
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Employment of electrostatic interactions for amperometric detection of carbon nanoparticles in a FIA system†
D.
Ogończyk
*a,
M.
Gocyla
ab and
M.
Opallo
*a
aInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka, 44/52, 01-224, Warsaw, Poland. E-mail: dogonczyk@ichf.edu.pl; mopallo@ichf.edu.pl
bFaculty of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland
First published on 29th April 2016
Abstract
The development of methods for nanoparticle detection is highly desirable due to their increasing presence in the environment. Recently, we have shown that the electrochemical detection in flow is one of the possible solutions. Here we demonstrate a dramatic improvement of analytical parameters of such detection. The significant enhancement of an amperometric signal resulting from the electrocatalytic oxidation of ascorbic acid (AA) in a negatively charged phenylsulphonated carbon nanoparticle suspension in the millifluidic flow injection analysis system as compared to earlier results (D. Ogończyk, et al., Electrochem. Commun., 2014, 43, 40) is presented. This effect results from the tailoring of electrostatic interactions, e.g. optimization of the supporting electrolyte and AA concentration and/or immobilization of positively charged functionalities at the electrode surface. The sensitivity is improved by almost three orders of magnitude and the limit of detection of carbon nanoparticles is decreased by two orders of magnitude down to 0.001 mg mL−1.
Introduction
Due to the unique size-dependent physicochemical properties, engineered nanoparticles (NPs) are extremely popular nanoobjects and their global production1,2 and applications3–5 increase every year. It is obvious that increasing amounts of NPs are released to the environment and their potential toxicity is becoming one of the most important issues related to nanotechnology. Therefore, the development of NP detection techniques is highly desirable. One of the possibilities is electrochemical detection.6 Compared to other techniques,7,8 it is cost-effective, simple and fast in realization. In the development of electrochemical methods for nanoparticle detection, one can distinguish three approaches: (i) direct electrochemical reduction or oxidation, (ii) electrode reactions of their functionalities and (iii) an employment of electrocatalytic reactions mediated by NPs.6 In the latter case the nanoparticles play a role as electron transfer mediators with specific surface modification. In other words, when catalytic nanoparticles collide with a non-catalytic electrode in the presence of a reaction substrate in solution, a significant overpotential decrease and a current increase, in comparison with bulk non-catalytic electrodes are observed.6Hitherto, almost all reports of the electrocatalytic studies in NP suspensions have been focused on experiments performed under quiescent conditions. However, we have recently reported the application of a flow technique for the detection of electrocatalytic amplification, due to carbon nanoparticle (CNP) collisions with an ITO (indium tin oxide) electrode in the presence of ascorbic acid (AA) as a reaction substrate.9 It is worth mentioning that nanoparticle-based assays have found application in flow systems, but contrary to our work, these research studies were oriented towards the detection of molecular analytes, not engineered nanoobjects.10 The electrocatalytic properties of CNPs towards AA are already well known, e.g. a significant decrease of the molecular analyte oxidation potential was observed on CNP modified electrodes.11–13 Among others, this phenomenon was employed for selective electrochemical dopamine sensing in the presence of AA as an interferent,12 or the construction of a self-powered AA sensor based on an ascorbic acid–dioxygen biofuel cell.13 On the contrary, in our earlier reported flow experiments9 we focused on the detection of CNPs and not AA. The flow injection analysis (FIA) system with electrochemical detection offered an increase of frequency of nanoparticle collisions with the electrode surface as compared to quiescent conditions and, therefore, a higher magnitude of the recorded current. However, the relationship between the amperometric signal and NP concentration was maintained in flow only if the fixed volumes of NP suspension samples were injected. Moreover, due to reproducible and well controlled dosing of the samples, the simple design of the system, as well as well-defined and reproducible experimental parameters, the FIA system could be successfully employed for experiments dedicated to nanoparticle detection. Our preliminary study pointed out the electrocatalytic effect of numerous experimental conditions such as: type and concentration of the supporting electrolyte, potential, the flow rate and the mixing system on the magnitude of the signal.9 The results demonstrated that CNP detection depends rather on experimental conditions than nanoparticle concentrations. Possibly, the charged CNPs underwent coagulation during the preparation and in the examined suspension due to a salting effect and the formation of aggregates/agglomerates that mostly conditioned the amperometric response. Accordingly, the use of the FIA system with wider channels as compared to recently popular microfluidic systems helped us to avoid the clogging. We also demonstrated that enhancement of amperometric signals in the FIA system resulted from CNP adsorption on the electrode surface.9 Nevertheless, the aggregate stage of CNPs seems to be predominant. We suppose that more precise control and steering of electrostatic interactions between nanoparticles (phenylsulphonate functionalized CNPs), the substrate (AA), the supporting electrolyte (NaH2PO4) and the electrode (ITO; +0.2 V vs. Ag|AgCl) can significantly improve the analytical parameters of the developed method. The study described in this manuscript is dedicated to these phenomena.
