
A long-wavelength quantum dot-concentric FRET configuration: characterization and application in a multiplexed hybridization assay†
Jia Jun
Li
and
W. Russ
Algar
*
Department of Chemistry, University of British Columbia, 2036 Main Mall, Vancouver, British Columbia V6T 1Z1, Canada. E-mail: algar@chem.ubc.ca
First published on 6th April 2016
Abstract
Quantum dot-based concentric Förster resonance energy transfer (cFRET) is a promising modality for the development of multifunctional fluorescent probes for bioanalysis and bioimaging. To date, the scope of cFRET has been largely limited to a prototypical configuration with a particular combination of quantum dot (QD) and fluorescent dyes linked through peptides. Expansion of the scope of cFRET is critical for its further development. Here, we expand the scope of cFRET in two capacities. First, we design and characterize a new long-wavelength cFRET configuration that combines red- and deep-red fluorescent dyes, Alexa Fluor 633 and Alexa Fluor 680, with an orange-emitting QD. Sequential and competitive energy transfer pathways are characterized through a rate analysis, where the balance of these rates more strongly favours competitive energy transfer in the new long-wavelength configuration versus sequential energy transfer in the previous prototypical configuration. Although the new cFRET configuration is more susceptible to photobleaching, its superior brightness and longer-wavelength excitation and emission provide an order of magnitude higher signal-to-background ratios in biological matrices (e.g., serum, blood) than the previous prototypical configuration. Second, we demonstrate that an oligonucleotide-linked, long-wavelength cFRET configuration has energy transfer similar to an analogous peptide-linked configuration, where the oligonucleotide-linked cFRET configuration can be combined with toehold-mediated strand displacement for the multiplexed detection of unlabeled nucleic acid targets as a single vector. Overall, this work establishes the general applicability of cFRET and introduces new strategies for its bioanalytical application.
Introduction
Semiconductor quantum dots (QDs) are arguably the best all-around photoluminescent materials. The combination of excellent brightness, spectrally broad absorption and spectrally narrow emission, resistance to photobleaching, and tunable optical properties has made these nanoparticles versatile and powerful tools for bioanalysis and bioimaging.1–3 Numerous reviews have highlighted the utility of QDs in these applications.2,4–6 The surface area of QDs adds further value to their optical properties by supporting chemical and biomolecular functionalization.7 Both the optical and interfacial properties of QDs are leveraged in Förster resonance energy transfer (FRET) configurations, where multiple FRET donors or acceptors can be arrayed around a QD that serves the opposite role. Several reviews have summarized the state-of-the-art in FRET-based assays, sensing, and other applications with QDs.8–10An emerging paradigm with QD-based FRET systems is the integration of multiple energy transfer pathways around a common QD scaffold. One such example is the development of multistep FRET and DNA-based photonic wires assembled to a QD, where the QD serves as a light harvester and initial donor,11,12 or as a relay point that enables initiation of the photonic wire FRET-cascade through bioluminescence.13 In another example, the central QD functions simultaneously as an acceptor for a luminescent lanthanide complex and a donor for a fluorescent dye.14–17 This configuration has been utilized for multiplexed bioassays16,17 and as the basis for bionanophotonic logic devices.14,15 A third example is a dual-colour photochromic FRET system with a central QD and energy transfer pathways between photochromic and non-photochromic dyes.18
In addition to the above examples, we have developed QD-based FRET systems with multiple energy transfer pathways that we call “concentric FRET” (cFRET) configurations.19–23 One exception notwithstanding,21 the QD in cFRET configurations serves as a central FRET donor for two assembled fluorescent acceptor dyes, between which energy transfer also occurs.19,20,22,23 cFRET probes have been used to assay protease activity and concentration,20,22 a cFRET imaging method has been developed,19 and the ensemble photophysics of cFRET have been characterized.23 The advantages of these cFRET probes are derived from their multifunctional design, which is expected to avoid challenges associated with the parallel use of two monofunctional probes, including the potential for multiplexing from the ensemble level down to the single-particle level, less spectral crosstalk between emission signals, and elimination of physicochemical differences between separate probes (e.g., brightness, size, stability, biological distribution and trafficking). However, the current scope of cFRET has been largely limited to the use of a prototypical system with a green-emitting QD, a yellow/orange-emitting dye such as Cyanine 3 (Cy3) or Alexa Fluor 555 (A555), and a deep-red emitting dye such as Alexa Fluor 647 (A647). Moreover, the bioanalytical applications of cFRET have focused solely on protease targets. As a potentially powerful method for bioanalysis and bioimaging, it is critical that the scope of cFRET be expanded.
