
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advancements in ion concentration polarization
Min
Li
and
Robbyn. K.
Anand
*
Department of Chemistry, Iowa State University, 1605 Gilman Hall, Ames, IA 50011-3111, USA. E-mail: rkanand@iastate.edu
First published on 4th March 2016
Abstract
In this minireview, we discuss advancements in ion concentration polarization (ICP)-based preconcentration, separation, desalination, and dielectrophoresis that have been made over the past three years. ICP as a means of controlling the distribution of the ions and electric field in a microfluidic device has rapidly expanded its areas of application. Recent advancements have focused on the development of ion-permselective materials with tunable dimensions and surface chemistry, adaptation to paper microfluidics, higher-throughput device geometries, and coupling ICP with other separation (isotachophoresis and dielectrophoresis) and fluidic (valve and droplet microfluidic) strategies. These studies have made great strides toward solving real-world problems such as low-cost and rapid analysis, accessible desalination technology, and single-cell research tools.
 Min Li | Min Li completed her BSc in Chemistry at China Agricultural University in 2012 and is currently in the third year of her Ph.D. at Iowa State University. Her research focuses on high-throughput cell sorting by dielectrophoresis (DEP), increasing the capability of faradaic ion concentration polarization (FICP), and the application of sheath-flow coulter counting for particle size sorting. |
 Robbyn K. Anand | Robbyn K. Anand joined the chemistry faculty in August 2015 as an Assistant Professor. She earned her Ph.D. in 2010 from the University of Texas at Austin under the guidance of Prof. Richard M. Crooks with the support of an NSF Graduate Research Fellowship. She developed microfluidic devices employing bipolar electrodes for electrokinetic focusing of charged species and membrane-free seawater desalination. Then, as an NIH Postdoctoral Fellow, she worked with Prof. Daniel T. Chiu at the University of Washington on the capture and analysis of circulating tumor cells. She pioneered a technique for dielectrophoretic manipulation of biological cells using extended electric field gradients. |
1 Introduction
Ion concentration polarization (ICP) is an electrokinetic phenomenon brought about by selective charge transport, such as occurs in a nanofluidic channel that links two microfluidic compartments (Scheme 1a). This asymmetric process results in the accumulation of charged species in one compartment (ion enrichment) and ion depletion in the other. A similar effect can be achieved via charge transfer reactions at an electrode, and therefore, a closely related technique called faradaic ICP (FICP), is also discussed in this review (Schemes 1b and c). ICP has found many areas of application owing to its unique advantage as a means of achieving sharp spatial variation in ion concentration and electric field strength. Examples include counter-flow focusing (CFF) (encompassing both concentration enrichment and separation of charged analytes), desalination, and dielectrophoresis.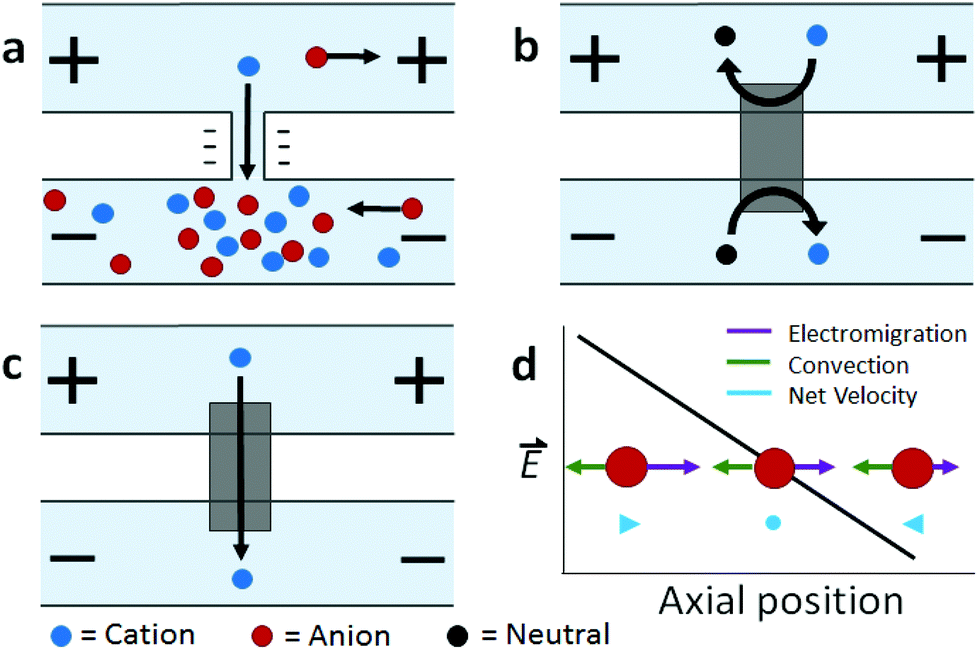 | ||
| Scheme 1 Illustration of (a) ICP, (b, c) FICP, and (d) ICP CFF mechanisms. | ||