A major challenge in handling nanoparticle suspensions is their aggregation due to van der Waals and electrostatic double layer (EDL) forces.14,15 In general, the dispersed NPs reveal a strong tendency to reduce the surface free energy via, among others, aggregation, agglomeration and attachment of molecules or molecular blocks of liquid media by NPs.16 NP aggregation depends on the type and composition of electrolyte, ionic strength, pH, their size and capping agent.6 It also strongly affects NP reactivity by changing reactive surface area geometry and transport properties. The ionic strength controls the size of the NP diffuse layer and causes NP attraction or repulsion. For example, NPs aggregate in the concentrated electrolyte, because ions screen the repulsive electrostatic forces. In a diluted electrolyte, this effect is negligible and NP suspension is stable.17 Electrostatic interactions may also influence the kinetics of reactions of charged NPs as it is reported for ionic reactants.18 The electrooxidation of negatively charged ascorbate at negatively charged CNPs is an example of such behaviour. Therefore, an addition of inert ions should boost the electrocatalysis, because of the increase of their electrostatic attraction. Moreover, the electron transfer between nanoparticles and the electrode substrate may be more efficient thanks to the modification of the electrode surface with positively charged functional groups attracting suspended catalysts and/or reactants. Last but not least, in electrochemical experiments the excess of inert ions decreases the resistance of the solution that affects the shape of the current–potential or current–time dependence.19
Here, we report the study of an effect of electrostatic interactions through (i) a composition of reaction medium and (ii) charged functional groups attached to the electrode surface on the electrocatalytic response of suspended carbon nanoparticles (CNPs) under flow conditions. The enhancement of the electrocatalytic signal is demonstrated with the example of electrooxidation of AA in the millifluidic FIA system with a bare and modified ITO serving as an electrode. The measurements were performed at a fixed pH 4.6. As a consequence, an excess of ascorbate ions, because pKa for AA are 4.2 and 11.6, respectively, was present and namely a single protonated form of AA undergoes electrooxidation.20
Results and discussion
The effect of electrolyte on CNP suspension stability in quiescent conditions
Taking into account that electrochemical experiments are performed in an electrolyte solution and knowing that ionic strength may affect the stability of nanoparticle suspensions and their aggregation stage, DLS (dynamic light scattering) and zeta potential measurements of CNP suspensions were executed (Fig. 1). The obtained hydrodynamic diameter of CNPs is close to 140 nm for NaH2PO4 concentration below 10 mM. This is larger than the value provided by the supplier by one order of magnitude, which already indicates the aggregation/agglomeration of nanoparticles. Further increase of salt concentration up to 50 mM results in the increase of dynamic diameter more than 3 times, indicating further aggregation/agglomeration. On the other hand, the decrease of the zeta potential for a larger NaH2PO4 concentration indicates the instability of the CNP suspensions (Fig. 1). These results confirm that the aggregation stage of CNPs is strongly determined by the concentration of the electrolyte solution. However, this dependence can be different and more complex, due to the different mass transport under flow conditions than in quiescent conditions. | ||
| Fig. 1 Hydrodynamic diameters and zeta potential (n = 3) measured in 0.01 mg mL−1 CNPs as a function of the CNP suspension composition in NaH2PO4. | ||
Electrolyte effect vs. CNP electrocatalytic properties in the forced convection conditions
In order to precisely study the effect of the NaH2PO4 electrolyte on electrocatalytic properties of CNP suspension, voltammetric experiments were performed in stirred (600 rpm) 1 mM AA in 25 mM NaH2PO4 aqueous solution. The choice of the conditions was motivated by a comparison between previous and received results.Shortly before the experiments were carried out, 10 mg mL−1 CNP suspensions in water or in 5 mM, 25 mM or 50 mM NaH2PO4 aqueous solutions were prepared. Then, 0.2 mL of these suspensions was dosed to the measuring cell. It has to be emphasized that the final compositions of the tested mixtures (20 mL) were almost identical (1 mM AA and 0.1 mg CNPs per mL in 25 mM NaH2PO4). Clearly, the shape of the voltammetric response and onset potential depend strongly on the composition of added CNP suspension (Fig. 2). When the electrolyte is absent in the added CNP suspension sample, a well-developed anodic peak appears. Moreover, the onset potential shift and the peak current are the largest as compared to the bare ITO (Fig. 2). Interestingly, the time of peak appearance is also the shortest in these conditions. This may be related to the smaller number of aggregates/agglomerates and the highest stability of the added suspension (Fig. 1). Therefore, CNPs suspended in pure water were used in further experiments.
