed. A. Amann and D. Smith, Elsevier, Amsterdam, 2013 Search PubMed.
- I. Horvath and J. E. De Jongste, European Respiratory Monograph, Number 49: Exhaled Biomarkers, European Respiratory Society, 2010 Search PubMed.
- C. M. Willis, S. M. Church, C. M. Guest, W. A. Cook, N. McCarthy, A. J. Bransbury, M. R. Church and J. C. Church, Br. Med. J., 2004, 329, 712 CrossRef PubMed.
- T. Khalid, P. White, B. Costello, R. Persad, R. Ewen, E. Johnson, C. S. Probert and N. Ratcliffe, PLoS One, 2013, 8, e69602 CAS.
- C. M. Weber, M. Cauchi, M. Patel, C. Bessant, C. Turner, L. E. Britton and C. M. Willis, Analyst, 2011, 136, 359–364 RSC.
- R. P. Arasaradnam, M. J. McFarlane, C. Ryan-Fisher, E. Westenbrink, P. Hodges, M. G. Thomas, S. Chambers, N. O'Connell, C. Bailey, C. Harmston, C. U. Nwokolo, K. D. Bardhan and J. A. Covington, PLoS One, 2014, 9, e108750 Search PubMed.
- S. Ganti and R. H. Weiss, Urol. Oncol.: Semin. Orig. Invest., 2011, 29, 551–557 CrossRef CAS PubMed.
- K. R. Elliker, B. A. Sommerville, D. M. Broom, D. E. Neal, S. Armstrong and H. C. Williams, BMC Urol., 2014, 14, 22 CrossRef PubMed.
- J. N. Cornu, G. Cancel-Tassin, V. Ondet, C. Girardet and O. Cussenot, Eur. Urol., 2011, 59, 197–201 CrossRef PubMed.
- A. D. Asimakopoulos, D. Del Fabbro, R. Miano, M. Santonico, R. Capuano, G. Pennazza, A. D'Amico and E. Finazzi-Agro, Prostate Cancer Prostatic Dis., 2014, 17, 206–211 CrossRef CAS PubMed.
- A. Roine, E. Veskimae, A. Tuokko, P. Kumpulainen, J. Koskimaki, T. A. Keinanen, M. R. Hakkinen, J. Vepsalainen, T. Paavonen, J. Lekkala, T. Lehtimaki, T. L. Tammela and N. K. Oksala, J. Urol., 2014, 192, 230–234 CrossRef PubMed.
- S. Smith, P. White, J. Redding, N. M. Ratcliffe and C. S. J. Probert, IEEE Sens. J., 2010, 10, 92–96 CrossRef CAS.
- P. Spanel, D. Smith, T. A. Holland, W. Al Singary and J. B. Elder, Rapid Commun. Mass Spectrom., 1999, 13, 1354–1359 CrossRef CAS PubMed.
- A. Pysanenko, T. Wang, P. Spanel and D. Smith, Rapid Commun. Mass Spectrom., 2009, 23, 1097–1104 CrossRef CAS PubMed.
- J. Rudnicka, P. Mochalski, A. Agapiou, M. Statheropoulos, A. Amann and B. Buszewski, Anal. Bioanal. Chem., 2010, 398, 2031–2038 CrossRef CAS PubMed.
- S. Bouatra, F. Aziat, R. Mandal, A. C. Guo, M. R. Wilson, C. Knox, T. C. Bjorndahl, R. Krishnamurthy, F. Saleem, P. Liu, Z. T. Dame, J. Poelzer, J. Huynh, F. S. Yallou, N. Psychogios, E. Dong, R. Bogumil, C. Roehring and D. S. Wishart, PLoS One, 2013, 8, e73076 CAS.
- G. A. Mills and V. Walker, J. Chromatogr. B: Biomed. Sci. Appl., 2001, 753, 259–268 CrossRef CAS.
- P. Mochalski, K. Krapf, C. Ager, H. Wiesenhofer, A. Agapiou, M. Statheropoulos, D. Fuchs, E. Ellmerer, B. Buszewski and A. Amann, Toxicol. Mech. Methods, 2012, 22, 502–511 CrossRef CAS PubMed.