Here, we expand the scope of cFRET probes by first creating and characterizing a new long-wavelength cFRET configuration with dye emission in the red through deep-red spectral regions, and, secondly, by developing a multiplexed nucleic acid hybridization assay with this cFRET configuration. As shown in Fig. 1A, the new long-wavelength cFRET system comprises an orange-emitting QD in combination with a red dye, Alexa Fluor 633 (A633), and a deep-red dye, Alexa Fluor 680 (A680). Energy transfer occurs from the QD to the A633, from the QD to the A680, and from the A633 to the A680. For characterization, this cFRET configuration is assembled with peptide linkers, as shown in Fig. 1B, and evaluated in terms of competitive and sequential energy transfer rates and efficiencies, photobleaching, and signal-to-background ratios in serum and blood matrices. These characteristics of the long-wavelength configuration are compared to the previously reported prototypical cFRET configuration with a green-emitting QD, A555, and A647. The format of a hybridization assay with this cFRET configuration is illustrated in Fig. 1C, where the orange-emitting QD is conjugated with multiple copies of two different probe oligonucleotide sequences. The probes are hybridized with three-quarter-length block oligonucleotides that are labeled with A633 and A680 to generate cFRET. Toeholds facilitate displacement of the blocks by complementary nucleic acid targets with corresponding changes in FRET, where measurement of the A633/QD and A680/QD photoluminescence (PL) intensity ratios enables quantitative detection of two target DNA sequences. The new long-wavelength cFRET configuration has both advantages and disadvantages versus the prototypical configuration and is very promising for applications in bioanalysis.
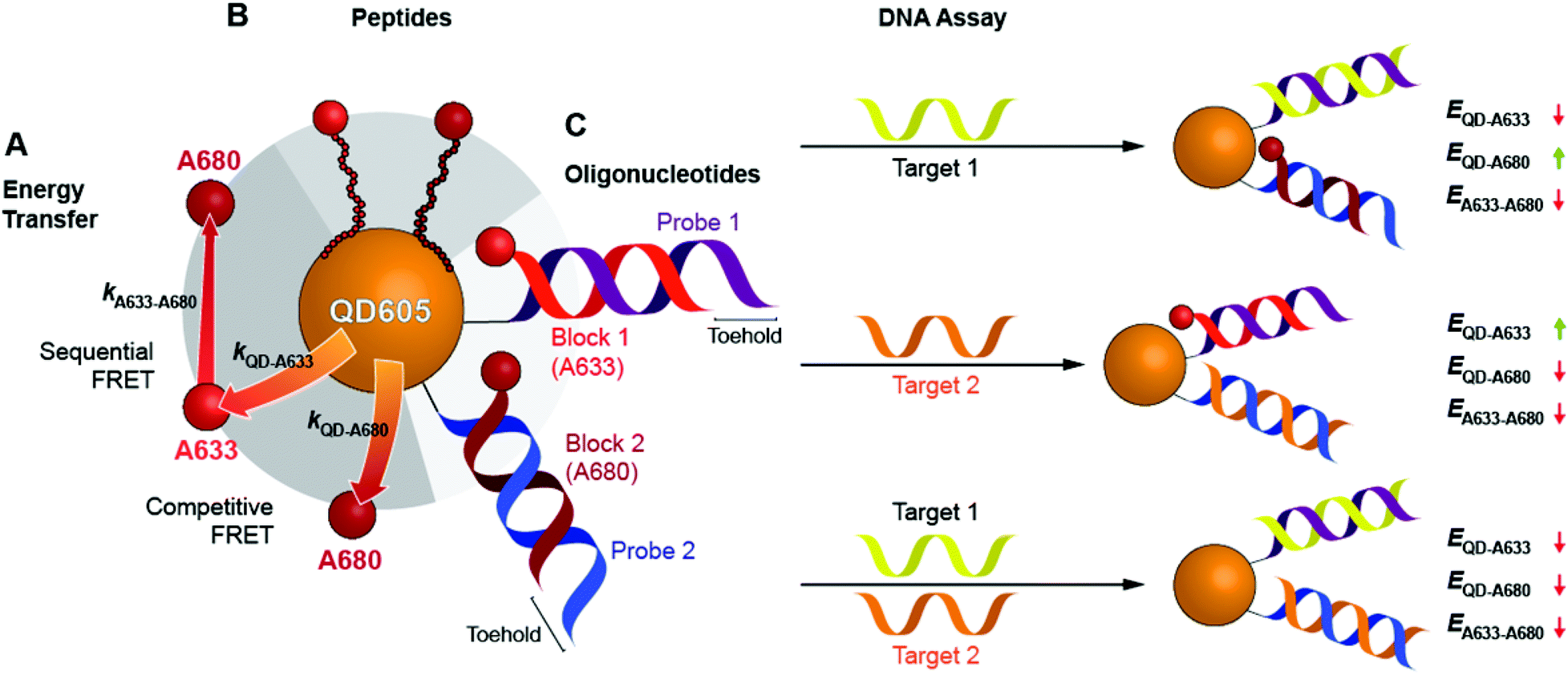 | ||
| Fig. 1 A long-wavelength cFRET configuration with QD605, A633, and A680, and its application in a toehold-mediated strand displacement hybridization assay. (A) Energy transfer pathways: the QD-to-A633 (kQD–A633) and QD-to-A680 (kQD–A680) pathways compete with one another, and the A633-to-A680 (kA633–A680) pathway can occur following QD-to-A633 energy transfer. (B) cFRET configuration assembled through peptide linkers between the QD605 and dyes. (C) Design of the cFRET hybridization assay. Probe oligonucleotides are conjugated to the QD605 and hybridized with block oligonucleotides that are labeled with either A633 or A680. Target DNA displaces one or both of the dye-labeled blocks with changes in the overall ensemble efficiencies, E, of the energy transfer pathways, which are tracked as the A633/QD605 and A680/QD605 PL ratios. | ||
Experimental section
Detailed experimental procedures can be found in the ESI.†Materials
CdSe/CdS/ZnS core/shell/shell QDs were synthesized using established methods.24,25 Alloyed CdSeS/ZnS QDs were from Cytodiagnostics (Burlington, ON, Canada). QDs were coated with glutathione (GSH) ligands for dispersion in aqueous media (see ESI† for details).26 Alexa Fluor 633 C5 maleimide and Alexa Fluor 680 C2 maleimide were from Thermo-Fisher Scientific (Carlsbad, CA, USA). Peptides were from Bio-Synthesis Inc. (Lewisville, TX, USA) and oligonucleotides were from Integrated DNA Technologies (Coralville, IA, USA). Table 1 lists the peptides and oligonucleotide sequences. Buffers were sterilized by either filtration or autoclaving. All other reagents were from Sigma-Aldrich (Mississauga, ON, Canada).| Name | Sequencea |
|---|---|
| a Peptide sequences (amino acids) are indicated with uppercase letters and written N-terminal to C-terminal (Ace = N-acetylation). Oligonucleotide sequences (nucleotides) are indicated with lowercase letters (DTL = dithiol linker; see ESI for details). | |
| Pep(A633) | Ace-HHHHHHSPPPPPPSGQGEGGNSDDDDKSGNGC-A633 |
| Pep(A680) | Ace-HHHHHHSPPPPPPSGQGEGGNSDDDDKSGNGC-A680 |
| Prb 1 | DTL-5′-atc tct tgg ccg tgt gga t-3′ |
| Blk 1(A633) | 5′-cac ggc caa gag at-3′-(A633) |
| Tgt 1 | 5′-atc cac acg gcc aag aga t-3′ |
| Prb 2 | DTL-5′-gct gtg tga gag acg gga aa-3′ |
| Blk 2(A680) | 5′-cgt ctc tca cac agc-3′-(A680) |
| Tgt 2 | 5′-ttt ccc gtc tct cac aca gc-3′ |
QD–peptide conjugates
Peptides were labeled with A633-maleimide or A680-maleimide, purified over nickel(II)–nitrilotriacetic acid (NTA), and desalted as described previously.27 Full details can be found in the ESI.† QD605–[Pep(A633)]M–[Pep(A680)]N samples were prepared by mixing M equivalents of Pep(A633) and N equivalents of Pep(A680) with GSH-coated QD605 with dilution in borate buffer (35 mM, pH 8.5, 35 mM NaCl, 30% v/v DMSO). Each peptide had a hexahistidine sequence that self-assembled to the ZnS shell of the QDs.28,29 The final concentration of QDs was 0.10 μM in a total volume of 100 μL.QD–oligonucleotide conjugates
Oligonucleotide blocks (Blk) were received with a thiol linker protected as a disulfide. The disulfide was reduced and labeled with A633-maleimide or A680-maleimide, and labeled block oligonucleotides were purified by size-exclusion chromatography. Oligonucleotide probes were received with an amine linker, which we modified to a dithiol-terminated linker (DTL) that self-assembled to the ZnS shell of the QDs. Full details on the linker synthesis and modification of the oligonucleotides can be found in the ESI.† To self-assemble the probe oligonucleotides to the QDs, P = 10 or 15 equiv. of each probe (Prb 1, Prb 2) were mixed with QD605 in tris-borate buffer (400 mM, pH 7.4, 50 mM NaCl), tris(2-carboxyethyl)phosphine was added as a reducing agent, and the ionic strength was increased partway through a 30 h incubation at room temperature. The QD–probe oligonucleotide conjugates were purified by size-exclusion chromatography. Full details can be found in the ESI.† The final concentration of QDs was determined by UV-visible spectrophotometry.30Hybridization and displacement assays