The foundational work preceding these ICP-based techniques described mass transport and electrokinetics in and around ion-permselective materials. Electrochemical measurements aided in quantifying these effects, such as the enrichment of counterions, mass transport, and Donnan exclusion in cation-exchange membranes1 and in individual conical nanopores.2 Highly spatially resolved fluorescence measurements later led to a clear and quantitative description of ICP, as is exemplified by the work of Leinweber and Tallarek in packed beds of mesoporous beads in a microfluidic channel.3 The development of such hierarchical devices, which comprise both nano- and microscale components, provided a means of controlled analyte transport,4 concentration enrichment,5 and ICP.6–8 Separate reviews provide an account of the theoretical and experimental advances in ICP9 and ICP-based concentrators10 that followed.
The majority of recent advancements in this field have been driven by the needs of existing ICP-based technologies – namely, to increase their breadth of applicability and scale. For example, the cost and difficulty of device fabrication has been reduced through the expansion of ICP-based focusing of charged analytes (such as DNA) to paper microfluidic platforms.11–13 In another area of application that benefits populations living in resource-limited settings, desalination by ICP presents an alternative to reverse osmosis and requires simpler infrastructure.14,15 However, the throughput of an individual desalting device is severely limited by the size of the ion depleted region that can be sustained. Therefore, new fabrication methods aim to augment selective charge transport, thus leading to a larger depletion zone and an enhanced rate of fresh water production.16 In addition to these advancements, enhanced performance and control have been achieved by combining ICP with other techniques. For example, the recent incorporation of constrictions and valves into microfluidic ICP devices has enabled post-enrichment recovery of focused analyte plugs.17 Further, the combination of ICP with isotachophoresis has greatly enhanced separation results.18 Before reviewing recent advancements, we provide a technical background of ICP as a foundation for the discussion that follows it.
2 Technical background
2.1 Ion concentration polarization
This discussion will focus on ICP at micro-/nano- fluidic junctions (Scheme 1a). An electrical double layer (EDL) forms at the solid–liquid interface between, for example, the wall of a fluidic channel and an electrolyte solution. If the channel wall is negatively charged, the electrical potential at the wall is more negative than that in the bulk solution. Donnan exclusion occurs when two such walls are brought into close proximity causing the potential profiles of the adjacent EDLs to overlap and to exclude anions from the channel formed between them. The characteristic length scale of the EDL (the Debye length) is less than 10 nm for a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 electrolyte at a concentration greater than 1 mM. Therefore, complete Donnan exclusion is limited to nanoscale channels.
:1 electrolyte at a concentration greater than 1 mM. Therefore, complete Donnan exclusion is limited to nanoscale channels.