 | ||
| Fig. 2 Difference in voltammograms of 1 mM AA in 25 mM NaH2PO4 depending on the initial composition of CNPs suspension: aqueous (A) or in 5 (B), 25 (C) and 50 mM NaH2PO4 (D). Cyclic voltammograms obtained with the ITO electrode in the absence (2nd scan—dotted lines) and after the addition of the prepared 10 mg mL−1 CNP suspensions to the final concentration of 0.1 mg mL−1 (2nd scan—red, 5th scan—blue, 10th scan—green, 15th scan—violet, 20th scan—grey) in stirred solution (600 rpm); scan rate 50 mV s−1. | ||
Improvement of CNP detection in the FIA system
Next, the chronoamperometric experiments were performed under similar conditions with CNP aqueous suspensions and 25 mM NaH2PO4 supporting electrolyte to provide the comparison with the earlier results.9 The peak currents were significantly higher (Fig. SI_2†), but results were also extremely irreproducible. Perhaps the behavior of samples of aqueous CNP suspensions in contact with the flowing electrolyte has a stochastic rather than a deterministic nature in contrast to CNP suspensions containing electrolyte. Therefore, in order to limit the variability of the gradient of ionic strength, we changed the method of AA dosing from injection to continuous pumping (Fig. 7A). This resulted in significantly improved repeatability (vide infra).
 | ||
| Fig. 3 Effect of various parameters on chronoamperometric measurements performed at 0.2 V in AA solution in 25 mM NaH2PO4 (pH 4.6) after the injection of aqueous suspension of 0.1 mg mL−1 CNPs in the FIA system (Fig. 7A). (A) Flow rate effect, 1 mM AA, a spiral system; (B) mixer effect, 1 mM AA, Vtotal = 6 mL min−1; (C) effect of AA concentration, a spiral system, Vtotal = 6 mL min−1; (D) effect of oxygen, 10 mM AA, a spiral system, Vtotal = 6 mL min−1. Other parameters are marked in the figure. | ||
Next, the effect of AA concentration (Fig. 3C) and oxygen presence (Fig. 3D) on the chronoamperometric response was studied. The other parameters (potential 0.2 V and 25 mM NaH2PO4 pH 4.6 as an electrolyte) were the same as in the earlier study.9 It is clear that the modification of a composition of injected CNP suspension and different methods of AA dosing yield considerable larger signals as compared to earlier results.9 Also the AA concentration has a slightly different effect on a recorded signal in comparison with the previous results.9 Here, with continuous pumping, the optimum AA concentration is 20 mM. As was shown earlier with the system based on simultaneous injection of samples, this concentration of AA yielded CNP precipitation. Probably the better stability of CNP aqueous suspension results from the differences in concentration gradient and electrostatic interactions of the medium during sample dispersion. As before, the spiral system and a flow rate of 6 mL min−1 (Fig. 3A and B) are optimal to observe the highest magnitude of the signal. The peak current is also appreciably higher for deoxygenated solutions (Fig. 3D) because of oxidation of fraction of AA by dissolved oxygen.21 Nevertheless, the new system provides an increase of the sensitivity for the CNP suspensions by almost three orders of magnitude and the detection limit is lowered by one order magnitude (LOD = 0.01 mg mL−1, Fig. 4, stars) in comparison with the results obtained with the earlier proposed experimental setup9 (Fig. 4, circles).