For hybridization assays, QD–[Prb 1]–[Prb 2] conjugates (final QD concentration: 50 nM in 40 μL) were mixed with the desired equivalents of Blk 1(A633) and Blk 2(A680) in tris-borate buffer (200 mM, pH 7.4, 200 mM NaCl) at room temperature for 1 h. For displacement assays, QD–[Prb 1]–[Prb 2] conjugates were pre-hybridized with Blk 1(A633) and Blk 2(A680), and this solution was mixed with an equal volume of solution with the desired equivalents of Tgt 1 and Tgt 2 (final QD concentration: 50 nM in 40 μL) at room temperature for 1.5–2 h. Full details can be found in the ESI.† A minimum of three hybridization and displacement assay experiments were done, and representative data for each is shown in the figures. Although there was batch-to-batch variation in absolute PL ratios, analogous trends were observed in every experiment.PL measurements
Absorption, PL excitation and emission spectra were measured with an Infinite M1000 Pro multifunction plate reader (Tecan Ltd, Morrisville, NC, USA) equipped with a xenon flash lamp, excitation and emission monochromators, and a photomultiplier tube detector. Samples were aliquoted into the wells of 96- or 384-well microtiter plates (Corning Inc., Corning, NY, USA). There was no evidence of inner filter effects in PL measurements and thus no such corrections were necessary. Photobleaching measurements were done on an IX83 inverted epifluoresence microscope (Olympus, Richmond Hill, ON, Canada). Samples were placed in a clear-bottom 96-well plate and covered with optical microplate sealing tape. Excitation was at 450/50 or 400/20 (center line/bandwidth in nm; Chroma Technology Crop., Bellow Falls, VT, USA) from an X-Cite 120XL metal–halide light (Excelitas Technologies, Mississauga, ON, Canada). Emission was collected by an objective lens (4.0×, 0.16 NA) and directed into a fiber-optic patch cable connected to a Green Wave CCD spectrometer (Stellar Net, Tampa, FL, USA).Data analysis
The PL intensities, XI, for X = QD605, A633, and A680 were used to calculate peak PL intensity ratios, ρdye/QD, according to eqn (1) and (2). The values of XI were calculated from the measured PL intensities, Iλ, measured at λ = 604, 648, and 704 nm, with corrections for crosstalk between different emitters (see ESI† for details).| ρA633/QD = A633I/QDI | (1) |
| ρA680/QD = A680I/QDI | (2) |
Results
QD and dyes
Fig. 2A shows the normalized absorption and PL emission spectra for the QD605, A633, and A680. There is significant overlap between the QD605 PL and the absorption of the A633 and A680, as is required for FRET. The absorption spectra for each of these emitters is re-plotted in terms of their wavelength-dependent molar absorption coefficients in Fig. 2B. The peak molar absorption coefficients for the dyes are comparable in magnitude to the molar absorption coefficient for the QD at its first exciton peak; however, the maximum absorption of the QD between ca. 400–475 nm is more than an order of magnitude larger than the maximum absorption of the dyes. The A633 and A680 have their weakest light absorption between ca. 450–500 nm, which corresponds with the minimum in their PL excitation spectra. In FRET experiments, efficient excitation of the QD donor with minimal direct excitation of the A633 and A680 acceptor(s) is desired. This criterion was met with excitation at 464 nm (indicated by the arrow in Fig. 2B). Table 2 summarizes some important properties of the QD605, A633, and A680, including the spectral overlap integrals and estimated Förster distances for the relevant FRET pairs.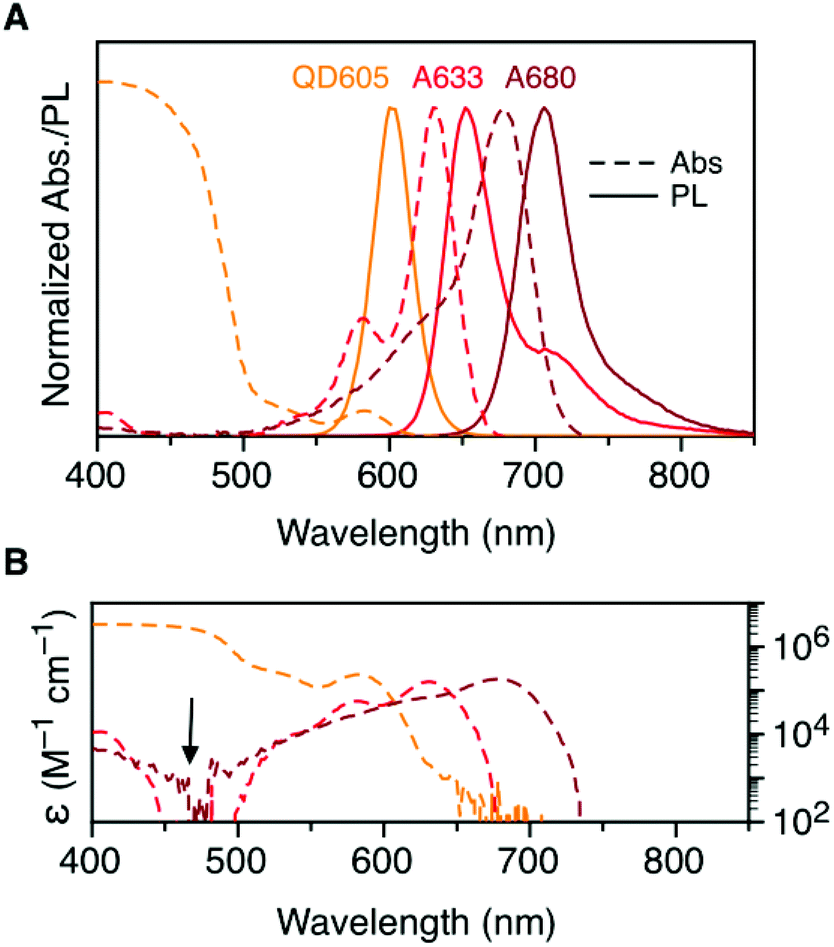 | ||
| Fig. 2 (A) Normalized absorption and PL emission spectra for QD605, A633, and A680. (B) Absorption spectra in panel A re-plotted as the wavelength-dependent molar absorption coefficient, ε(λ). The arrow indicates the preferred excitation wavelength for the QD605–A633–A680 cFRET system. Note the logarithmic scale in panel B. | ||
| Property | QD605 | A633 | A680 |
|---|---|---|---|
| a First exciton peak for the QD605 and absorption maximum for the dyes. b Peak molar absorption coefficient. c Peak PL wavelength. d Quantum yield of the QD or dye–oligonucleotide conjugate. e PL lifetime (amplitude weighted average lifetime for QD605). f Spectral overlap integral. g Förster distance. See ESI (Fig. S1 and S2) for τ and Φ data. | |||
| λ Abs (nm) | 584 | 630 | 678 |
| ε (103 M−1 cm−1) | 232 | 159 | 183 |
| λ PL (nm) | 604 | 648 | 704 |
| Φ | 0.23 | 0.45 | 0.31 |
| τ (ns) | 12.7 | 1.0 | 0.4 |
| J X–A633 (10−10 cm6 mol−1) | 8.3 | — | — |
R
0, X–A633![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) g (nm) g (nm) |
5.7 | — | — |
|
J
X–A680f (10−10 cm6 mol−1) |
6.4 | 19 | — |
|
R
0, X–A680g (nm) |
5.5 | 7.4 | — |
cFRET spectra and PL ratios
To characterize the energy transfer pathways and PL for the QD605–A633–A647 cFRET configuration, an array of QD605–[Pep(A633)]M–[Pep(A680)]N samples were prepared for 0 ≤ M ≤ 12 and 0 ≤ N ≤ 12, and the corresponding PL emission spectra were measured. These configurations are abbreviated as (M, N), where M and N are the average number of Pep(A633) and Pep(A680), respectively, per QD across the ensemble. Assembly of the dye-labeled peptides to the QD605 was confirmed via gel electrophoresis (see ESI, Fig. S3†). Representative PL spectra are shown in Fig. 3A for selected samples, including the subsets of (M, 0), (0, N), (12, N), and (M, 12). The spectra for all (M, N) combinations can be found in the ESI (Fig. S4†). The (M, 0) and (0, N) spectra show that both A633 and A680 were effective FRET acceptors for the QD605 donor, as evident from quenching of the QD605 PL and sensitization of the dye PL. The PL contribution from direct excitation of both dyes was between 1–2 orders of magnitude smaller than with FRET sensitization (see insets in Fig. 3A). The (12, N) data shows how addition of A680 further quenched the QD605 via the introduction of a second net energy transfer pathway, and quenched the A633 by both A633-to-A680 energy transfer and by competing for energy transfer from the QD605. The (M, 12) spectra show that both QD605 and A680 PL were quenched as M increased, and indicated that the competition between the QD605-to-A633 and QD605-to-A680 energy transfer pathways had a larger effect on the system than A633-to-A680 energy transfer. These (M, N)-dependent trends in the energy transfer dynamics of the cFRET system were reflected in the A633/QD605 and A680/QD605 PL intensity ratios, which are summarized in Fig. 3B. The A633/QD605 PL ratio increased as M increased and decreased as N increased, whereas the A680/QD605 PL ratio increased as both M and N increased. These ratios are useful as analytical signals in assays because each (M, N) combination yields a unique combination of A633/QD605 and A680/QD605 PL ratios, as shown by the 25 resolved data points plotted in Fig. 3C.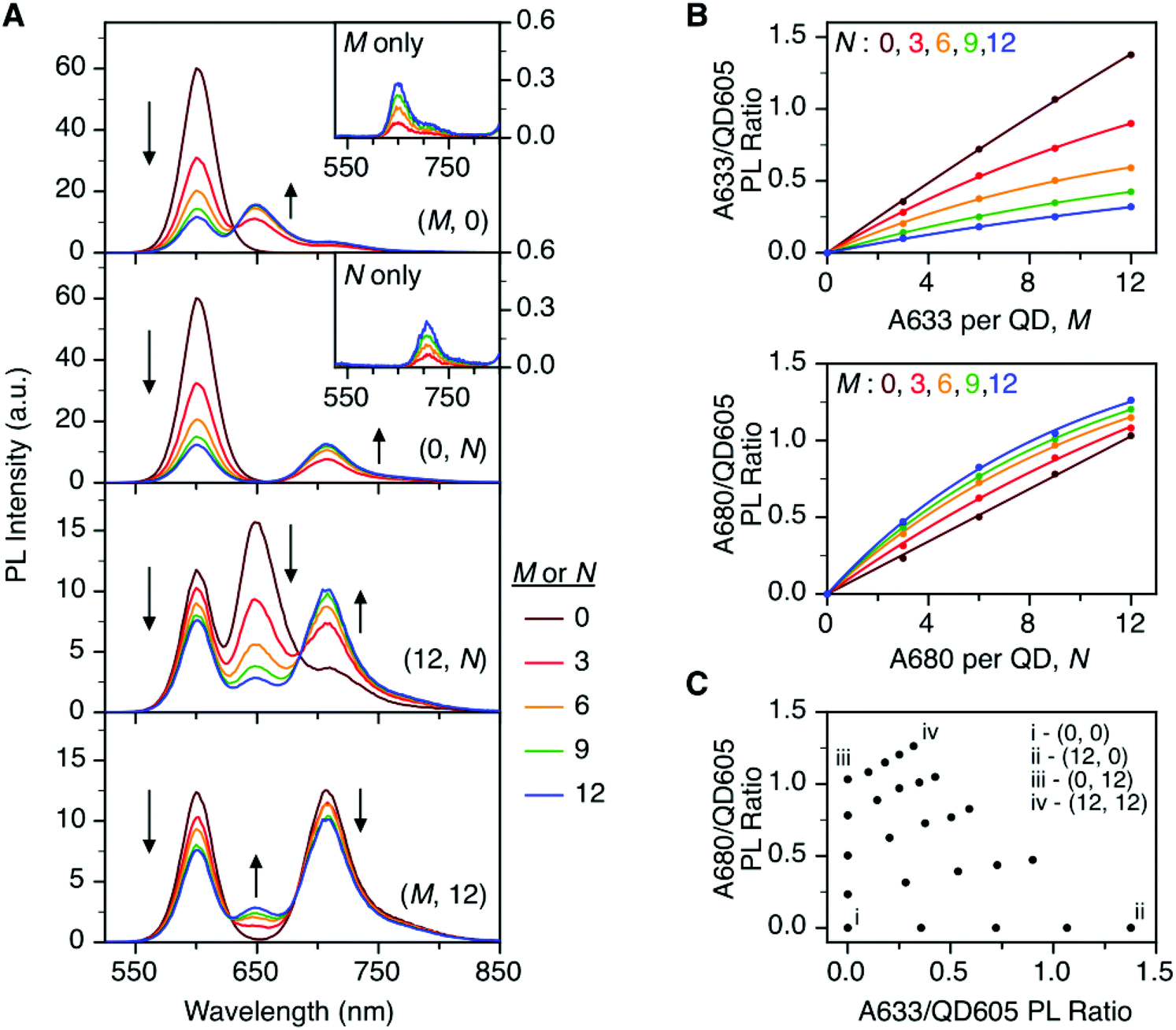 | ||