If a voltage is applied across such a nanochannel, ICP results (Scheme 1a). The key feature of this process is that charge transport is selective – in the example depicted in Scheme 1a, only cations are able to carry charge across the pore. An ion depletion zone (IDZ) develops (upper channel, Scheme 1a) as cations are removed via the pore, and the migration of anions, away from this end of the pore is enhanced by the locally high electric field strength (due to decreased ionic conductivity). At the other end of the pore (bottom channel, Scheme 1a), cations accumulate and anions migrate inward, creating an ion enrichment zone (IEZ). A key feature of any ICP device is the current due to selective charge transport, which is directly proportional to the rate of IDZ and IEZ growth. In general, more rapid growth is achieved when more nanochannels are present. ICP devices frequently employ nanoscale features that are too large to cause complete EDL overlap. It is important to note that ICP occurs even at larger length scales, and only partial charge selectivity is required, which is accomplished whenever a significant proportion of the current through the pore is carried through the EDL. In fact, studies involving a related technique, faradaic ICP (FICP), demonstrated that the two microcompartments do not have to be completely separated. Both ICP and FICP have been shown to occur in a single open microfluidic channel with a nanoporous membrane or electrode located along a portion of the channel floor.19,20
2.2 Faradaic ion concentration polarization
Like ICP, FICP results from selective depletion and enrichment of charged species in a confined environment, such as a microfluidic channel (Schemes 1b and c).21 In this system, ion depletion is caused by neutralization of a freely diffusing ionic species via a faradaic electrochemical route. Likewise, ion enrichment results from electron transfer leading to an increase in the magnitude of charge of a chemical species. Scheme 1b depicts these two processes occurring at either end of a bipolar electrode (BPE), and the net result (Scheme 1c) closely mimics ICP. FICP has been employed to accomplish many of the same experimental goals as ICP and has the following distinct advantages: (1) ease of fabrication of electrodes versus nanofeatures, (2) different selection rules and rate for charge removal/injection, (3) possibility for ion depletion to occur in both microfluidic compartments simultaneously. FICP has been discussed in more detail elsewhere.21,222.3 Electric field profile in the ion depletion zone
Of the two poles of ICP, the ion depletion zone (IDZ) has, by far, produced the most compelling experimental results owing to its intense enhancement of the local electric field strength. In all ICP-based techniques, a component of the electric field is applied axially along the microchannel that contains the depletion zone. The resulting axial electric field peaks where ion depletion is greatest and forms an extended electric field gradient as it tapers to the edge of the zone. The recent advancements in ICP-based techniques reviewed here all depend on the way by which this electric field gradient is formed, maintained, and applied to scientific problems. In the following sections, we review recent advancements in ICP-based focusing (concentration enrichment and separation), desalination, and dielectrophoresis.3 Preconcentration of trace analytes
3.1 Introduction
The preceding section described how ICP occurs when a voltage bias is applied across a device feature that is capable of facilitating selective charge transport. Counter-flow focusing (CFF) can occur if opposing convective and migrational velocities of a charged analyte are balanced at an axial location along the electric field gradient at the IDZ boundary. In the example depicted in Scheme 1d, convection (cathodic EOF or pressure driven flow) is from right to left, and electromigration of an anion (toward the anodic end of the channel) is from left to right. The location of the balance point for a specific analyte is determined by its electrophoretic mobility. As a result, distinct analytes can be separated on the basis of differing charge-to-drag ratios. While focusing of an anionic analyte is depicted, schemes have been devised for the enrichment of cations or both cations and anions simultaneously.23,24The use of ICP CFF opens up many new possibilities in the preconcentration of analytes. First, the limit of detection of low abundance analytes can be significantly improved after preconcentration while using only a small amount of sample. Enrichment of up to one-million fold has been reported.25 Second, this electrokinetic approach is broadly applicable to any charged species. Third, this electrically-driven system allows enrichment to be achieved rapidly – within a few minutes or even a few seconds.26–28 Fourth, ICP CFF is robust, operating under diverse conditions brought about by nanochannel or membrane materials (e.g., polydimethylsiloxane (PDMS), Nafion, charged hydrogel, and other modified nanopores),10,29–31 channel geometry,32–34 and integration with other lab-on-chip (LOC) components (e.g. sensors35 and droplet microfluidics36). Moreover, ICP CFF compares favorably to competing enrichment methods. For example, ICP does not suffer from clogging and loss of collected molecules as do morphology-based concentrators.37 Additionally, unlike affinity-based devices, ICP can achieve high sensitivity without the need to functionalize ligands.38 Current research in this field aims to improve performance, to increase scale, and to develop customized ICP systems for new platforms, analytes, and materials.