 | ||
| Fig. 4 The effect of the substrate concentration on sensitivity and LOD for CNP detection. Left: The concentrations of injected aqueous CNP suspensions were: 0.001 (blue), 0.005 (red), 0.01 (gray), 0.1 and 1 mg mL−1 (data no shown). Chronoamperometric measurements were performed at 0.2 V in the FIA system (Fig. 7A) with the use of deoxygenated 1 (green squares), 10 (cyan triangles) and 20 mM (black stars) AA solutions in 25 mM NaH2PO4 (pH 4.6) with a spiral system, a bare ITO and a total flow rate fixed at 6 mL min−1. The vertical bars are 10 nA (left). Right: The corresponding calibration plots for deoxygenated 1 (green squares), 10 (cyan triangles) and 20 mM (black stars) AA prepared as peak current (ΔI) in the function of CNP concentration of aqueous suspensions is presented. For comparison the earlier results9 are given (violet circles). | ||
Moreover, the LOD is further improved (decreased) by one order of magnitude by the decrease of AA concentration without any significant loss of sensitivity (Fig. 4, triangles and squares). However, for the lower AA concentration (Fig. 4, triangles and squares), the signal for CNPs was smaller than in the previously established optimal conditions when 20 mM AA was used (Fig. 4, stars). It is clear that the higher substrate concentration provides higher efficiency of the catalytic reaction, but simultaneously the higher AA concentration yields an increase of the undesirable aggregate stage of detected CNPs that interrupts detection of their lower concentrations. Consequently, due to enhancement of sensitivity and lowered detection limits the system may need a re-calibration–alteration of experimental conditions in the function of nanoparticle concentrations. Such an operation may be important for the quantitative CNP detection and can be easily executed by means of the millifluidic system.
To summarize, the CNP detection limit seems to be strongly dependent on the electrostatic interactions, which are induced by the presence of both ionic substances: the supporting electrolyte and AA. A higher concentration of AA is desirable because of the magnitude of the electrocatalytic current. However, it cannot be too high because of CNP precipitation. These results also suggest that the CNP aggregation state is more important than the effect of kinetics of the reactions between ions: negatively charged NPs and the single protonated form of AA.
Additionally, we tested the effect of the concentration of a supporting electrolyte in the context of changing resistance (Fig. 5) with a modified FIA system consisting of a separate line with an electrolyte in front of the detector (Fig. 7B).
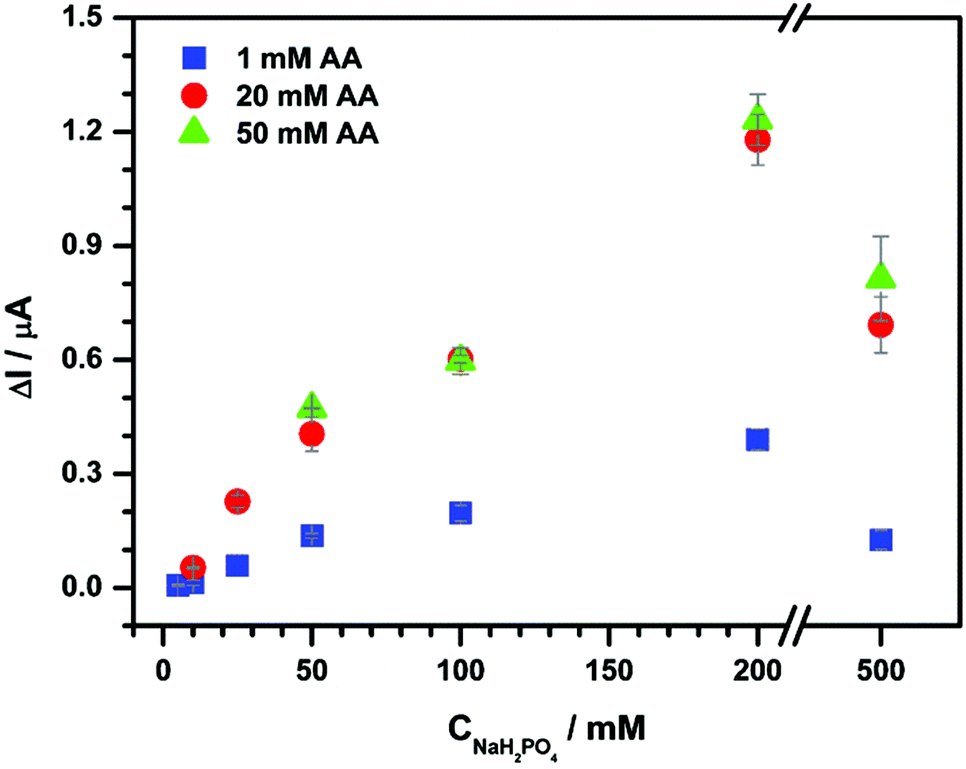 | ||
| Fig. 5 The effect of the supporting electrolyte concentration on chronoamperometry performed with 0.1 mg mL−1 aqueous CNP suspensions and deoxygenated 1 (blue squares), 20 (red circles) and 50 mM (green triangles) AA in NaH2PO4 at 0.2 V. In the experiment a three line FIA system (Fig. 7B) was used with a total flow rate fixed at 6 mL min−1 and unmodified ITO. Each assay was repeated twice. | ||
This approach enables the suppression of the CNP aggregation effect because of the relatively short time of contact of the reactants. The peak current resulting from AA oxidation after addition of 0.1 mg mL−1 aqueous suspension of CNP is proportional to NaH2PO4 concentration up to 200 mM (Fig. 5). Moreover, the increase of AA concentration at a fixed concentration of NaH2PO4 produces an ionic strength increase and yields higher current signals. This behavior is expected, because of lower solution resistance and higher efficiency of the catalytic reaction by a higher substrate concentration (see above). However, in these conditions, the increase of sensitivity is not accompanied by improvement of detection limits (LOD = 0.01 mg mL−1; Fig. SI_3†).
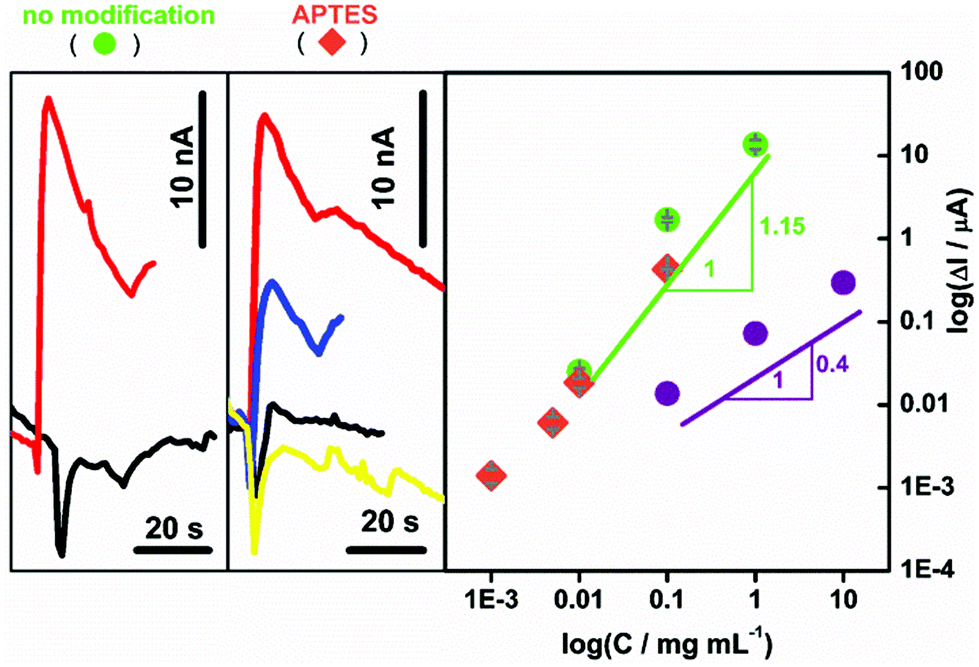 | ||
| Fig. 6 Left: The effect of electrode modification on chronoamperometry performed at 0.2 V in the FIA system (Fig. 7A) with deoxygenated 20 mM AA in 25 mM NaH2PO4 (pH 4.6) with a spiral system and a total flow rate fixed at 6 mL min−1. The concentrations of aqueous CNP suspensions were: 0.0001 (yellow), 0.001 (black), 0.005 (blue), 0.01 (red), 0.1 and 1 mg mL−1 (data no shown). The electrodes are marked in the figure. Right: The corresponding calibration graph for: unmodified ITO (green circles) and APTES-modified ITO (orange diamonds) as the function of different concentrations of aqueous CNP suspensions is presented. For comparison earlier results9 are presented (violet circles). | ||
Conclusions
We demonstrated a significant improvement of the parameters of electrochemical detection of phenylsulphonated functionalized carbon nanoparticles: (i) magnitude of the electrochemical signal, (ii) sensitivity, and (iii) detection limit by manipulation of electrostatic parameters. The steering of ionic strength in the FIA system improves the sensitivity by almost three orders of magnitude and lowers the limit of detection of CNPs by two orders of magnitude. Moreover, the employment of electrostatic interactions between CNPs and the modified electrode surface (among others introduction of favourable conditions for adsorption of CNPs) enables the detection of CNPs in a wider range of concentrations without system recalibration.The presented results also indicate how the manipulation of ionic strength may increase the efficiency of the electrocatalytic process in nanoparticles suspension, what may be applied not only in their electroanalysis, but also in energy conversion systems.
Experimental
Chemicals and materials