- S. Smith, H. Burden, R. Persad, K. Whittington, B. de Lacy Costello, N. M. Ratcliffe and C. S. Probert, J. Breath Res., 2008, 2, 037022 CrossRef CAS PubMed.
- H. G. Wahl, A. Hoffmann, D. Luft and H. M. Liebich, J. Chromatogr. A, 1999, 847, 117–125 CrossRef CAS PubMed.
- B. De Lacy Costello and N. M. Ratcliffe, in Volatile Biomarkers. Non-Invasive Diagnosis in Physiology and Medicine, ed. A. Amann and D. Smith, Elsevier, Amsterdam, 2013, ch. 22, pp. 405–462 Search PubMed.
- A. Takeuchi, S. Yamamoto, R. Narai, M. Nishida, M. Yashiki, N. Sakui and A. Namera, Biomed. Chromatogr., 2010, 24, 465–471 CAS.
- S. Fustinoni, R. Giampiccolo, S. Pulvirenti, M. Buratti and A. Colombi, J. Chromatogr. B: Biomed. Sci. Appl., 1999, 723, 105–115 CrossRef CAS.
- A. Brega, P. Villa, G. Quadrini, A. Quadri and C. Lucarelli, J. Chromatogr., 1991, 553, 249–254 CrossRef CAS PubMed.
- Q. Y. Liu, Y. J. Liu and X. X. Wang, Microchem. J., 2011, 99, 352–355 CrossRef CAS.
- H. J. Jun, K. G. Lee, Y. K. Lee, G. J. Woo, Y. S. Park and S. J. Lee, Food Chem. Toxicol., 2008, 46, 1753–1759 CrossRef CAS PubMed.
- T. Satoh, K. Omae, T. Takebayashi, H. Nakashima, T. Higashi and H. Sakurai, Int. Arch. Occup. Environ. Health, 1995, 67, 131–134 CrossRef CAS PubMed.
- L. Perbellini, A. Princivalle, M. Cerpelloni, F. Pasini and F. Brugnone, Int. Arch. Occup. Environ. Health, 2003, 76, 461–466 CrossRef CAS PubMed.
- A. P. Anton, A. M. C. Ferreira, C. G. Pinto, B. M. Cordero and J. L. P. Pavon, J. Chromatogr. A, 2014, 1367, 9–15 CrossRef CAS PubMed.
- A. Jordan, S. Heidacher, G. Hanel, E. Hartungen, J. Herbig, L. Maerk, R. Schottkowsky, H. Seehauser, P. Siulzer and T. D. Maerk, Int. J. Mass Spectrom., 2009, 286, 32–38 CrossRef CAS.
- I. Kohl, J. Beauchamp, F. Cakar-Beck, J. Herbig, J. Dunkl, O. Tietje, M. Tiefenthaler, C. Boesmueller, A. Wisthaler, M. Breitenlechner, S. Langebner, A. Zabernigg, F. Reinstaller, K. Winkler, R. Gutmann and A. Hansel, J. Breath Res., 2013, 7, 017110 CrossRef PubMed.
- J. King, A. Kupferthaler, B. Frauscher, H. Hackner, K. Unterkofler, G. Teschl, H. Hinterhuber, A. Amann and B. Hogl, Physiol. Meas., 2012, 33, 413–428 CrossRef PubMed.
- J. King, P. Mochalski, A. Kupferthaler, K. Unterkofler, H. Koc, W. Filipiak, S. Teschl, H. Hinterhuber and A. Amann, Physiol. Meas., 2010, 31, 1169–1184 CrossRef CAS PubMed.
- J. King, K. Unterkofler, G. Teschl, S. Teschl, H. Koc, H. Hinterhuber and A. Amann, J Math. Biol., 2011, 63, 959–999 CrossRef PubMed.
- J. King, K. Unterkofler, S. Teschl, A. Amann and G. Teschl, Conf. Proc. IEEE Eng. Med. Biol. Soc., 2011, 2011, 1001–1004 Search PubMed.
- D. Smith and P. Spanel, Mass Spectrom. Rev., 2005, 24, 661–700 CrossRef CAS PubMed.
- D. Smith, P. Spanel, J. Herbig and J. Beauchamp, J. Breath Res., 2014, 8, 027101 CrossRef PubMed.
- W. Huber, Accredit. Qual. Assur., 2003, 8, 213–217 CAS.