The main goal in sample preconcentration using ICP CFF devices is to achieve a high enrichment factor with low voltage and short residence time. Enrichment factor (EF), the ratio between the concentration of analytes before and after ICP CFF, is determined by the combined effect of sequestering and dispersive mechanisms experienced by charged molecules. To maximize enrichment, several parameters need to be considered. For example, a steep electric field gradient yields high contrast in the electrophoretic velocity of an analyte between neighboring locations, hence narrowing a focused band. The slope of the electric field gradient, in turn, is influenced by parameters such as the buffer/electrolyte conditions, applied voltage, electrode configuration and the nanoporous materials employed. Of particular importance is the geometry of the microchannel and nanoscopic features. For example, a converging microchannel design can draw in analyte from a large area and sequester it into a more confined space. Finally, for any of these experimental parameters, there is an optimum level past which ICP CFF degenerates. For a defined ICP device, increasing voltage differences between sample channels and buffer channels,39 and extending residence time11,12 are favorable for reaching high EF. However, Joule heating eventually becomes significant, which broadens the focused band.13 Hence, extensive effort has gone into optimization of all of these factors to enhance EF.
While high EF is a key figure of merit, devices that meet more specific requirements needed in special circumstances are critical. Point-of-care (POC) applications increasingly demand customized ICP preconcentration systems. For instance, the use of POC devices in resource-limited settings requires low-power consumption, cost-effectiveness and simplified design and operation. Other applications demand simultaneous enrichment and separation of multiple targets, for which the main challenge may be resolution and not EF. Finally, some ICP devices are being designed to recover larger quantities of enriched materials for downstream analysis. For these devices, the geometry is optimized for scale.
3.2 New approaches to make nano-features in PDMS-based microfluidic devices
The geometry, surface chemistry, and number density of nanofeatures impact the formation of an ion depletion zone. Therefore, this subsection will highlight recent advances in nanofeature fabrication.In recent years, interest in preconcentrators having nanofeatures fabricated in PDMS has been rapidly expanding.21,40,41 PDMS allows convenient bonding and rapid replication, while wet etching and photolithography using silicon and glass are time-consuming.42–44 However, with a low Young's modulus (360–870 KPa), nanochannels in PDMS substrate are prone to collapse and deformation. Hence, extensive efforts have been made to create nanochannels/nanojunctions with PDMS-based non-soft lithography methods. Examples include the nanoscale-fracturing method of oxidized PDMS,45 the wrinkling process,46 mechanical cutting of PDMS by razor blades47 and the “roof-collapse” method.48 Although all of these reports have achieved flexible, simple and low-cost fabrication processes, the nanochannels are limited to two dimensions (2D) which greatly limits the throughput of PDMS-based microfluidic devices.
Recently, stacking and rolling methods have enabled the extension of topological configurations of nanochannels to three dimensions (3D).49 For the stacking scheme, multiple PDMS layers, fabricated using traditional photolithography, were repeatedly treated with oxygen plasma and permanently bonded together to form multilayered stacks (Fig. 1a). As shown in Fig. 1c and d, the density of the 3D polymeric nanochannel stack arrays (PNSAs) was directly controlled by the master mold – bump arrays patterned onto a silicon wafer. On the other hand, the size and shape of the nanochannel arrays were controlled via elongation of the PDMS films (Fig. 1g). Stretching and releasing the films created irreversible changes in nanochannel dimensions. For instance, the volume of the nanochannels shrinks irreversibly when an applied tensile force is removed. PDMS molded from a bump pattern of 2 μm or 4 μm in height were first stretched to 0, 25, 40, 55 and 70% of their original volume. Depending on the degree of elongation, various shapes such as circular and elliptical nanochannels could be accessed, while the size of nanochannels continuously decreased (Fig. 1g). Therefore, nanochannel structures with various sizes, shapes and densities could be achieved by the synergy of stacking PNSAs and elongation.