AA and NaH2PO4 were obtained from Riedel-de Haën and Chempur, respectively. APTES was produced by Alfa Aesar. These chemicals were pure and analytical grade and were used without further purification. CNPs with phenylsulfonate functionalities (13 nm average diameter, Emperor 2000) were bought from Cabot Corporation. Nanoparticle agglomerates were broken down and dispersed using an ultrasonic cleaner (Emmi 30 HC, EMAG) in a fixed time (20 min) and temperature (4 °C). This allows obtaining repeatable deagglomeration and suppression of temperature effects. Selection of time and initial temperature was dictated by excessive CNP heating in the ultrasonic cleaner. Experiment-ready CNP suspensions were always heated to room temperature. For the preparation of the aqueous solutions Milli-Q water was used. ITO coated glass (resistivity 8–12 Ω sq−1) was purchased from Delta Technologies.Cleaning and modification of the ITO surface
Commercial ITO substrates were sonicated in acetone, ethanol and water. Every cleaning step lasted 15 min. Then, the ITO electrodes were heated to 500 °C for 20 min or modified with APTES as following. First, the cleaned ITO electrodes were activated in ‘base piranha’ solution (NH4OH/H2O2/H2O 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:5, 60 °C) for 20 min. Then, APTES coating was produced by a ‘T-BAG’ method.24
:1:5, 60 °C) for 20 min. Then, APTES coating was produced by a ‘T-BAG’ method.24
This procedure includes the self-assembly of APTES on an activated ITO by slow evaporation of ethanolic APTES solution for 24 h, and subsequent thermal annealing (140 °C, 24 h). The concentration range of APTES solution used for modification was inspected in the range of 10−11–10−3 mol L−1 (Fig. SI_4†). The optimal concentration of APTES was selected on the basis of the magnitude of the recorded current in the previous FIA system.9 It was equal to 10−6 mol L−1 and it was selected for other experiments.
Instrumentation
Cyclic voltammetry (CV) and chronoamperometry (CA) were performed with a microAutolab electrochemical system (Eco Chemie) with dedicated software in a three-electrode configuration. ITO electrode, a platinum wire (0.5 mm diameter) and Ag|AgCl|KClsat. were used as the working, counter and reference electrodes, respectively. CV was performed in a stirred solution (600 rpm; RCT basic magnetic stirrers, IKA), whereas chronoamperometry was done in the FIA setups (Fig. 7). They consist of one or two digitally controlled syringe pumps (PHD Ultra, Harvard Apparatus), a rotary injection valve (35 μL, 6 port 2-positional, Vici), polyethylene tubing (BD) and a homemade wall-jet electrochemical detector. These FIA systems are modified in comparison with the previously reported work.9 The Zetasizer Nano ZS (Malvern) for measuring zeta potential and size distribution of nanoparticles was used. | ||
| Fig. 7 The schemes of the used FIA systems (A and B) consisting of a mixing system (M), an electrochemical wall-jet detector (D) and waste (W). | ||
Acknowledgements
This work was partially sponsored by the National Centre of Science 2011/03/B/ST4/02620.References
- C. O. Robichaud, A. E. Uyar, M. R. Darby, L. G. Zucker and M. R. Wiesner, Environ. Sci. Technol., 2009, 43, 4227 CrossRef CAS PubMed.