| Fig. 3 (A) Representative PL emission spectra for (M, N) = (M, 0), (0, N), (12, N), and (M, 12) QD605–[Pep(A633)]M–[Pep(A680)]N cFRET configurations. The insets show PL spectra for control samples of Pep(A633) (M only) and Pep(A680) (N only) without QD605. (B) Summary of the A633/QD605 and A680/QD605 PL ratios as a function of M and N. (C) Plot showing that each (M, N) yields a unique combination of A633/QD605 and A680/QD605 PL ratios. | ||
cFRET efficiencies
The foregoing trends in PL intensities were analyzed quantitatively in terms of relative energy transfer rates and efficiencies. In a cFRET configuration, eqn (3) should predict the QD605 quenching efficiency, QQD, for all non-zero (M, N) configurations, where kQD–A633 is the rate of QD-to-A633 energy transfer, kQD–A680 is the rate of QD-to-A680 energy transfer, and kQD is the intrinsic relaxation rate of the QD605.23 The values of kQD–A633kQD−1 and kQD–A680kQD−1 can be determined from the (M, 0) and (0, N) data subsets, respectively, as shown in Fig. 4A(i–ii). The predicted and measured values of the QD605 quenching efficiency in the (M, N) configurations were in good agreement, as indicated by the correlation with a near-unity slope in Fig. 4A(iii).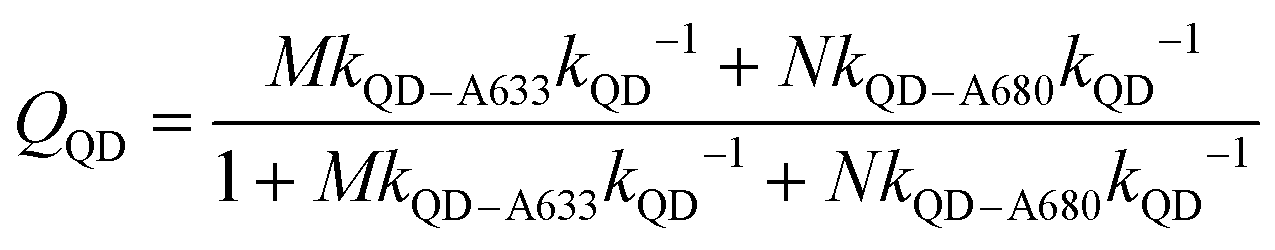 | (3) |
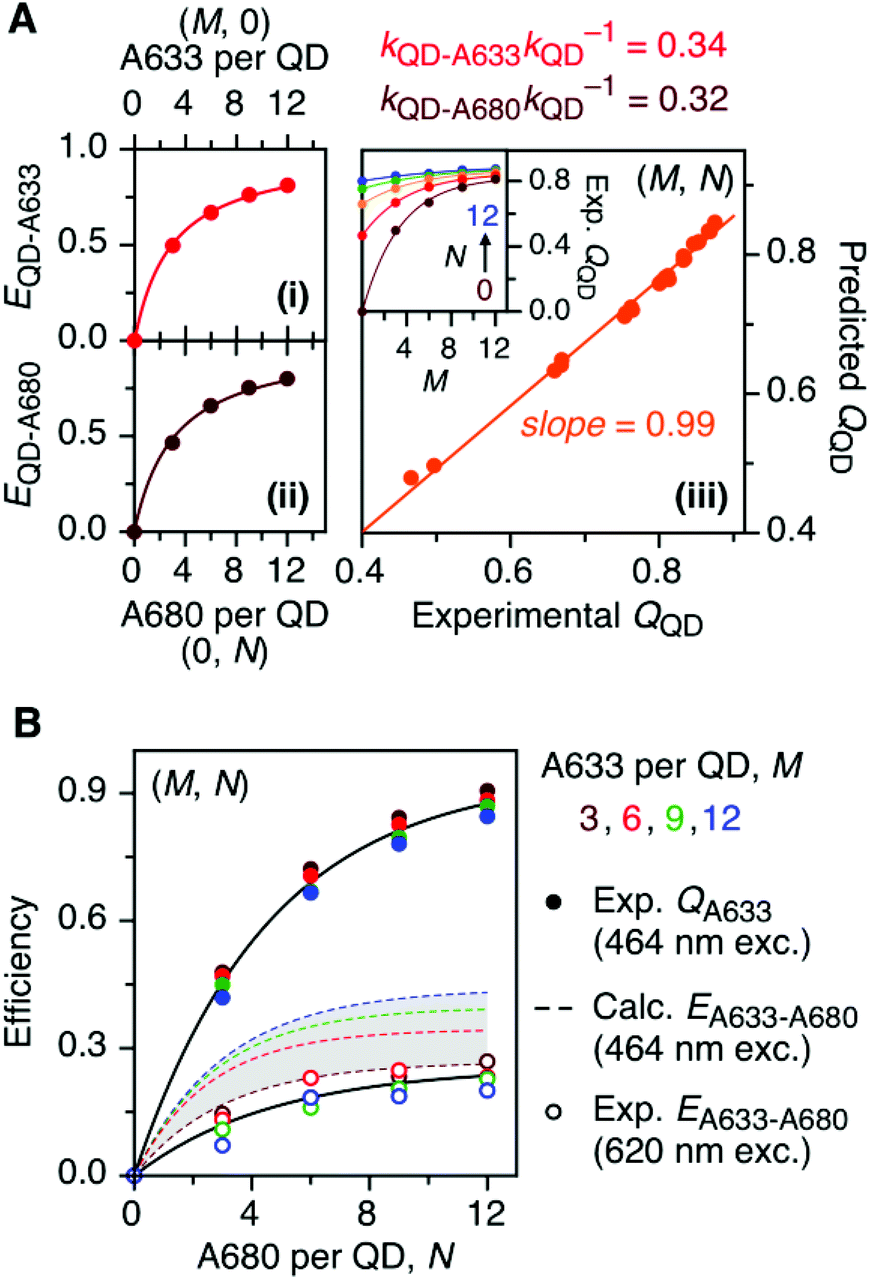 | ||
| Fig. 4 (A) Efficiencies, EQD–X, for (i) QD-to-A633 FRET measured from (M, 0) configurations and (ii) QD-to-A680 FRET measured from (0, N) configurations. The relative energy transfer rates, kQD–XkQD−1, derived from this data are listed and can be used to predict the efficiency of QD PL quenching, QQD, for (M, N) configurations, which (iii) show a good correlation with the experimentally measured quenching efficiencies. The inset shows the experimental QQD as a function of M and N. (B) Characterization of A633-to-A680 energy transfer. The solid circles plot the measured quenching of A633 PL in (M, N) configurations, QA633, relative to the corresponding (M, 0) configuration, with predominant excitation of the QD605 at 464 nm. This quenching is from a combination of A633-to-A680 energy transfer and the competition between A633 and A680 to accept energy from the QD605. The latter can be calculated from the data in panel A and used to estimate the efficiencies of A633-to-A680 energy transfer, EA633–A680 (calc.), which are shown as dashed coloured lines within the shaded region. The open circles plot the measured A633-to-A680 energy transfer efficiency, EA633–A680 (exp.), with direct excitation of the A633 (and, to a lesser extent, A680) at 620 nm. | ||
Another consideration is the A633-to-A680 energy transfer efficiency. This pathway can be probed directly by exciting the A633 at 620 nm, a wavelength longer than the first exciton peak of the QD605, which largely bypasses the QD-to-dye FRET pathways. Fig. 4B shows the measured A633-to-A680 FRET efficiencies with direct excitation of the A633 (and, by necessity, some collateral direct excitation of A680; see Fig. 2). The energy transfer efficiency, EA633–A680 (exp.), reaches a maximum of ca. 23 ± 3% at N = 12, with only small variations in the trend for different values of M. Also shown in Fig. 4B is the apparent quenching of A633 PL, QA633, with increasing N and excitation at 464 nm, where the excitation of the QD sensitizes both the A633 and A680 through FRET. The magnitude of QA633 spans ca. 40–90%, which is much larger than with its direct excitation at 620 nm, but represents quenching due to a combination of energy transfer from A633-to-A680 and, more prominently, the competition between A633 and A680 to accept energy from the QD605. Quenching of A633 PL due to competitive energy transfer, QA633-c, can be predicted from eqn (4), and the difference between the observed quenching and that predicted for competitive FRET is the quenching due to A633-to-A680 energy transfer. The resulting envelope (for different M) for the expected A633-to-A680 FRET efficiencies, EA633–A680 (calc.), with excitation at 464 nm are shown in Fig. 4B (shaded region with dashed lines). The observed A633-to-A680 efficiencies from direct excitation of the A633, EA633–A680 (exp.), are near the bottom of this envelope, which can be attributed to a decrease in the effective number of A680 acceptors caused by unwanted direct excitation of A680. Only ground state A680 is an acceptor, and the molar absorption coefficient for A680 at 620 nm is ∼63000 M−1 cm−1versus 117000 M−1 cm−1 for A633, such that A680 is approximately half as likely to be excited as A633. Accounting for a decrease in the effective number of A680 acceptors will shift the experimental data for the directly excited A633-to-A680 efficiency within the predicted envelope for the same pathway following sensitization by QD605-to-A633 energy transfer.
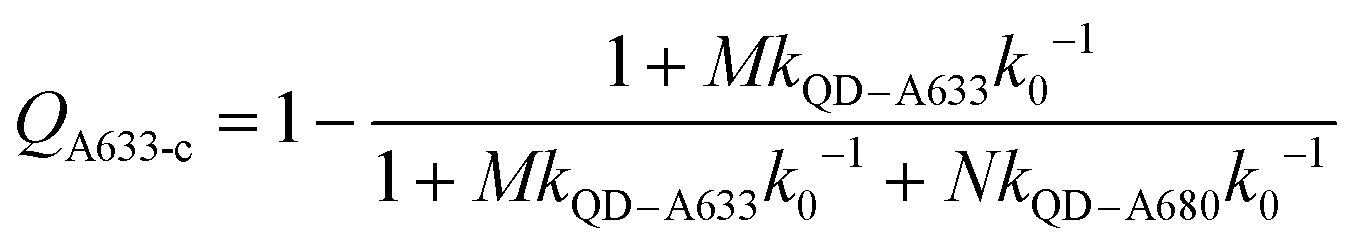 | (4) |
The cumulative results in Fig. 4 confirm that the net energy transfer pathways in the QD605–A633–A680 cFRET system behave as depicted in Fig. 1A, which is analogous to the energy transfer pathways in the previously studied and prototypical QD520–A555–A647 cFRET system, but with a different balance of competitive and sequential energy transfer (vide infra).