- P. Spanel, Y. Ji and D. Smith, Int. J. Mass Spectrom. Ion Processes, 1997, 165, 25–37 CrossRef.
- P. Mochalski, K. Unterkofler, P. Spanel, D. Smith and A. Amann, Int. J. Mass Spectrom., 2015, 386, 42–46 CrossRef CAS.
- P. Mochalski, K. Unterkofler, P. Spanel, D. Smith and A. Amann, Rapid Commun. Mass Spectrom., 2014, 28, 1683–1690 CrossRef CAS PubMed.
- V. Walker and G. A. Mills, Clin. Chim. Acta, 2001, 306, 51–61 CrossRef CAS.
- N. J. Loftus, B. H. Woollen, G. T. Steel, M. F. Wilks and L. Castle, Food Chem. Toxicol., 1994, 32, 1–5 CrossRef CAS PubMed.
- J. C. Burnell, T. K. Li and W. F. Bosron, Biochemistry, 1989, 28, 6810–6815 CrossRef CAS PubMed.
- C. C. Ditlow, B. Holmquist, M. M. Morelock and B. L. Vallee, Biochemistry, 1984, 23, 6363–6368 CrossRef CAS PubMed.
- D. W. Crabb, M. Matsumoto, D. Chang and M. You, Proc. Nutr. Soc., 2004, 63, 49–63 CrossRef CAS PubMed.
- S. J. Crosbie, P. G. Blain and F. M. Williams, Hum. Exp. Toxicol., 1997, 16, 131–137 CAS.
- J. E. Edwards, R. L. Rose and E. Hodgson, Chem.-Biol. Interact., 2005, 151, 203–211 CrossRef CAS PubMed.
- J. Bahima, A. Cert and M. Menendezgallego, Toxicol. Appl. Pharmacol., 1984, 76, 473–482 CrossRef CAS PubMed.
- P. Mochalski, J. King, M. Klieber, K. Unterkofler, H. Hinterhuber, M. Baumann and A. Amann, Analyst, 2013, 138, 2134–2145 RSC.
- K. Schwarz, A. Pizzini, B. Arendacka, K. Zerlauth, W. Filipiak, A. Schmid, A. Dzien, S. Neuner, M. Lechleitner, S. Scholl-Burgi, W. Miekisch, J. Schubert, K. Unterkofler, V. Witkovsky, G. Gastl and A. Amann, J. Breath Res., 2009, 3, 027003 CrossRef CAS PubMed.
- C. Turner, P. Spanel and D. Smith, Physiol. Meas., 2006, 27, 321–337 CrossRef PubMed.
- P. Mochalski, J. King, K. Unterkofler, H. Hinterhuber and A. Amann, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2014, 959, 62–70 CrossRef CAS PubMed.
- M. Gallagher, C. J. Wysocki, J. J. Leyden, A. I. Spielman, X. Sun and G. Preti, Br. J. Dermatol., 2008, 159, 780–791 CrossRef CAS PubMed.
- K. Musa-Veloso, S. S. Likhodii and S. C. Cunnane, Am. J. Clin. Nutr., 2002, 76, 65–70 CAS.
- N. Gupta and T. D. Porter, J. Nutr., 2001, 131, 1662–1667 CAS.
- A. Tangerman and E. G. Winkel, J. Breath Res., 2010, 4, 017003 CrossRef CAS PubMed.
- A. Tangerman, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2009, 877, 3366–3377 CrossRef CAS PubMed.
- T. A. Glauser, A. L. Kerremans and R. M. Weinshilboum, Drug Metab. Drug Dispos., 1992, 20, 247–255 CAS.
- O. Zoller, Food Addit. Contam., 2007, 24(S1), 91–107 CrossRef CAS PubMed.
- W. Filipiak, V. Ruzsanyi, P. Mochalski, A. Filipiak, A. Bajtarevic, C. Ager, H. Denz, W. Hilbe, H. Jamnig, M. Hackl, A. Dzien and A. Amann, J. Breath Res., 2012, 6, 036008 CrossRef CAS PubMed.
- Y. Y. Broza, P. Mochalski, V. Ruzsanyi, A. Amann and H. Haick, Angew. Chem., Int. Ed., 2015, 54, 11036–11048 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |