
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceToward food analytics: fast estimation of lycopene and β-carotene content in tomatoes based on surface enhanced Raman spectroscopy (SERS)†
Andreea Ioana
Radu
ab,
Oleg
Ryabchykov
ab,
Thomas Wilhelm
Bocklitz
 ab,
Uwe
Huebner
b,
Karina
Weber
ab,
Dana
Cialla-May
*ab and
Jürgen
Popp
ab
ab,
Uwe
Huebner
b,
Karina
Weber
ab,
Dana
Cialla-May
*ab and
Jürgen
Popp
ab
aFriedrich Schiller University Jena, Institute of Physical Chemistry and Abbe Center of Photonics, Helmholtzweg 4, 07745 Jena, Germany
bLeibniz Institute of Photonic Technology Jena, Albert-Einstein-Str. 9, 07745 Jena, Germany. E-mail: dana.cialla-may@uni-jena.de
First published on 12th May 2016
Abstract
Carotenoids are molecules that play important roles in both plant development and in the well-being of mammalian organisms. Therefore, various studies have been performed to characterize carotenoids’ properties, distribution in nature and their health benefits upon ingestion. Nevertheless, there is a gap regarding a fast detection of them at the plant phase. Within this contribution we report the results obtained regarding the application of surface enhanced Raman spectroscopy (SERS) toward the differentiation of two carotenoid molecules (namely, lycopene and β-carotene) in tomato samples. To this end, an e-beam lithography (EBL) SERS-active substrate and a 488 nm excitation source were employed, and a relevant simulated matrix was prepared (by mixing the two carotenoids in defined percentages) and measured. Next, carotenoids were extracted from tomato plants and measured as well. Finally, a combination of principal component analysis and partial least squares regression (PCA-PLSR) was applied to process the data, and the obtained results were compared with HPLC measurements of the same extracts. A good agreement was obtained between the HPLC and the SERS results for most of the tomato samples.
Introduction
For the last several centuries, the scientific focus has been directed toward characterizing the functioning and necessities of the body. Moreover, currently, the role different molecules have, their pathway upon ingestion and their daily intake necessity is being documented. Among others, carotenoids have attracted much attention because of their large bioavailability1–4 and their important roles in the mammalian organism.5–10 Nevertheless, it has been found that only 50 out of more than 600 different known carotenoids1–4 are actually present in the human diet, and only 5–6 of them are detectable in human plasma (α- and β-carotene, β-cryptoxanthin, lycopene, lutein and zeaxanthin).5,11 The functions each of these carotenoids play in the body range from pro-vitamin A activity and antioxidant activity to radical scavenging. For instance, all-trans-β-carotene is the only carotenoid capable of oxidative cleavage into two all-trans-retinal molecules, and this process appears to have a feedback regulation property.2,8,11,12 That is, β-carotene absorption and conversion to retinol partially depends on the individual's vitamin A availability. Moreover, according to different published statistics the necessary intake of vitamin A based on dietary sources of animal origin (e.g., fatty fish, liver and eggs) is often not reached.9–11,13 Further on, out of the 6 carotenoids detectable in the human plasma, lycopene was found to have the highest efficiency as an antioxidant capable of neutralizing reactive oxygen species (ROS)5,14,15 and reducing both cell-division at the G0–G1 cell cycle phase and insulin-like growth of mitogens in various cancer cell lines.5,16,17 There are, however, also negative effects of excessive carotenoid uptake in combination with smoking and alcohol drinking.9,18 It is, accordingly, important from a health point of view to have a balanced dietary regime. Still, to achieve such a regime, information regarding the quality and composition of the food is needed.The golden standard in carotenoid analysis is high performance liquid chromatography (HPLC).19,20 The drawbacks of this method are high costs and limited specificity (because of co-elution).21 Thus, there is potential for alternative analytical methods. Among others, Raman spectroscopy and surface enhanced Raman spectroscopy (SERS) were tested for analyzing mixtures of carotenoids in various matrices.22–26 However, previous Raman studies have failed to obtain sufficient differentiation between the two carotenoids at lower concentrations, with HPLC coupled with UV-VIS or MS detection remaining the better option for reliable food analytics.27–29 Here, we present the first results of using SERS to differentiate between lycopene and β-carotene in tomatoes at different ripening stages. However, at this stage of the research, no advantage of the potential the method has towards being non-invasive was applied. Instead, a significant, but small amount of the full-food-batch production sample was taken apart, and an established extraction protocol was used to obtain analyte solutions that were measured by both SERS and HPLC. Regarding the SERS measurements, we designed a step-by-step experimental procedure that reaches towards developing a possible protocol for analyzing carotenoids from tomatoes by employing a relatively simple spectroscopic technique in combination with a statistical analytical tool. For this, the first step consisted of the characterization of the pure carotenoids. Next, different mixtures of β-carotene/lycopene solutions were prepared and measured to create a database and a statistical model that could be used for analysis of food extract samples. The available literature providing information about the amounts of the two carotenoids present in natural products was consulted to decide on the actual mixtures. Finally, a tomato extraction protocol was applied, and the tomato-extracts were measured and analyzed by applying the already existing statistical model. To verify the results of the proposed SERS approach, all tomato samples were also measured by HPLC.
Experimental
Chemicals and reagents
All reagents were of analytical or HPLC reagent grade. Lycopene (≥90% pure), β-carotene (≥95% pure) and 2,6-di-tert-butyl-4-methylphenol (BHT, ≥99% pure) were purchased from Sigma Aldrich (Steinheim, Germany). Methanol (≥99.5% pure) was purchased from Carl Roth (Karlsruhe, Germany). Tetrahydrofuran (THF, ≥99.9% pure) was purchased from Merck KGaA (Darmstadt, Germany).Cherry tomatoes at different ripening stages were provided by local producers from the area of Jena, Germany. First, a series of tomatoes (series A) exhibiting different degrees of ripeness (yellow to red) were taken from the same tomato plant. The tomatoes were immediately frozen and stored at −20 °C until analysis. A second series of four tomatoes (all of them exhibiting the same degree of ripeness – all yellow) were gathered from one plant (series B). One tomato from this batch was frozen immediately. The remaining tomatoes were illuminated with an 11 W lamp for various periods of time leading to increasing degrees of ripeness. After illumination the tomatoes were frozen and stored at −20 °C.
SERS active substrates
For the development of the SERS active substrates e-beam lithography combined with ion-beam etching were used according to the protocol described by Huebner et al.30,31 More exactly, a 4′′ fused silica wafer was cleaned using a peroxymonosulfuric acid solution, and then a thin undercoating (hexamethyldisilazane – HMDS) and a 260 nm thick positive tone electron beam resist ‘AR6200.09’ (ALLRESIST GmbH) were spun on the wafer. Further on, the resist was baked for 3 min at 150 °C on a hotplate, and a 10 nm gold layer was evaporated on top of the resist. The electron beam exposure, which was performed by using the unique character projection-based electron beam technique,31 of the shaped beam writer SB350OS (from Vistec Electron Beam GmbH) resulted in the formation of 48 chips per wafer (5 × 10 mm2). Each of the obtained chips contains 4 gratings with a size of 1 × 1 mm2 for the SERS investigations. The exposure and the removing of the gold layer were followed by the development of the resist in an AR 600-546 developer for 60 s and the IPA rinsing for 30 s. Next, the etching into the fused silica surface was performed with a CHF3–SF6–ICP etching process (Inductively Coupled Plasma – ICP) by using an ICP power of 300 W. The etch depth of the 2D gratings with a period of 436 nm is approximately 100 nm. Last, the residual resist was removed using an oxygen plasma, and the wafer was separated into single chips.Silver films were deposited freshly (at the beginning of every measurement day) by means of thermal evaporation at an oil-free background pressure in the lower 10−7 mbar range. For this, the chips were mounted line of sight to the evaporation boat to let the vapor strike the substrate normal to the surface. High-purity 99.999% silver granules were used as raw material. The thickness as well as the deposition rate was controlled in situ using a quartz microbalance. The thickness of the silver layer was 40 nm. A scanning electron microscopy (SEM) image of the measuring fields used throughout the experiments is presented in Fig. 1. The image was obtained using a JEOL JSM-6700F system.
 | ||
| Fig. 1 SEM image of the measurement field of the SERS active substrate (grating pitch: 436 nm): a quartz-grating as the template without a silver film (A) and the quartz-grating covered with 40 nm silver as a SERS-substrate (B). | ||
Sample preparation
For all of the experiments discussed in this study a mixed solvent of methanol and THF stabilized with 0.1% BHT (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v) was used.
:1, v/v) was used.
For the concentration dependent SERS measurements of the two analytes, stock solutions of 106 μM β-carotene and lycopene were prepared by dissolving the appropriate analyte quantities in MeOH/THF. Measuring solutions were prepared by dilution in MeOH/THF immediately prior to use. Final concentrations of 106, 90, 74, 58, 42, 26, 10, 9, 7.4, 5.8, 4.2, 2.6 and 1 μM were obtained for both analytes. All named solutions were used for the SERS measurements shortly after preparation. For each measurement, a new SERS substrate was used.
For the SERS measurements of the β-carotene/lycopene mixtures two stock solutions of 100 μM of each analyte were prepared, and the two analytes were mixed to obtain the analyte percentages shown in Table S1.† Subsequently, the resulting mixtures were measured. However, recording the full data set took a couple of hours, and the mixtures were stored at −20 °C for the needed time. For each mixture a different substrate was used.
The food samples were homogenized to obtain a puree. 2–5 g of each pure sample were mixed with 30 ml methanol/THF of a solution (1:1, v/v) and 200 mg magnesium bicarbonate. The resulting mixture was stirred using an ultra turrax and filtered with a Buchner funnel. The procedure was repeated two times. The combined filtrates were evaporated to dryness and then dissolved in a defined volume of the extracting agent. The resulting sample was measured by both SERS and HPLC. For the SERS measurements a new substrate was used for each extract.
Spectroscopic measurements
The extinction spectra of the analytes were recorded using a Jasco V650 diode-array spectrophotometer.For the SERS measurements, the substrates were incubated in the analyte solutions for 30 min and then dried in an Ar stream, which was chosen due to the instability of the carotenoid molecules under normal lab conditions. SERS spectra were recorded using a commercially available WITec confocal Raman system (WITec alpha 300 SR, WITec GmbH, Ulm, Germany) equipped with a 488 nm laser. The light was focused onto the sample via a 100× objective (NA 0.9), and the Raman scattered light was collected with the same microscope objective. An optical grating of 1800 g mm−1 was used resulting in a spectral resolution of ∼2 cm−1. Scans consisting of 100 point measurements were recorded with an integration time of 0.5 s per point. The power at the surface of the sample was adjusted to 20 μW. For each measured analyte (standard solution or sample extract) 13, scans were recorded.
Data analysis
All of the presented spectra were analyzed using R (version 3.0.2)32 and plotted using Origin 8.5. For data analysis, spectra were first averaged over a 50-point measurement. The resulting spectra were wavenumber calibrated, cut to the relevant spectral range of 500–1700 cm−1, background corrected using the sensitive nonlinear iterative peak (SNIP) algorithm,33 spike corrected and, for the analysis of the mixtures and food extracts, normalized for the whole spectral range. For the statistical analysis, a principal component analysis (PCA)34,35 (using a different number of components) was performed and followed by a partial least squares regression (PLSR)34,35 (using a different number of components) analysis. Two types of cross-validation were performed. First, to build up the training data set, all values representing one concentration were removed, which was repeated for all of the applied concentrations (further referred to as M1). Second, 1% of the total number of measurements was randomly taken out for training (further referred to as M2). The optimal number of principal components for PCA and PLS was chosen, and a model was built using all of the measured data. The obtained model was applied to the test data to predict the food composition. A PCA was applied prior to the PLS regression because performing a regression of high-dimensional data within each repetition of the cross-validation loop would dramatically increase the processing time. On the other side, PCA is an unsupervised method that can be used for the dimensionality reduction of the data outside of the cross-validation loop. Consequently, the PLS regression was performed for low-dimensional data, so the time required for the model construction and evaluation was significantly decreased.Limit of detection (LOD) values were defined according to the IUPAC norms and are equal to the signal of the blank plus three times the standard deviation of the blank.
HPLC measurements
The HPLC system consisted of a Shimadzu binary gradient system with a DGU-20A3R degassing unit, SIL-20AC auto-sampler, CTO-20AC column oven and SPD-20A UV/VIS detector. The injection volume was 50 μl, and the separation was performed on a 250 × 4.6 mm S-5 μm YMC 30 HPLC column. The mobile phase consisted of methanol (solvent A) and tertiary butyl methyl ether (tBME, solvent B). The total flow was 1.3 ml min−1, and the column temperature was adjusted to 29 °C. The gradient started with 90% solvent A and 10% solvent B. A linear gradient was applied up to 55% solvent A and 45% solvent B (45 min) followed by another linear gradient up to 45% solvent A and 55% solvent B (5 min). This ratio was held constant for 5 min before returning to the starting conditions (90% solvent A) within 2 min.The peaks were evaluated at 450 nm and 470 nm. The quantification was performed by an external calibration with standard solutions taking into account the internal standard. The limit of quantification (LOQ) determined by the signal to noise ratio was 0.03 μg ml−1.
Results and discussion
Plants naturally produce carotenoids to color their flowers and fruits, to attract animals, to gather the light needed for photosynthesis and to protect chlorophyll from photo-damage.1 According to the available literature1,2,36,37 lycopene and β-carotene are two carotene molecules (out of 600 known) that can be found in the same plants, in different ratios, depending on the maturity/age of the plant. As depicted in Fig. S1† and largely presented in the literature,1,2,23,36 in the plant biosynthesis, lycopene is first formed, and upon cyclization, it converts to either β-carotene or α-carotene, which further undergo conversions to other carotenoid molecules. Accordingly, at the different stages of fruit ripening the amount of lycopene and β-carotene also differs. Considering this and keeping in mind the different roles the two carotenoids have in the mammalian organism, it is important to be able to differentiate which plants contain high amounts of each of them. On the other hand, lycopene and β-carotene are very similar from a chemical and spectroscopic point of view (Fig. 2), making the differentiation rather difficult. Analyzing the extinction profile of the analytes (see Fig. 2A), a gain from the resonance contribution by using a 488 nm laser as an excitation source is expected. In a further step, SERRS measurements of the two analyte solutions having a concentration of 106 μM were performed, and the obtained data are shown in Fig. 2B. By analyzing these spectra, a number of differences in the two analytes concerning band intensities and band positions are identified. The ratio of the bands centered at 1526 and 1155 cm−1 and assigned to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C in-phase stretching and C–C stretching vibrations of the polyene chain of the two molecules change when comparing the case of β-carotene with that of lycopene.38,39 Further on, in the spectral range of 1230–1330 cm−1, two different small bands can be observed. That is, the band centered at 1270 cm−1 and assigned to the C–H rocking vibration (also belonging to the polyene chain), and the one at 1287 cm−1 assigned to the ring methylene twist.38 The ratio of these two also changes for the different molecules. Additionally, a 5 cm−1 shift of the band centered at approximately 1190 cm−1 (and assigned to the C–C stretching vibration) from one molecule to the other can be observed.38
C in-phase stretching and C–C stretching vibrations of the polyene chain of the two molecules change when comparing the case of β-carotene with that of lycopene.38,39 Further on, in the spectral range of 1230–1330 cm−1, two different small bands can be observed. That is, the band centered at 1270 cm−1 and assigned to the C–H rocking vibration (also belonging to the polyene chain), and the one at 1287 cm−1 assigned to the ring methylene twist.38 The ratio of these two also changes for the different molecules. Additionally, a 5 cm−1 shift of the band centered at approximately 1190 cm−1 (and assigned to the C–C stretching vibration) from one molecule to the other can be observed.38
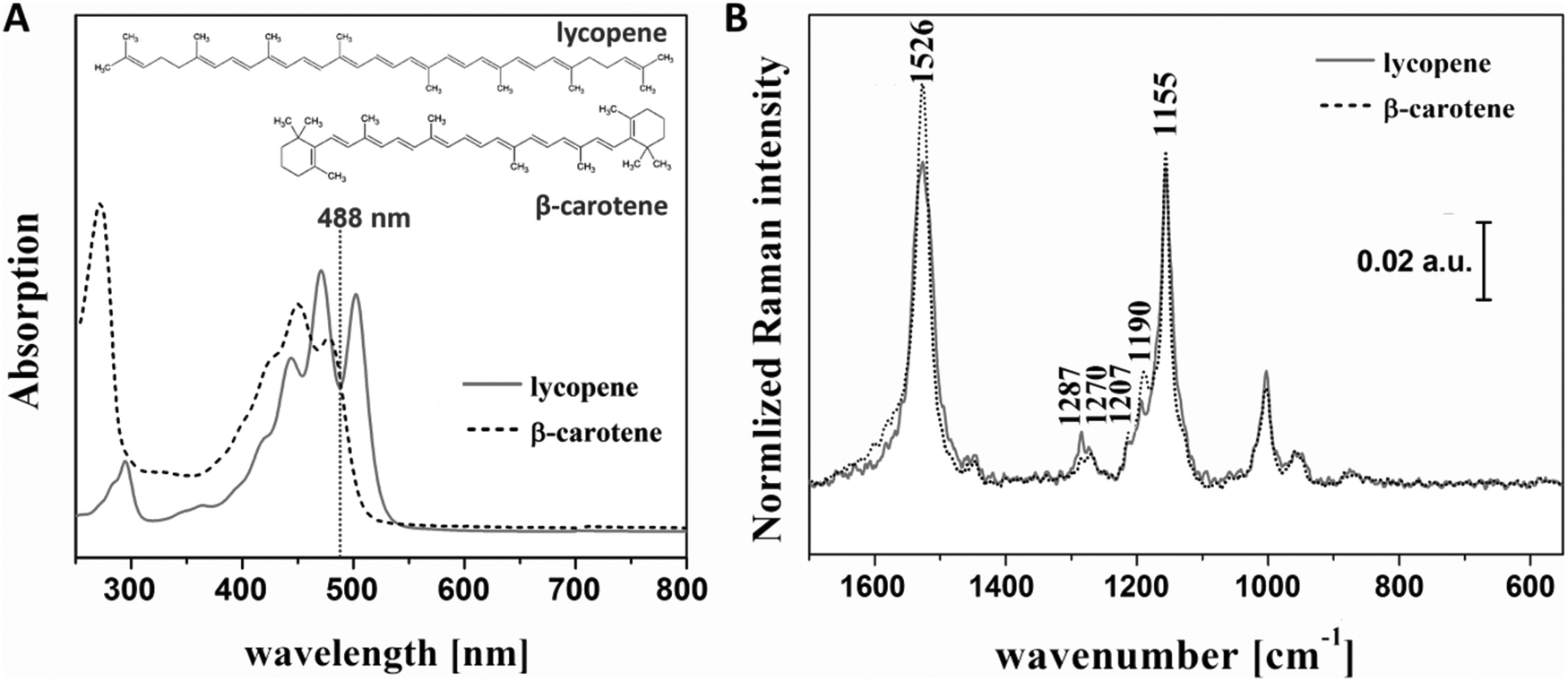 | ||
| Fig. 2 Chemical structure and extinction spectra of lycopene, 18.7 nM and β-carotene, 6 μM (A) and SERRS spectra of the two analytes, 100 μM (B). | ||
As already mentioned, this study is directed towards the detection of β-carotene and lycopene out of a food matrix. To do so, different experimental steps were designed and performed. First, different concentrations of the independent pure analytes were measured before mixing them in different ratios (see Table S1† for the exact percentages). Upon performing these measurements and the analysis, a calibration curve was generated and used for estimating the presence of the two analytes in the studied tomatoes. The results were then compared with the current gold standard, HPLC.
SERRS spectra of different concentrations of the analytes ranging from 106 μM to 1 μM were measured to establish an understanding of the technique's sensitivity. As observed from the plots in Fig. S2,† detection down to a concentration of 10 μM and 26 μM were achieved for lycopene and β-carotene, respectively. Keeping this in mind and considering the already discussed plant-carotenoid transformation path (see Fig. S1†), different lycopene/β-carotene mixtures were prepared. Information regarding the individual percentages of these two analytes in each solution and the different individual concentrations are included in Table S1,† while Fig. 3 depicts the obtained SERRS spectra. As already mentioned, a change in the ratio of the bands centered at 1270 and 1287 cm−1 is observed in the case of the SERRS spectra of the pure analytes (Fig. 2B). The same observation is also valid for the spectra in Fig. 3, where a gradual change of the two bands’ intensities occurs with a variation in the amount of the two analytes in the solution. Additionally, a change in the shapes of the bands centered at 1190 and 1207 cm−1 as the solution composition changes is observed. That is, the band centered at 1190 cm−1 becomes more defined with increasing in β-carotene fractions. To better comprehend and visualize these features and to show the potential of the SERRS technique in food analytics, the SERRS data were further analyzed by applying statistical methods. As mentioned, this consisted of a PCA-PLSR analysis considering the optimal number of components, selected from two different cross validation procedures (see Data analysis section of Experimental for further information). Nevertheless, before applying the PCA-PLSR analysis, each group of 50 spectra were averaged, resulting in a total number of 30 spectra per mixture that were further used for the statistical analysis. This step was performed to compensate for the widely discussed SERRS drawbacks regarding the chemical binding of the analyte to the substrate and the reproducibility of the larger scale SERRS measurements.40,41 Additionally, when analyzing this result, one should consider that β-carotene percentages lower than 26% are lower than the lowest detectable analyte concentration achieved for measuring the pure analyte. The same is valid for percentages of lycopene lower than 10%. This was expected to negatively influence the root mean square error (RMSE)34 value of the obtained regression results. The RMSE values obtained for the different considered PCA-PLS component numbers are depicted in Fig. 4. When analyzing this data, one realizes that by using more than 4 PCA components and 2 PLS components a saturation of the RMSE for both cross-validation approaches is achieved. Accordingly, to avoid overfitting, the chosen PCA-PLS combination for further analysis was limited to 4 PCA and 2 PLS components, having a RMSE value of 11.7%. The different cross-validated regression results are depicted in Fig. 5. The expected accuracy of the proposed SERRS method in predicting the concentrations of the two investigated carotenoids in food samples is also around the same level. To assess the potential of our SERS approach as well as its limitations by employing real food samples, cherry tomatoes in different ripening stages were investigated.
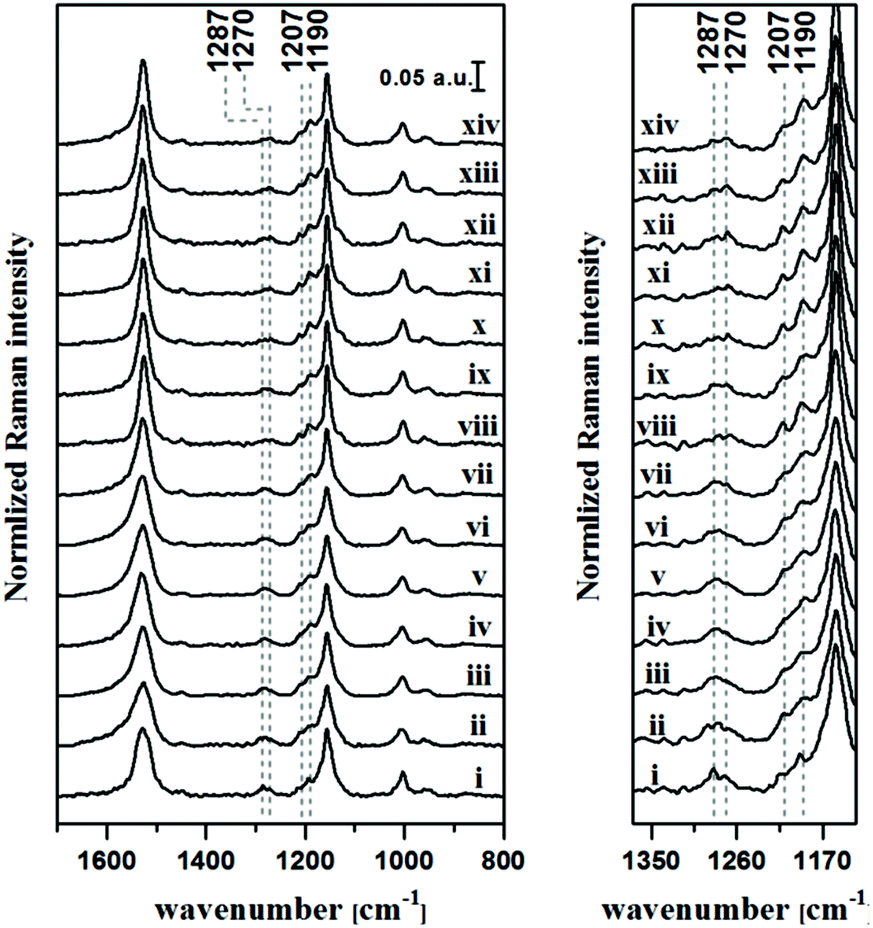 | ||
| Fig. 3 SERRS spectra of the β-carotene/lycopene mixtures. The spectra are arranged based on the variation of the two analyte percentages: (i) 0% β-carotene and 100% lycopene (0% βc and 100% lyc), (ii) 8% βc and 92% lyc, (iii) 16% βc and 84% lyc, (iv) 24% βc and 76% lyc, (v) 32% βc and 68% lyc, (vi) 40% βc and 60% lyc, (vii) 48% βc and 52% lyc, (viii) 56% βc and 44% lyc, (ix) 64% βc and 36% lyc, (x) 72% βc and 28% lyc, (xi) 80% βc and 20% lyc, (xii) 88% βc and 12% lyc, (xiii) 96% βc and 4% lyc, and (xiv) 100% βc and 0% lyc. The graph on the right side depicts the spectral range between 1330 and 1160 cm−1 for better visualization. | ||
 | ||
| Fig. 4 RMSE values for various numbers of components used for PCA and for PLS. | ||
 | ||
| Fig. 5 Cross-validation analysis results obtained for the case of the 4-component PCA and 2-component PLS analysis. | ||
To do so, the data already presented was used in an attempt to analyze two different series of cherry tomatoes differing in their degree of ripeness. Tomatoes in the A series were picked from the tomato plant upon reaching the different ripening stages, and tomatoes from the B series were picked from the tomato plant at the same early ripening stage (yellow). In the case of the B series, the different investigated ripening stages were achieved in simulated lab-ripening conditions consisting of illuminating the vegetable with an 11 W lamp for the needed period of time. Upon reaching a different ripening stage under the lab illumination conditions, one tomato out of the batch, corresponding to the new ripening stage, was frozen and stored at −20 °C until the extraction and measurement were performed. The analysis protocol depicted in Fig. 6 consists of 4 easy steps. First, an HPLC extraction protocol (described in the Sample preparation section) was performed individually for each of the tomatoes studied (i) to obtain the solutions (ii) used for incubating the SERRS substrates for 30 min (iii). Upon incubation, the substrates were dried using Ar (iv) and measured by applying a 488 nm excitation source. The obtained SERRS spectra are depicted in Fig. 7 together with pictures of the 4 different ripening stages considered throughout this study. As observed, the spectra are similar to the ones already discussed (see Fig. 2B). Nevertheless, there are a few differences in the spectral range of 1100–1300 cm−1. First, a difference in the intensity of the band centered at 1190 cm−1 can be observed in both Fig. 7A and B as the color of the tomato changes from yellow (spectra A1 and B1) to red (spectra A4 and B4). The band intensity is higher in the case of the yellow tomato and comparably lower for the red tomatoes. By comparing this alone to the spectra in Fig. 2B one would expect that the red tomato has higher lycopene content than the yellow one. Further on, the shoulder that can be identified at approximately 1207 cm−1 in the spectra of the pure analytes (and that is assigned to the C–C stretching vibration)39 develops into a well-defined band in the case of the tomato samples. This can also be observed in the case of the mixed carotenoid spectra (Fig. 3) and could be a result of the interaction of the different carotenoid molecules via the polyene chain. Additionally, the band centered at 1526 cm−1 in the case of the pure analytes (Fig. 2B) is blue shifted in the case of the tomato extracts by 6 cm−1 for the yellow tomatoes and 9 cm−1 for the red tomatoes (Fig. 7A and B). All of the named spectral changes can be caused by the interaction of the carotenoid molecules among themselves and with the SERRS active substrate. However, one should keep in mind that in the case of the tomato extracts the analyzed matrix has a higher degree of complexity than the one used for creating the analytical model. More exactly, other carotenoids, such as phytoene, phytofluene, ζ-carotene, γ-carotene and neurosporene, can also be found in the tomato fruit matrix and could have spectral contributions.42 Nevertheless, according to the literature, the predominant molecule in the tomato fruit is lycopene followed by β-carotene.26,42
 | ||
| Fig. 6 Schematic representation of the analysis chain. As depicted, the first step consists of the preparation of the analytes to be measured. To this end, the described extraction protocol was applied (i) and the resulting solutions (ii) were used for incubating the SERRS active substrate (iii). Upon incubation the substrates were dyed with N2 (iv) and measured by means of SERRS. | ||
 | ||
| Fig. 7 SERRS spectra of the garden-ripening tomato batch (A) and the lab-ripening tomato batch (B), as well as sample pictures of the colors the tomatoes had when analyzed. | ||
The preprocessing of the tomato SERRS spectra was performed by following the same steps performed in the case of the studied pure analyte. The value obtained for the RMSE is approximately 18.9%. The results (in the form of PLS scores obtained by applying the PCA-PLS regression analysis) are presented in Table 1 together with the HPLC measurements’ results. For the latter, the exact same extract was measured as in the case of the SERRS experiments. As observed from the table, a quite good agreement was obtained for the two analytical methods. In the case of the samples B1 and B2, however, the lycopene and β-carotene content predicted by SERRS and measured by HPLC presented different levels of the two carotenoids. This might be due to miss-assignments of other carotenoids present in the extract (i.e., phytoene, phytofluene, ζ-carotene, γ-carotene and neurosporene) by SERRS, as at this ripening stage their presence in the plant is expected. An improved prediction of the tomato composition is expected when employing all of the mentioned carotenoids for building the model. This prediction is, however, beyond the aim of this study. A further observation that can be made by analyzing Table 1 is related to the variation in the lycopene composition in an adult tomato fruit. Upon reaching the red ripening stage, further storage of the tomato before consumption leads to a decrease of the lycopene content in favor of other carotenoids. This is important when deciding on a dietary regime building towards a health-improving result. A last, interesting observation, first noted during experimentation, confirms that a tomato's lycopene content still increases when the fruit ripening is achieved in lab/shop conditions. This is important regarding the transportation time needed from the actual plantations to the commercializing facilities.
| SERRS resultsa | HPLC results | ||||||
|---|---|---|---|---|---|---|---|
| lyc | βc | lyc | βc | lycb | βcb | ||
| % | % | μM | μM | % | % | ||
| lyc – lycopene. βc – β-carotene.a PLS score value.b The % calculation was performed by considering that lycopene and β-carotene are the only two carotenoids present in the extract. | |||||||
| Plant-ripening | A1 | 41.5 ± 12.9 | 58.5 ± 12.9 | 10.7 ± 1.1 | 7.8 ± 0.8 | 57.9 | 42.1 |
| A2 | 70.1 ± 6.4 | 29.9 ± 6.4 | 30.0 ± 3.0 | 10.6 ± 1.1 | 74.0 | 26.0 | |
| A3 | 67.2 ± 9.4 | 32.8 ± 9.4 | 51.5 ± 5.1 | 13.4 ± 1.3 | 79.3 | 20.7 | |
| A4 | 59.6 ± 15.6 | 40.4 ± 15.6 | 20.8 ± 2.1 | 12.0 ± 1.2 | 63.4 | 36.6 | |
| Lab-ripening | B1 | 11.3 ± 10.4 | 88.7 ± 10.4 | 13.1 ± 1.3 | 10.7 ± 1.1 | 55.1 | 44.9 |
| B2 | 48.1 ± 9.0 | 51.9 ± 9.0 | 27.9 ± 2.8 | 13.2 ± 1.3 | 67.9 | 32.1 | |
| B3 | 86.6 ± 5.7 | 13.4 ± 5.7 | 99.7 ± 9.9 | 14.6 ± 1.5 | 87.2 | 12.8 | |
| B4 | 63.9 ± 4.8 | 36.1 ± 4.8 | 54.8 ± 5.5 | 18.0 ± 1.8 | 75.3 | 24.7 | |
Conclusions