cFRET photobleaching
To evaluate the resistance of the QD605–[Pep(A633)]M–[Pep(A680)]N cFRET configuration toward photobleaching, samples for (0, 0), (12, 0), (0, 12), and (12, 12) were continuously illuminated with 450 nm excitation light under a fluorescence microscope and PL spectra were recorded at regular time intervals. As shown in Fig. 5A, the (0, 0) configuration (i.e., QD605 alone) was very resistant to photobleaching, exhibiting no decrease in its PL intensity over 30 min at the full lamp intensity (∼98 mW at the sample). For all other configurations, the lamp power had to be reduced by ∼90% (∼11 mW at the sample) to slow the rate at which the dyes photobleached. With the (12, 0) configuration, there was a short induction period (∼1.5 min) over which the QD and A633 PL were stable before photobleaching of the A633 with an approximate first-order rate constant of 0.66 min−1. The QD PL intensity increased in parallel with photobleaching of the A633, consistent with the loss of QD605-to-A633 energy transfer. With the (0, 12) configuration, the A680 started bleaching immediately upon illumination, with an approximate first-order rate constant of 0.75 min−1. With the full cFRET configuration, (12, 12), the A680 once again started to photobleach immediately upon illumination. As the A680 photobleached, there was a four-fold increase in the A633 PL intensity and a modest 60% increase in the QD PL intensity. The increase in A633 PL intensity was consistent with (i) more efficient QD605-to-A633 energy transfer with loss of the QD605-to-A680 energy transfer upon photobleaching of A680, and (ii) parallel loss of A633-to-A680 energy transfer. After the A680 had photobleached, the A633 started to photobleach with a parallel increase in the QD PL intensity, consistent with final loss of QD605-to-A633 energy transfer. This overall photobleaching behavior of the (12, 12) configuration was further evidence of the energy transfer network depicted in Fig. 1A.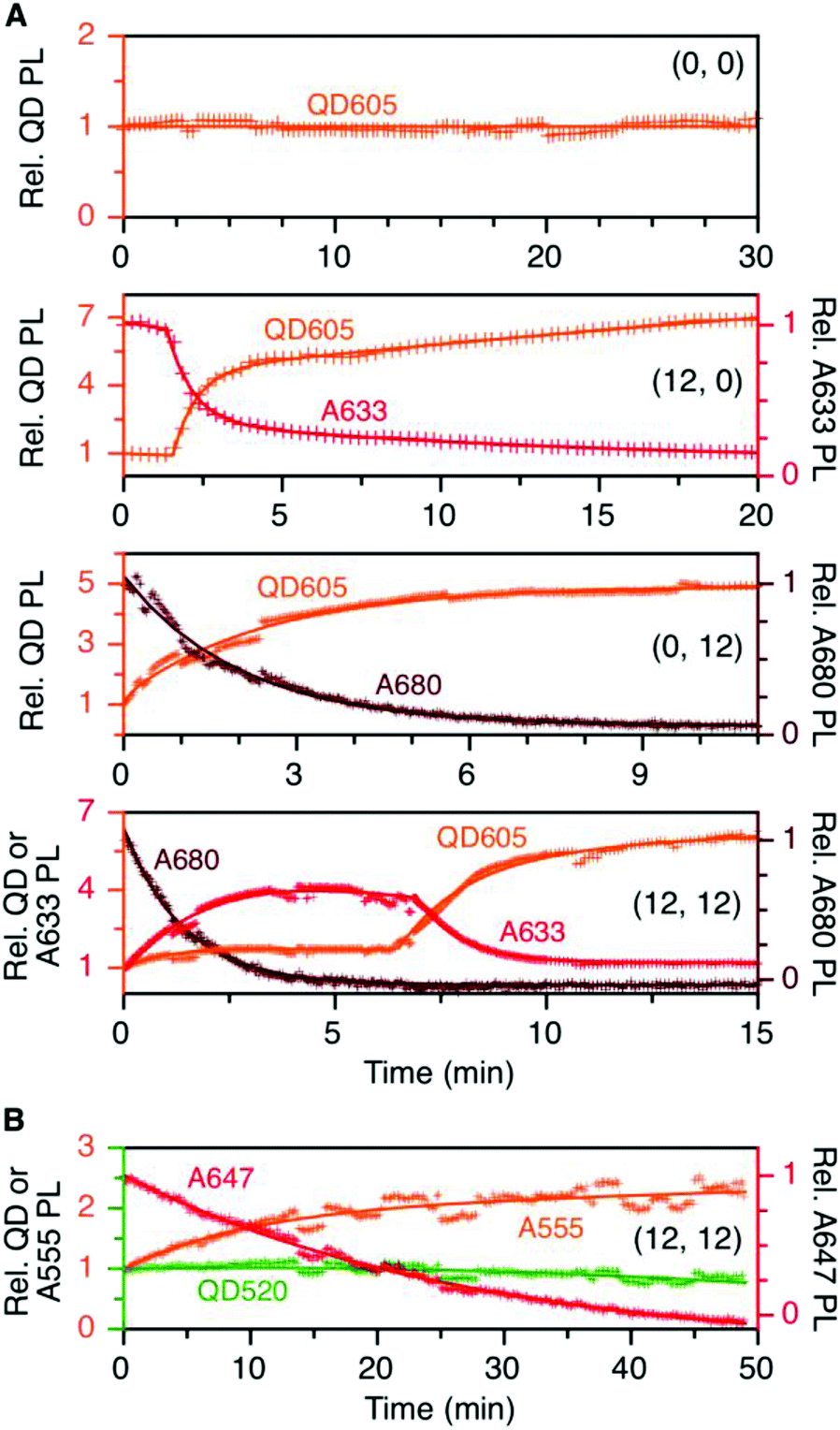 | ||
| Fig. 5 (A) Relative stabilities of QD605, A633, and A680 PL (as applicable) in the (0, 0), (12, 0), (0, 12), and (12, 12) permutations of the QD605–[Pep(A633)]M–[Pep(A680)]N cFRET configuration with continuous illumination at 450 nm. The measured power at the sample was ∼11 mW, except for the (0, 0) sample, which was ∼98 mW. The PL intensities are normalized to an initial value of unity. Note the two different y-axis scales (colour-coded to match the QD, A633, or A680). (B) Relative stabilities of the QD, A555, and A647 PL in the (12, 12) permutation of the prototypical QD520–[Pep(A555)]M–[Pep(A647)]N cFRET configuration. The power at the sample was ∼10 mW. Data for other permutations can be found in the ESI.† | ||
In our previous QD520–A555–A647 cFRET configuration, the A647 was found to be the weak link and exhibited the fastest rate of photobleaching, whereas the A555 resisted photobleaching to a degree similar to the QD520. For comparison, we reassessed the photobleaching of QD520–[Pep(A555)]M–[Pep(A647)]N samples under conditions similar to those for the QD605–A633–A680 cFRET configuration. Full results can be found in the ESI (see Fig. S5†), and Fig. 5B shows the results for the (12, 12) permutation. Consistent with previous measurements, the QD520 and A555 were resistant to photobleaching, with the QD exhibiting ±10% variation over 50 min of continuous illumination at 400 nm (∼50 mW at the sample). To evaluate bleaching of the A647, the excitation was reduced by 80% (∼10 mW at the sample), whereupon the A647 was observed to bleach at a rate between 0.04–0.08 min−1. The A555 PL intensity increased with bleaching of the A647 and otherwise tracked in parallel with changes in QD520 PL intensity. The increase in the A555 PL intensity with bleaching of A647 in the (12, 12) sample was similar to that observed with A633 and A680, with the exception that the A633 subsequently bleached and the A555 did not.
Serum and blood matrices
As noted earlier, an anticipated benefit of the long-wavelength cFRET configuration was better signal-to-background (S/B) ratios when working with biological samples. To test this hypothesis, QD605–[Pep(A633)]M–[Pep(A680)]N samples were prepared in matrices of buffer, 50% and 90% v/v serum, and 50% and 90% v/v blood. Analogous QD520–[Pep(A555)]M–[Pep(A647)]N samples at the same molar concentration were prepared for comparison. Each configuration was excited at its optimal wavelength for minimization of direct excitation of acceptors (464 nm for QD605–A633–A680, 400 nm for QD520–A555–A647).Fig. 6A and B shows spectra in buffer for the (0, 0), (12, 0), (0, 12), and (12, 12) samples of both cFRET configurations, and high average S/B ratios (103–104) were easily obtained. In 50% v/v serum (see ESI, Fig. S6A and B†), the average S/B ratios were still high for QD605–A633–A680 samples (∼102), but decreased dramatically for QD520–A555–A647 (<5). With respect to the latter, the QD520 PL was difficult to resolve when strongly quenched in the (12, 0) and (12, 12) samples, as was the quenched A555 PL in the (12, 12) sample. With dilution to 90% v/v serum, the S/B ratios decreased even further, such that the QD520 PL and both dye PL signals were largely lost in the fluorescence background of the serum. As shown in Fig. 6C, the average S/B ratios ranged between ca. 0.1–1 across all wavelengths. Substitution of brighter CdSeS/ZnS QD525a for the QD520 resulted in improved S/B ratios in serum (see ESI, Fig. S6C†), with a range of 0.4–4 across all wavelengths in 90% v/v serum. In contrast, clear PL signals were still observed in 90% v/v serum with the QD605–A633–A680 samples, with average S/B ratios between ca. 4–30, as shown in Fig. 6C. The wavelength-dependent S/B ratios can be compared to the average PL spectra in buffer (normalized to peak QD PL intensity) that are also shown in Fig. 6C. For all of the cFRET configurations, S/B ratios were better retained between buffer and 90% v/v serum samples at longer wavelengths, as indicated by the difference between the shapes of the average PL spectra in buffer and the average S/B ratio plots in 90% v/v serum.
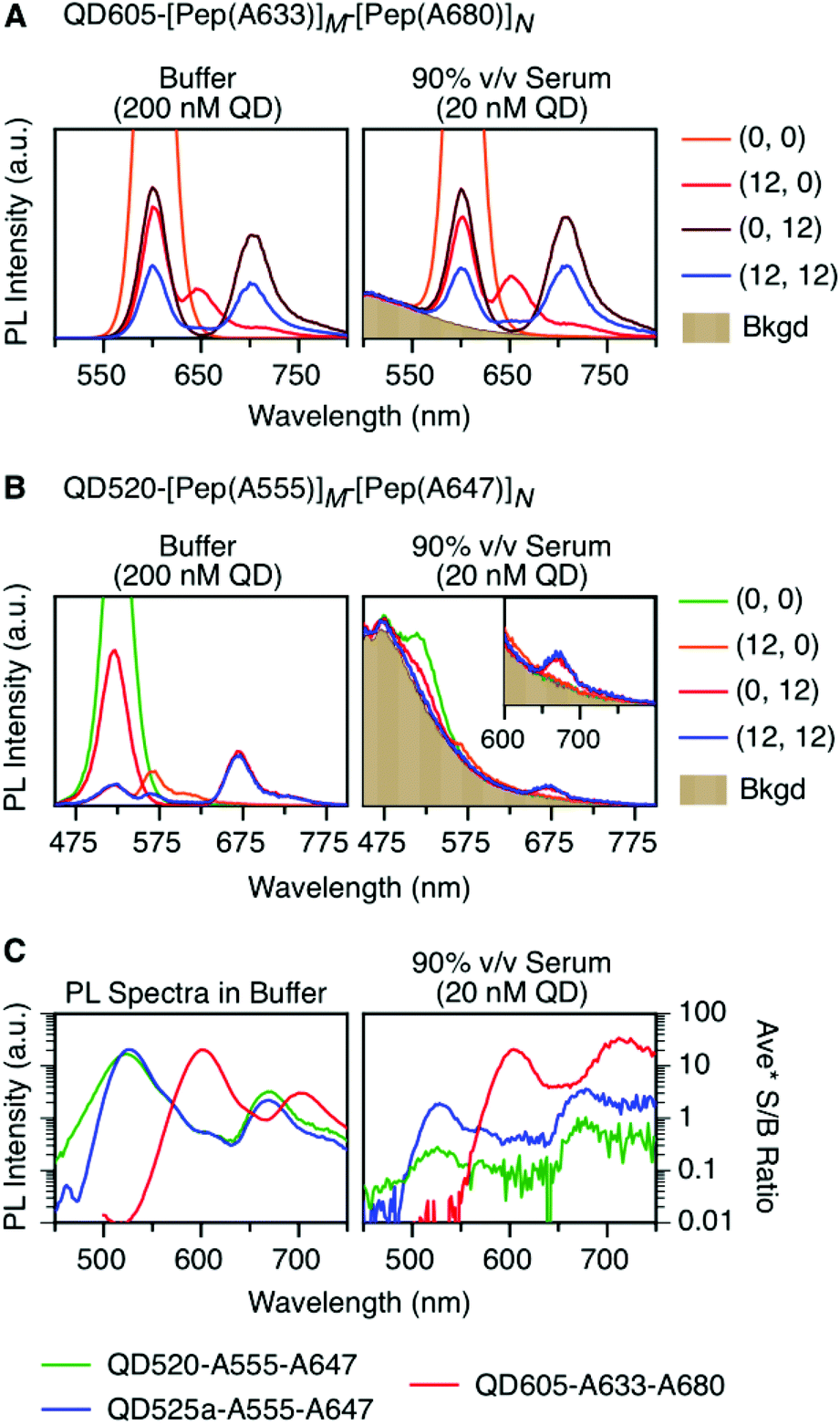 | ||
| Fig. 6 PL emission spectra for (0, 0), (12, 0), (0, 12), and (12, 12) samples of (A) QD605–[Pep(A633)]M–[Pep(A680)]N and (B) QD520–[Pep(A555)]M–[Pep(A647)]N in buffer (left plot) and diluted into 90% v/v serum (right plot). The intensity axis has been set so that the QD PL peaks for the (0, 0) samples are off the scale. The excitation wavelengths were 464 nm for QD605–A633–A680 and 400 nm for QD520–A555–A647. Numerical PL intensity scales are not shown in panels A and B because different gain settings were used. (C) Average PL spectra in buffer, plotted on a logarithmic scale, for QD520–[Pep(A555)]M–[Pep(A647)]N, QD525a–[Pep(A555)]M–[Pep(A647)]N, and QD605–[Pep(A633)]M–[Pep(A680)]N (left plot). The average signal-to-background (S/B) ratio as a function of wavelength for these cFRET systems in 90% v/v serum is shown for comparison (right plot). | ||
Serum is an example of a sample matrix that has significant background autofluorescence. Another aspect to consider is attenuation of excitation light by sample matrices, regardless of background autofluorescence. For this purpose, we selected whole blood as an example of a sample matrix with deep colour and strong light scattering, and prepared dilutions of cFRET configurations in 50% and 90% v/v whole blood (see Fig. S7† for spectra). The average S/B ratios for QD605–A633–A680 were between 5–60 in 90% v/v blood, and those for QD525a–A555–A647 were between 0.5–4. These values are very similar to those for 90% v/v serum, and can be attributed to a combination of very low background autofluorescence from the blood and very high attenuation of absolute PL intensities. With 50% v/v blood, the PL intensities from QD520 and QD525a were reduced by ∼93% versus buffer, and the PL intensity of QD605 was reduced ∼82% versus buffer. PL intensities were only reduced between 20–40% in 50% v/v serum.
It is clear that the QD605–A633–A680 configuration offers higher PL intensities and S/B ratios in serum and blood than the QD520/QD525a–A555–A647 configuration. The data show that these advantages are derived from a combination of higher intrinsic brightness for the QD605, less background autofluorescence at wavelengths longer than 600 nm, and less attenuation of 464 nm excitation light versus 400 nm excitation light.
Hybridization assay
Previous work has thoroughly demonstrated the utility of peptide-linked cFRET configurations for multiplexed detection of protease activity.19–23 Here, the goal was to demonstrate an oligonucleotide-linked cFRET configuration for multiplexed detection of nucleic acid targets. Oligonucleotide probes (19–20 nt in length), Prb 1 and Prb 2, were modified with a dithiol-terminated linker and conjugated to QD605 via self-assembly with subsequent purification to remove unassembled oligonucleotides and reducing agent. Assembly was confirmed via gel electrophoresis (see ESI, Fig. S8†). These conjugates were hybridized with different numbers of equivalents of partially complementary block sequences (15–16 nt), either Blk 1(A633) or Blk 2(A680). The FRET efficiency and A633/QD or A680/QD PL ratio increased as the number of equivalents of block increased, up until saturation of the probes (see ESI, Fig. S9†). The time required for full hybridization was ca. 20–30 min. Next, conjugates were hybridized with mixtures of both Blk 1(A633) and Blk 2(A680) at different ratios. As shown in Fig. 7A, the FRET efficiency tracked with the number of A633 and A680 per QD, M and N, with trends analogous to the peptide-linked configuration in Fig. 4A(iii). The trends in the A633/QD605 and A680/QD605 PL ratios (Fig. 7A(iii)) were also similar between the oligonucleotide- and peptide-linked configurations. Overall, these results demonstrated assembly of an oligonucleotide-linked cFRET configuration with spectroscopic behaviour similar to the peptide-linked configuration.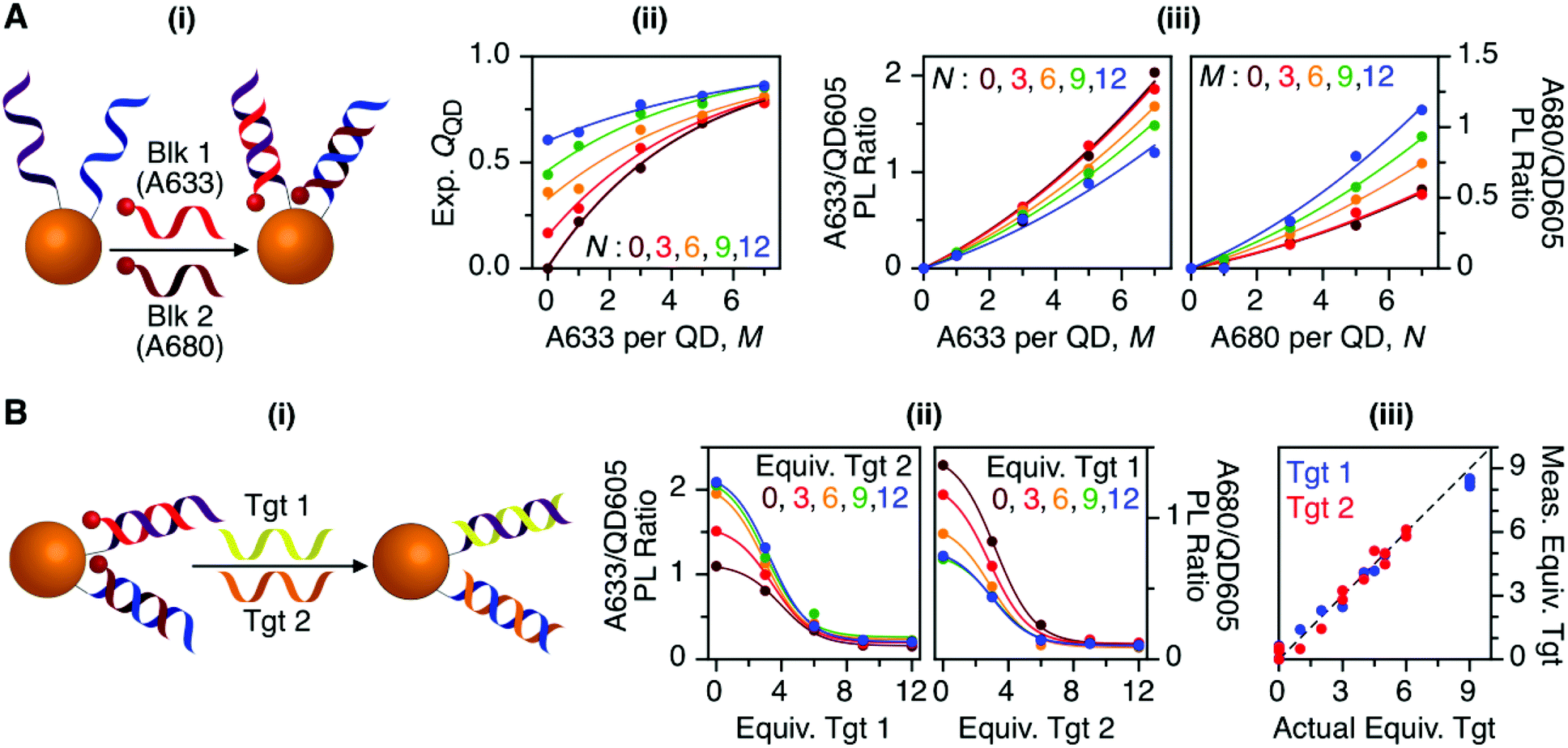 | ||
| Fig. 7 (A) cFRET hybridization: (i) schematic; (ii) experimental QQD as a function of M and N; and (iii) summary of the A633/QD605 and A680/QD605 PL ratios as a function of M and N. The results are similar to those in Fig. 3 and 4. (B) cFRET toehold-mediated strand displacement hybridization assay: (i) schematic; (ii) summary of the A633/QD605 and A680/QD605 PL ratios as a function of the number of equivalents of Tgt 1 and Tgt 2 added; and (iii) correlation between the measured and actual numbers of equivalents of targets when the calibration data in (ii) was applied to blind samples. The dashed line has a slope of unity. | ||
To demonstrate the potential for a multiplexed hybridization assay, QD605 were conjugated with Prb 1 and Prb 2 and hybridized with Blk 1(A633) and Blk 2(A680). Fig. 7B shows changes in the A633/QD605 and A680/QD605 PL ratios when different amounts of Tgt 1 and Tgt 2, the full-length complements of the probe oligonucleotides, were added. The targets displaced the dye-labeled block sequences with progressive loss of cFRET until all of the blocks had been displaced. This assay format and the expected changes in cFRET are illustrated in Fig. 1C. The toeholds created by having shorter block sequences than probe sequences facilitated displacement of the blocks by full-length complementary target.31–33 A set of 25 different combinations of equivalents of Tgt 1 and Tgt 2 were used to generate a calibration data set. This calibration was then used to test a series of blind samples, which had different amounts of Tgt 1 alone, Tgt 2 alone, or a combination of different amounts of both Tgt 1 and Tgt 2 (see ESI, Fig. S11† for details on how the data in Fig. 7B(ii) was used to calculate the number of equivalents of target). Fig. 7B(iii) shows the correlation between the measured and actual number of equivalents of Tgt 1 and Tgt 2 in the blind samples. A good correlation was observed, demonstrating that an oligonucleotide-linked cFRET configuration can be used for a quantitative multiplexed hybridization assay.
Discussion
While the unique physical and optical properties of QDs enable the assembly of cFRET configurations, the properties of the fluorescent dye acceptors are also critical to their function. The prototypical QD520–A555–A647 configuration19,20,22,23 has been effective, in no small part, because the A555 and A647 satisfy several key properties: large molar absorption coefficients, sufficiently similar quantum yields, spectral overlap with one another and the QD, spectrally resolved emission peaks, a common excitation minimum, good aqueous solubility, a low tendency toward self-quenching, and availability as reactive derivatives suitable for labeling biomolecules. This combination of properties is not trivial to achieve. The new QD605–A633–A680 combination reported here also satisfies these criteria to a large degree, and demonstrated that cFRET is not restricted to one unique combination of QD and fluorescent dyes. With careful design and selection of dyes, cFRET is a general strategy.A comparison of the spectral properties of the QD520–A555–A647 and QD605–A633–A680 cFRET systems reveals both similarities and differences (see Table 2 for the properties of the latter system). The molar absorption coefficient of A633 is equivalent to that of A555 (155000 M−1 cm−1), and although the molar absorption coefficient of A680 is smaller than that of A647 (270000 M−1 cm−1), it is still quite large. Like A555 and A647, A633 and A680 both have a common excitation minimum (∼464 nm), which is at a longer wavelength than for A555 and A647 (∼400 nm). The spectral separation between the QD605, A633, and A680 emission peaks are each ∼50 nm, which is similar to the ∼50 nm spectral separation between QD520 and A555, but smaller than the ∼100 nm separation between A555 and A647. The separation between the absorbance peaks of the dyes are also ∼50 nm for A633 and A680 versus ∼100 nm for A555 and A647. These cumulative characteristics combine to yield differences in the FRET spectral overlap integrals between the two cFRET systems. The integral for the QD605–A633 FRET-pair is a modest 40–50% larger than the integral for the QD520–A555 FRET-pair (∼5–7 × 10−10 cm6 mol−1); however, the integral for QD605–A680 is notably 12-fold larger than for QD520–A647 (∼0.4–0.7 × 10−10 cm6 mol−1).22,23 The integral for A633–A680 is almost 3-fold larger than for A555–A647 (6–8 × 10−10 cm6 mol−1). The larger integrals for the QD605–A633–A680 system arise from both the smaller spectral separation between the dye absorbance peaks and the general shift to longer wavelengths (the overlap integral is proportional to λ4).
The differences in spectral overlap integrals between the QD605–A633–A680 and QD520–A555–A647 cFRET configurations, particularly QD605–A680 overlap versus QD520–A647 overlap, resulted in fundamental differences in the balance of the three net energy transfer pathways. In the prototypical QD520–A555–A647 configuration, the relative rates of energy transfer between a donor and acceptor, kD–AkD−1, scaled as QD520–A555 (0.37) > A555–A647 (0.09) ≈ QD520–A647 (0.08). In the new QD605–A633–A680 configuration, the scaling was QD605–A633 (0.34) ≈ QD605–A680 (0.32) > A633–A680 (0.03). Consequently, quenching of A633 was driven mostly by the competition with A680 to accept energy from the QD605, whereas quenching of A555 was driven more by A555-to-A647 energy transfer.
Here, the QD605–A633–A680 system was advantageous with respect to measurements in biological sample matrices such as serum and blood, providing much higher S/B ratios than the prototypical QD520–A555–A647 (and QD525a–A555–A647) system. This advantage can be primarily attributed to the greater brightness of the QD605 versus the QD520 (or QD525a), which arises from the 20-fold (or 5-fold) larger molar absorption coefficient of the QD605 at its optimal excitation wavelength. Two secondary factors that contribute to the higher S/B ratios are emission at longer wavelengths where the background matrix autofluorescence is less intense, and/or less attenuation of 464 nm excitation light versus 400 nm excitation light by the sample matrix. A trade-off for greater S/B ratios with biological sample matrices was that the QD605–A633–A680 system was less resistant to photobleaching than the prototypical QD520–A555–A647 system. An inspection of the data in Fig. 5 would suggest that A633 and A680 photobleached approximately ten-fold faster than A647; however, the excitation rate of the A633 and A680 was greater than that of A647 because of the greater excitation rate of the QD605 versus QD520. Our analysis (see ESI†) suggests that this faster excitation rate may have accounted for much of the difference in photobleaching rates. Consequently, the main disadvantage of the QD605–A633–A680 system was that both A633 and A680 were susceptible to photobleaching, whereas only A647 was susceptible in the QD520–A555–A647 system.
Beyond the new cFRET combination of QD and dyes, a second important aspect of this study was demonstration of an oligonucleotide-linked cFRET configuration and hybridization assay. Unlike peptides, oligonucleotides cannot be directly synthesized with polyhistidine for self-assembly to QDs. Although the ligation of oligonucleotides with polyhistidine has been reported,34,35 we opted to modify our oligonucleotides with a small molecule linker that could be chemically reduced to a dithiol, which also has strong affinity for the ZnS shell of the QDs. As described in the ESI,† this strategy was effective, but not quantitative. The number of oligonucleotide equivalents assembled per QD was always less than the number mixed with the QDs (see Fig. S9†), and it was necessary to purify the conjugates by size-exclusion chromatography. Another key difference between the peptide- and oligonucleotide-linked configurations was the oligonucleotides themselves, which had notably different physical properties. Oligonucleotides are polyanions that repel one another when non-complementary, whereas no strong repulsion is expected for the peptides, which are also smaller in size (∼1.2 nm dia. × 4 nm length for peptides versus ∼2 nm × 6.6 nm for oligonucleotides). The increase in probe oligonucleotide conjugation efficiency with salt aging, which is well-known and widely utilized with gold nanoparticle–oligonucleotide conjugates36 but generally unnecessary with QD–peptide conjugates, was one indication of these physical differences. Perhaps surprisingly, the cFRET trends were similar between the peptide-linked and oligonucleotide-linked cFRET configurations. The physical differences between the oligonucleotides and peptides may have been largely mitigated by the proximal position of the dye relative to the QD in the oligonucleotide-linked configuration versus the distal position of the dye in the peptide-linked configuration (see Fig. 1B and C).
Analytically, a peptide-linked cFRET configuration has been shown to be valuable for detection of protease activity.19,20,22 In principle, nuclease activity could be detected using the same general configuration by replacing the distally-labeled peptides with distally-labeled oligonucleotides, as demonstrated with non-concentric QD-FRET probes;37–39 however, the detection of specific nucleic acid sequences (e.g. genes) is of greater interest. A practical requirement of this latter application is the detection of unlabeled targets. One possible strategy is a sandwich assay format, where target hybridization mediates assembly of a third, dye-labeled reporter oligonucleotide to the QD.40,41 This format necessarily places the dyes at a position equivalent to the distal terminus of the QD-conjugated probe oligonucleotides, and the long distance (more than 6 nm plus the radius of the QD for an 18 nt probe) will decrease FRET efficiency and the effectiveness of cFRET. The alternative approach, which we adopted here, was pre-hybridization of a dye-labeled oligonucleotide with the QD-conjugated oligonucleotide probe. The dye was placed proximal to the QD, thereby minimizing the distance between the QD and acceptor dyes, and maximizing the effectiveness of cFRET. Another advantage of a displacement format over a sandwich format is that the former functions as a single entity, and is thus more practical for future cellular sensing applications (a sandwich probe would require introduction of three components: the QD–probe conjugate and two separate reporter oligonucleotides). In practice, toehold-mediated strand displacement is a strategy for achieving rapid displacement of a shorter block oligonucleotide from a probe oligonucleotide by a full-length complementary target.31–33 Even when destabilized by mismatches, the displacement of a full-length block sequence from a probe oligonucleotide by fully complementary target is inefficient.42 Times for full hybridization and displacement were less than 30 min for our oligonucleotide-linked cFRET probes, and detection of between 1–2 equiv. of target per QD was possible. The detection limit will scale with the concentration of QDs used in an assay, which, in turn, will depend on the sensitivity of the instrumentation utilized. Given the brightness of QDs, nanomolar and sub-nanomolar detection limits should be readily achievable with most commercial instruments. cFRET probes are anticipated to be competitive with recently developed multiplexed gold nanoparticle probes that have two attached oligonucleotide probes, where multiplexed signaling arises from quenching of fluorescent dyes in displacement or molecular beacon formats.43,44 The added advantage of QD-based cFRET probes is ratiometric detection afforded by the QD PL signal.