- M. C. Stensberg, Q. Wei, E. S. McLamore, D. M. Porterfield, A. Wei and M. S. Sepúlveda, Nanomed., 2011, 6, 879 CrossRef CAS PubMed.
- Applications of carbon nanoparticles in the automotive sector, Carboninspired - Blog at http://carboninspired.com/blog/?p=561&lang=en.
- T. Tsuzuki, Int. J. Nanotechnol., 2009, 6, 567 CrossRef CAS.
- G. Schmid, Nanoparticles: From Theory to Application, Wiley-VCH, 2010 Search PubMed.
- W. Cheng and R. G. Compton, TrAC, Trends Anal. Chem., 2014, 58, 79 CrossRef CAS.
- M. Hassellöv, J. W. Readman, J. F. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344 CrossRef PubMed.
- D. Baer, D. Gaspar, P. Nachimuthu, S. Techane and D. Castner, Anal. Bioanal. Chem., 2010, 396, 983 CrossRef CAS PubMed.
- D. Ogończyk, M. Gocyla and M. Opallo, Electrochem. Commun., 2014, 43, 40 CrossRef.
- M. L. C. Passos, P. C. A. G. Pinto, J. L. M. Santos, M. L. M. F. S. Saraiva and A. R. T. S. Araujo, Anal. Chim. Acta, 2015, 889, 22 CrossRef CAS PubMed.
- M. Amiri, S. Sarokhian and F. Marken, Electroanalysis, 2007, 19, 1032 CrossRef CAS.
- A. Celebanska, D. Tomaszewska, A. Lesniewski and M. Opallo, Biosens. Bioelectron., 2011, 26, 4417 CrossRef CAS PubMed.
- A. Zloczewska, A. Celebanska, K. Szot, D. Tomaszewska, M. Opallo and M. Jonsson-Niedziolka, Biosens. Bioelectron., 2014, 54, 455 CrossRef CAS PubMed.
- E. M. Hotze, T. Phenrat and G. V. Lowry, J. Environ. Qual., 2010, 39, 1909 CrossRef CAS PubMed.
- J. C. Lees, J. Ellison, C. Batchelor-McAuley, K. Tschulik, C. Damm, D. Omanović and R. G. Compton, ChemPhysChem, 2013, 14, 3895 CrossRef CAS PubMed.
- W. Jeżewski, Phys. Chem. Chem. Phys., 2015, 17, 8828 RSC.
- D. McFarland, C. L. Haynes, C. A. Mirkin, R. P. Van Duyne and H. A. Godwin, J. Chem. Educ., 2004, 81, 544A CrossRef.
- P. W. Atkins and J. de Paula, Atkins’ Physical Chemistry, Oxford University Press, 2014 Search PubMed.
- A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, Wiley, 2000 Search PubMed.
- J. Du, J. J. Cullen and G. R. Buettner, Biochim. Biophys. Acta, 2012, 1826, 443 CAS.
- A. O. Dekker and R. G. Dickinson, J. Am. Chem. Soc., 1940, 62, 2165 CrossRef CAS.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322 CrossRef CAS PubMed.
- B. De Boer, A. Hadipour, M. M. Mandoc, T. van Woudenbergh and P. W. M. Blom, Adv. Mater., 2005, 17, 621 CrossRef CAS.
- S. P. Pujari, L. Scheres, A. T. M. Marcelis and H. Zuilhof, Angew. Chem., Int. Ed., 2014, 53, 6322 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S4. See DOI: 10.1039/c6an00752j |
| This journal is © The Royal Society of Chemistry 2016 |