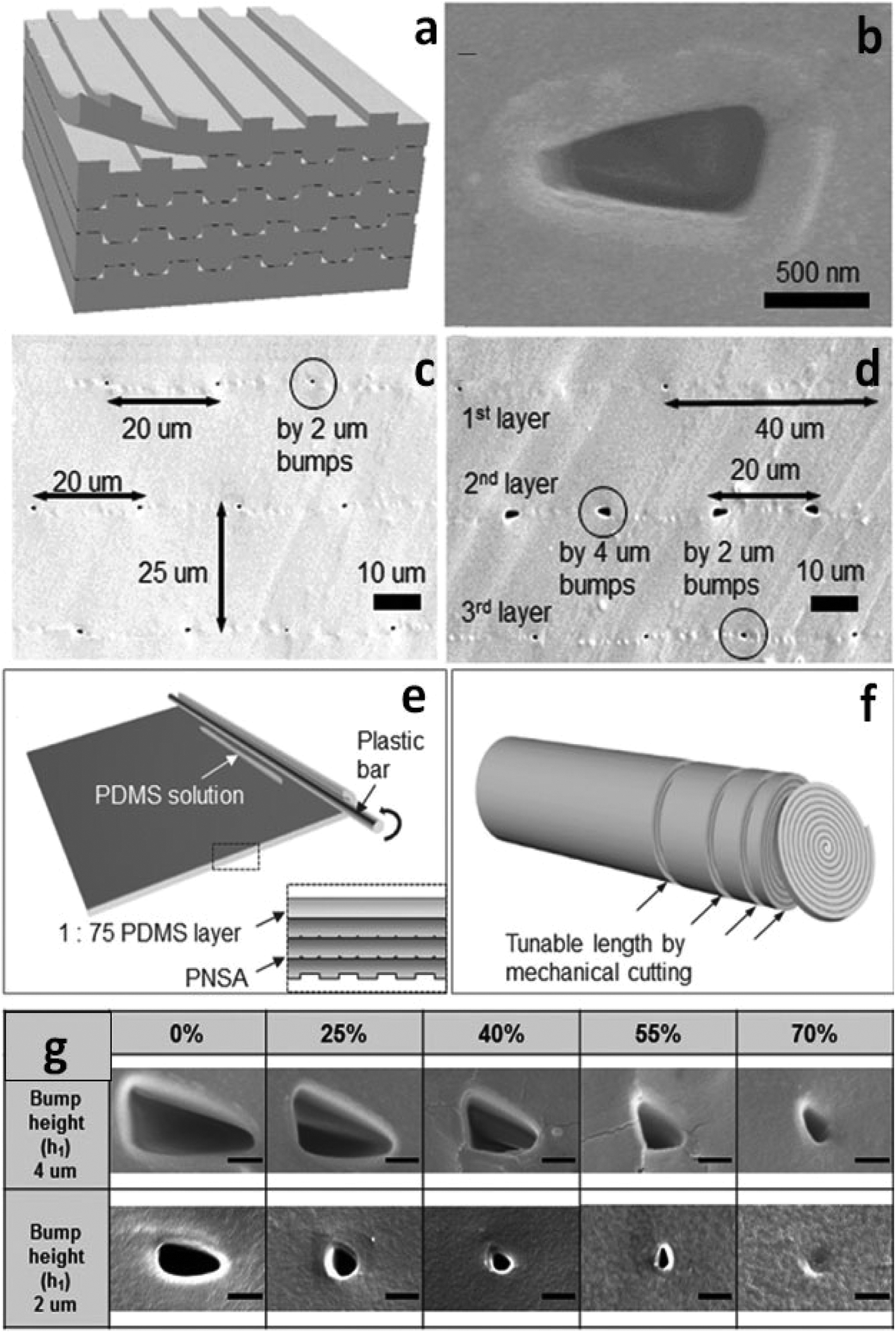 | ||
| Fig. 1 Fabrication and results of the 3D PNSAs. (a) Schematic of the stacking method to form a PNSA; (b) scanning electron microscopy (SEM) image of nanochannel opening created by layer-to-layer bonding (bump height = 2 μm); (c) SEM image of PNSAs which feature a constant pitch of 20 μm, and a constant layer thickness of 25 μm; (d) SEM image of PNSAs exhibiting variable pitches and sizes at each inner layer; (e) schematic of the rolling technique using PDMS. The magnified image is a cross-sectional view before rolling; (f) the resulting PNSAs after the rolling process; (g) investigation of nanochannel size and shape control by various elongation rates (scale bar: 500 nm). | ||
Due to the tediousness of manual stacking cycles, the authors introduced a fabrication method of simply rolling the PNSA film from its edge with a plastic bar to realize large-area integration (Fig. 1e). The number of layers were governed by the rolling cycles, while sheets of a specified thickness could be created by cutting the PNSA cylinder (Fig. 1f). A PNSA sheet was then sandwiched in between two PDMS layers containing two orthogonal microchannels for ICP investigation. The authors used current–voltage (i–v) measurements to demonstrate the three characteristic regions of ICP in the PNSA device. In region I (Ohmic region), the linear relationship between voltage and current indicates the constant conductance of the nanochannels. In region II (limiting current region), the unchanged current as voltage increases suggests the formation of an IDZ. In region III (overlimiting region), the conductance increases significantly due to the formation of vortices trigged by electro-convection in the depletion zone.50–52 These results indicate that the ICP phenomena was driven by the perm-selectivity of the PNSAs. The strategy demonstrated using stacking and rolling techniques provides an opportunity to prepare new 3D PDMS-based nanofeatures with high throughput and good control over size and shape. The ease of preparation and straightforwardness of the approach facilitate the design of nanochannels for the preconcentration of charged analytes.