Conclusions
We have developed a new long-wavelength QD605–A633–A680 cFRET configuration and demonstrated its analytical utility in a multiplexed hybridization assay. The configuration assembled red- and deep red-fluorescent A633 and A680 dyes around a central orange-emitting QD605 through either peptide or oligonucleotide linkers. Importantly, this new configuration showed that our previously reported prototypical QD520–A555–A647 cFRET configuration was not a special case, and that the cFRET strategy is generally applicable. Competitive and sequential energy transfer efficiencies were characterized and shown to be predictable through a rate analysis formalism calibrated to conventional QD–A633 and QD–A680 FRET configurations. This analysis revealed differences in the balance of competitive and sequential energy transfer rates for the new long-wavelength QD605–A633–A680 configuration versus the prototypical QD520–A555–A647 system, consistent with the different absorbance and fluorescence properties of the dyes. The new long-wavelength configuration was advantageous in that it was brighter than the prototypical configuration and also provided an order of magnitude better signal-to-background ratios in serum and blood. The latter was not only a function of the brightness of the QD605, but also the longer emission and excitation wavelengths of the new configuration. The main disadvantage for the QD605–A633–A680 configuration was its greater susceptibility to photobleaching than the prototypical configuration. In addition to the novel long-wavelength configuration, this work expanded the scope of cFRET by demonstrating that a cFRET configuration can be assembled through oligonucleotide linkers and hybridization, whereas previous cFRET configurations were assembled through peptide linkers. The long-wavelength cFRET configuration was then adapted to a toehold-mediated strand displacement assay format for multiplexed, quantitative detection of unlabeled nucleic acid targets. Overall, the long-wavelength cFRET configuration is very promising for future applications in bioanalysis and sensing.Acknowledgements
The authors acknowledge support for this research from the Natural Sciences and Engineering Research Council of Canada (NSERC), the Canada Foundation for Innovation (CFI), and the University of British Columbia. J.J.L. is grateful for support from NSERC through the CREATE NanoMat training program. W.R.A. is grateful for a Canada Research Chair (Tier 2) and a Michael Smith Foundation for Health Research Scholar Award.References
- U. Resch-Genger, M. Grabolle, S. Cavaliere-Jaricot, R. Nitschke and T. Nann, Nat. Methods, 2008, 5, 763–775 CrossRef CAS PubMed.
- E. Petryayeva, W. R. Algar and I. L. Medintz, Appl. Spectrosc., 2013, 67, 215–252 CrossRef CAS PubMed.
- I. L. Medintz, H. T. Uyeda, E. R. Goldman and H. Mattoussi, Nat. Mater., 2005, 4, 435–446 CrossRef CAS PubMed.
- H. Mattoussi, G. Palui and H. B. Na, Adv. Drug Delivery Rev., 2012, 64, 138–166 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- T. R. Pisanic, Y. Zhang and T. H. Wang, Analyst, 2014, 139, 2968–2981 RSC.
- J. B. Blanco-Canosa, M. Wu, K. Susumu, E. Petryayeva, T. L. Jennings, P. E. Dawson, W. R. Algar and I. L. Medintz, Coord. Chem. Rev., 2014, 263–264, 101–137 CrossRef CAS.
- W. R. Algar, H. Kim, I. L. Medintz and N. Hildebrandt, Coord. Chem. Rev., 2014, 263–264, 65–85 CrossRef CAS.
- W. R. Algar, A. J. Tavares and U. J. Krull, Anal. Chim. Acta, 2010, 673, 1–25 CrossRef CAS PubMed.
- I. L. Medintz and H. Mattoussi, Phys. Chem. Chem. Phys., 2009, 11, 17–45 RSC.
- K. Boeneman, D. E. Prasuhn, J. B. Blanco-Canosa, P. E. Dawson, J. S. Melinger, M. Ancona, M. H. Stewart, K. Susumu, A. Huston and I. L. Medintz, J. Am. Chem. Soc., 2010, 132, 18177–18190 CrossRef CAS PubMed.
- C. M. Spillmann, M. G. Ancona, S. Buckhout-White, W. R. Algar, M. H. Stewart, K. Susumu, A. L. Huston, E. R. Goldman and I. L. Medintz, ACS Nano, 2013, 7, 7101–7118 CrossRef CAS PubMed.
- C. L. Dwyer, S. A. Diaz, S. A. Walper, A. Samanta, K. Susumu, E. Oh, S. Buckhout-White and I. L. Medintz, Chem. Mater., 2015, 27, 6490–6494 CrossRef CAS.
- J. C. Claussen, N. Hildebrandt, K. Susumu, M. G. Ancona and I. L. Medintz, ACS Appl. Mater. Interfaces, 2014, 6, 3771–3778 CAS.
- J. C. Claussen, W. R. Algar, N. Hildebrandt, K. Susumu, M. G. Ancona and I. L. Medintz, Nanoscale, 2013, 5, 12156–12170 RSC.
- W. R. Algar, A. P. Malanoski, K. Susumu, M. H. Stewart, N. Hildebrandt and I. L. Medintz, Anal. Chem., 2012, 84, 10136–10146 CrossRef CAS PubMed.
- W. R. Algar, D. Wegner, A. L. Huston, J. B. Blanco-Canosa, M. H. Stewart, A. Armstrong, P. E. Dawson, N. Hildebrandt and I. L. Medintz, J. Am. Chem. Soc., 2012, 134, 1876–1891 CrossRef CAS PubMed.
- S. A. Diaz, L. Giordano, J. C. Azcarate, T. M. Jovin and E. A. Jares-Erijman, J. Am. Chem. Soc., 2013, 135, 3208–3217 CrossRef CAS PubMed.
- M. Wu and W. R. Algar, Anal. Chem., 2015, 87, 8078–8083 CrossRef CAS PubMed.
- M. Wu, E. Petryayeva and W. R. Algar, Anal. Chem., 2014, 86, 11181–11188 CrossRef CAS PubMed.
- H. Kim, C. Y. W. Ng and W. R. Algar, Langmuir, 2014, 30, 5676–5685 CrossRef CAS PubMed.
- W. R. Algar, M. G. Ancona, A. P. Malanoski, K. Susumu and I. L. Medintz, ACS Nano, 2012, 6, 11044–11058 CrossRef CAS PubMed.
- M. Wu, M. Massey, E. Petryayeva and W. R. Algar, J. Phys. Chem. C, 2015, 119, 26183–26195 CAS.
- J. J. Li, Y. A. Wang, W. Z. Guo, J. C. Keay, T. D. Mishima, M. B. Johnson and X. G. Peng, J. Am. Chem. Soc., 2003, 125, 12567–12575 CrossRef CAS PubMed.
- W. W. Yu and X. Peng, Angew. Chem., Int. Ed., 2002, 41, 2368–2371 CrossRef CAS.
- E. Petryayeva and W. R. Algar, Anal. Chem., 2014, 86, 3195–3202 CrossRef CAS PubMed.
- W. R. Algar, J. B. Blanco-Canosa, R. L. Manthe, K. Susumu, M. H. Stewart, P. E. Dawson and I. L. Medintz, Methods Mol. Biol., 2013, 1025, 47–73 CAS.
- K. E. Sapsford, T. Pons, I. L. Medintz, S. Higashiya, F. M. Brunel, P. E. Dawson and H. Mattoussi, J. Phys. Chem. C, 2007, 111, 11528–11538 CAS.
- F. Aldeek, M. Safi, N. Q. Zhan, G. Palui and H. Mattoussi, ACS Nano, 2013, 7, 10197–10210 CrossRef CAS PubMed.
- W. W. Yu and X. Peng, Chem. Mater., 2003, 15, 2854–2860 CrossRef CAS.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- N. Srinivas, T. E. Ouldridge, P. Sulc, J. M. Schaeffer, B. Yurke, A. A. Louis, J. P. K. Doye and E. Winfree, Nucleic Acids Res., 2013, 41, 10641–10658 CrossRef CAS PubMed.
- R. R. F. Machinek, T. E. Ouldridge, N. E. C. Haley, J. Bath and A. J. Turberfield, Nat. Commun., 2014, 5, 5324 CrossRef CAS PubMed.
- L. Berti, P. S. D'Agostino, K. Boeneman and I. L. Medintz, Nano Res., 2009, 2, 121–129 CrossRef CAS.
- I. L. Medintz, L. Berti, T. Pons, A. F. Grimes, D. S. English, A. Alessandrini, P. Facci and H. Mattoussi, Nano Lett., 2007, 7, 1741–1748 CrossRef CAS PubMed.
- S. J. Hurst, A. K. R. Lytton-Jean and C. A. Mirkin, Anal. Chem., 2006, 78, 8313–8318 CrossRef CAS PubMed.
- M. Suzuki, Y. Husimi, H. Komatsu, K. Suzuki and K. T. Douglas, J. Am. Chem. Soc., 2008, 130, 5720–5725 CrossRef CAS PubMed.
- S. Huang, Q. Xiao, Z. K. He, Y. Liu, P. Tinnefeld, X. R. Su and X. N. Peng, Chem. Commun., 2008, 5990–5992 RSC.
- R. Gill, I. Willner, I. Shweky and U. Banin, J. Phys. Chem. B, 2005, 109, 23715–23719 CrossRef CAS PubMed.
- C. Y. Zhang, H. C. Yeh, M. T. Kuroki and T. H. Wang, Nat. Mater., 2005, 4, 826–831 CrossRef CAS PubMed.
- W. R. Algar and U. J. Krull, Langmuir, 2010, 26, 6041–6047 CrossRef CAS PubMed.
- C. H. Vannoy, L. Chong, C. Le and U. J. Krull, Anal. Chim. Acta, 2013, 759, 92–99 CrossRef CAS PubMed.
- W. Pan, Y. Li, M. Wang, H. Yang, N. Li and B. Tang, Chem. Commun., 2016, 52, 4569–4572 RSC.
- A. E. Prigodich, P. S. Randeria, W. E. Briley, N. J. Kim, W. L. Daniel, D. A. Giljohann and C. A. Mirkin, Anal. Chem., 2012, 84, 2062–2066 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental methods including peptide and oligonucleotide labeling, linker synthesis and modification of oligonucleotides, GSH ligand exchange on QDs, assay methods, details of crosstalk corrections and data analysis, additional results including PL lifetimes, quantum yields, gel electrophoresis, full sets of PL spectra, additional photobleaching data and hybridization experiments. See DOI: 10.1039/c6an00492j |
| This journal is © The Royal Society of Chemistry 2016 |