The current paper presents the work performed toward analyzing cherry tomato fruits by means of SERRS. To this end, a rather simple but relevant simulated matrix was prepared. This matrix consisted of different mixtures of the two most prevalent carotenoids found in tomatoes, namely, β-carotene and lycopene. The percentages of the two carotenoids were varied to simulate possible compositions in vegetables, such as tomatoes. Upon statistical analysis, a regression curve was obtained and used to analyze the tomato samples. Further on, two tomato series representative of garden-ripening and laboratory-ripening conditions were considered to test the developed analytical method and to comparatively access the lycopene/β-carotene abundance of market-available tomatoes. Accordingly, upon acquiring the needed tomatoes for the designed experiments, they were subjected to the same carotenoid-extraction protocol and measured by both SERRS and HPLC. The SERRS measurements were performed under the same conditions as the ones employed for the lycopene/β-carotene mixtures. Upon analyzing the data, we were able to estimate the abundance of the two carotenoids investigated in the tomato samples. Moreover, a good agreement was obtained between the HPLC and the SERRS results for most of the tomato samples. Additionally, both measurement methods registered a gradual increase of the lycopene content independent of the tomato ripening conditions investigated.Acknowledgements
The funding of research projects “QuantiSERS” (03IPT513A) and “Jenaer Biochip Initiative 2.0” (03IPT513Y) within the framework of “InnoProfile Transfer – Unternehmen Region” of the Federal Ministry of Education and Research, Germany (BMBF) is gratefully acknowledged. Tobias Voigt, Dr Bianca Braha and Dr Bernd Giese from Food GmbH (Jena, Germany) are gratefully acknowledged for their expertise concerning carotenoid extraction from food and for providing the HPLC measurements. Sabine Schmidt and Torsten May are gratefully acknowledged for providing the different cherry tomato patches used throughout the experiments.References
- J. Hirschberg, Curr. Opin. Plant Biol., 2001, 4, 210–218 CrossRef CAS PubMed.
- E. H. Harrison, C. dela Sena, A. Eroglu and M. K. Fleshman, Am. J. Clin. Nutr., 2012, 96, 1189S–1192S CrossRef CAS PubMed.
- R.-X. Yu, W. Köcher, M. E. Darvin, M. Büttner, S. Jung, B. N. Lee, C. Klotter, K. Hurrelmann, M. C. Meinke and J. Lademann, J. Biophotonics, 2014, 7, 926–937 CrossRef PubMed.
- M. E. Darvin, H. Richter, S. Ahlberg, S. F. Haag, M. C. Meinke, D. Le Quintrec, O. Doucet and J. Lademann, J. Biophotonics, 2014, 7, 735–743 CrossRef CAS PubMed.
- A. Gajowik and M. M. Dobrzyńska, Rocz. Panstw. Zakl. Hig., 2014, 65, 263–271 CAS.
- M. Dizdaroglu, Free Radicals Biol. Med., 1991, 10, 225–242 CrossRef CAS PubMed.
- J. E. Klaunig and L. M. Kamendulis, Annu. Rev. Pharmacol. Toxicol., 2004, 44, 239–267 CrossRef CAS PubMed.
- J. D. Ribaya-Mercado, F. S. Solon, M. A. Solon, M. A. Cabal-Barza, C. S. Perfecto, G. Tang, J. A. A. Solon, C. R. Fjeld and R. M. Russell, Am. J. Clin. Nutr., 2000, 72, 455–465 CAS.
- M. Jenab, S. Salvini, C. H. van Gils, M. Brustad, S. Shakya-Shrestha, B. Buijsse, H. Verhagen, M. Touvier, C. Biessy, P. Wallstrom, K. Bouckaert, E. Lund, M. Waaseth, N. Roswall, A. M. Joensen, J. Linseisen, H. Boeing, E. Vasilopoulou, V. Dilis, S. Sieri, C. Sacerdote, P. Ferrari, J. Manjer, S. Nilsson, A. A. Welch, R. Travis, M. C. Boutron-Ruault, M. Niravong, H. B. Bueno-de-Mesquita, Y. T. van der Schouw, M. J. Tormo, A. Barricarte, E. Riboli, S. Bingham and N. Slimani, Eur. J. Clin. Nutr., 0000, 63, S150–S178 CrossRef PubMed.
- A. Agudo, N. Slimani, M. C. Ocké, A. Naska, A. B. Miller, A. Kroke, C. Bamia, D. Karalis, P. Vineis, D. Palli, H. B. Bueno-de-Mesquita, P. H. M. Peeters, D. Engeset, A. Hjartåker, C. Navarro, C. M. Garcia, P. Wallström, J. X. Zhang, A. A. Welch, E. Spencer, C. Stripp, K. Overvad, F. Clavel-Chapelon, C. Casagrande and E. Riboli, Public Health Nutr., 2002, 5, 1179–1196 CrossRef CAS PubMed.
- T. Grune, G. Lietz, A. Palou, A. C. Ross, W. Stahl, G. Tang, D. Thurnham, S.-A. Yin and H. K. Biesalski, J. Nutr., 2010, 140, 2268S–2285S CrossRef CAS PubMed.
- H. Bachmann, A. Desbarats, P. Pattison, M. Sedgewick, G. Riss, A. Wyss, N. Cardinault, C. Duszka, R. Goralczyk and P. Grolier, J. Nutr., 2002, 132, 3616–3622 CAS.
- D. R. Tennant, J. Davidson and A. J. Day, Br. J. Nutr., 2014, 112, 1214–1225 CrossRef CAS PubMed.
- P. Di Mascio, S. Kaiser and H. Sies, Arch. Biochem. Biophys., 1989, 274, 532–538 CrossRef CAS PubMed.
- L. G. Rao, E. Guns and A. V. Rao, Agric. Food Industry Hi-Tech, 2003, 14, 25–30 CAS.
- J. Levy, E. Bosin, B. Feldman, Y. Giat, A. Miinster, M. Danilenko and Y. Sharoni, Nutr. Cancer, 1995, 24, 257–266 CrossRef CAS PubMed.
- R. Matsushima-Nishiwaki, Y. Shidoji, S. Nishiwaki, T. Yamada, H. Moriwaki and Y. Muto, Lipids, 1995, 30, 1029–1034 CrossRef CAS PubMed.
- T. Tanaka, M. Shnimizu and H. Moriwaki, Molecules, 2012, 17, 3202 CrossRef CAS PubMed.
- S. F. P. R. H. C. P. P. S. D. K. Lloyd, in Liquid Chromatography Applications, Elsevier, Amsterdam, 2013, p. 667, DOI:10.1016/B978-0-12-415806-1.01001-9.
- Trends Food Sci. Technol., 2003, 1410, 438.
- M. W. Dong, LCGC North Am., 2013, 31(6), 472–479 CAS.
- A. M. Nikbakht, T. T. Hashjin, R. Malekfar and B. Gobadian, J. Agric. Sci. Technol., 2011, 13, 517–526 CAS.
- R. Baranski, M. Baranska and H. Schulz, Planta, 2005, 222, 448–457 CrossRef CAS PubMed.
- M. Køcks, S. O. Banke, B. Madsen, T. Vaz, M. Carvalheira, N. Pandega, I. Sousa and S. D. Nygaard, Appl. Spectrosc., 2013, 67, 681–687 CrossRef PubMed.
- K. Hesterberg, S. Schanzer, A. Patzelt, W. Sterry, J. W. Fluhr, M. C. Meinke, J. Lademann and M. E. Darvin, J. Biophotonics, 2012, 5, 33–39 CrossRef CAS PubMed.
- J. Qin, K. Chao and M. S. Kim, Postharvest Biol. Technol., 2012, 71, 21–31 CrossRef CAS.
- J. Trebolazabala, M. Maguregui, H. Morillas, A. de Diego and J. M. Madariaga, Spectrochim. Acta, Part A, 2013, 105, 391–399 CrossRef CAS PubMed.
- M. Baranska, W. Schütze and H. Schulz, Anal. Chem., 2006, 78, 8456–8461 CrossRef CAS PubMed.
- W. Liu, Z. Wang, Z. Zheng, L. Jiang, Y. Yang, L. Zhao and W. Su, Chin. J. Chem., 2012, 30, 2573–2580 CrossRef CAS.
- U. Huebner, K. Weber, D. Cialla, H. Schneidewind, M. Zeisberger, H. G. Meyer and J. Popp, Microelectron. Eng., 2011, 88, 1761–1763 CrossRef CAS.
- U. Huebner, M. Falkner, U. D. Zeitner, M. Banasch, K. Dietrich and E.-B. Kley, 30th European Mask and Lithography Conference, Proc. SPIE, 2014, 9231, 92310E Search PubMed.
- R. C. Team, R: A language and environment for statistical computing, R Foundation for Statistical Computing, Vienna, Austria, 2014 Search PubMed.
- M. Omer, H. Negm, R. Kinjo, Y.-W. Choi, K. Yoshida, T. Konstantin, M. Shibata, K. Shimahashi, H. Imon, H. Zen, T. Hori, T. Kii, K. Masuda and H. Ohgaki, in Zero-Carbon Energy Kyoto 2012, ed. T. Yao, Springer, Japan, 2013, ch. 27, pp. 245–252. DOI:10.1007/978-4-431-54264-3_27.
- R. Wehrens, Chemometrics with R. Multivariate data analysis in the natural sciences and life Sciences, 2011, DOI:10.1007/978-3-642-17841-2.
- T. Bocklitz, A. Walter, K. Hartmann, P. Rösch and J. Popp, Anal. Chim. Acta, 2011, 704, 47–56 CrossRef CAS PubMed.
- L. Arab, S. Steck-Scott and P. Bowen, Participation of lycopene and beta-carotene in carcinogenesis: defenders, aggressors, or passive bystanders?, 2001 Search PubMed.
- S. Schlücker, A. Szeghalmi, M. Schmitt, J. Popp and W. Kiefer, J. Raman Spectrosc., 2003, 34, 413–419 CrossRef.
- N. Tschirner, M. Schenderlein, K. Brose, E. Schlodder, M. A. Mroginski, C. Thomsen and P. Hildebrandt, Phys. Chem. Chem. Phys., 2009, 11, 11471–11478 RSC.
- M. R. López-Ramírez, S. Sanchez-Cortes, M. Pérez-Méndez and G. Blanch, J. Raman Spectrosc., 2010, 41, 1170–1177 CrossRef.
- C. Wang, C. J. Berg, C.-C. Hsu, B. A. Merrill and M. J. Tauber, J. Phys. Chem. B, 2012, 116, 10617–10630 CrossRef CAS PubMed.
- V. R. Salares, N. M. Young, P. R. Carey and H. J. Bernstein, J. Raman Spectrosc., 1977, 6, 282–288 CrossRef CAS.
- F. Khachik, L. Carvalho, P. S. Bernstein, G. J. Muir, D.-Y. Zhao and N. B. Katz, Exp. Biol. Med., 2002, 227, 845–851 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00390g |
| This journal is © The Royal Society of Chemistry 2016 |