The conditions under which ICP can be initiated are limited by the ratio of surface conductivity to bulk conductivity (Dukhin number, Du) in nanofeatures. Small channels with a high surface charge and low ionic strength electrolytes favor increasing Du53,54 due to greater overlap of the EDL. Therefore, to broaden the applicability of ICP, simple methods are required for fabrication of sub-50 nm nanofeatures with controlled surface functionalization.
Song et al. introduced a self-assembly process that employed colloidal silica beads to achieve size control of nanochannels, as well as selectivity via surface coating.55 The strategy is based on the concept that the close-packed structure of colloidal crystals after evaporation has pores with diameters about 15% of the colloidal particle diameter.56 Further, the authors coated the silica beads to alter their zeta potential and as a result, observed changes in their ability to initiate ICP.55 Silica beads with diameters of 300 nm, 500 nm, and 900 nm were used to form pore sizes of ∼45 nm, 75 nm and 135 nm, respectively. PSS (poly(styrenesulfonic acid)) and PAH (poly(allylamine)) were applied as coatings to study the effect of surface functionalization on charge selectivity. An array of lithographically patterned nanochannels (depth: 700 nm, width: 2 μm) were fabricated on both sides of the sample channel. These nanochannels served to trap silica beads in the delivery channel while maintaining a connection between sample and auxiliary (buffer) channels (Fig. 2). This self-assembly process allows for nanoscale-size control completely outside of a sealed microfluidic device. Considering the highly developed coating processes available for silica beads, the preparation of nanochannels sensitive to temperature,57 pH,58 and ionic strength59 can be expected.
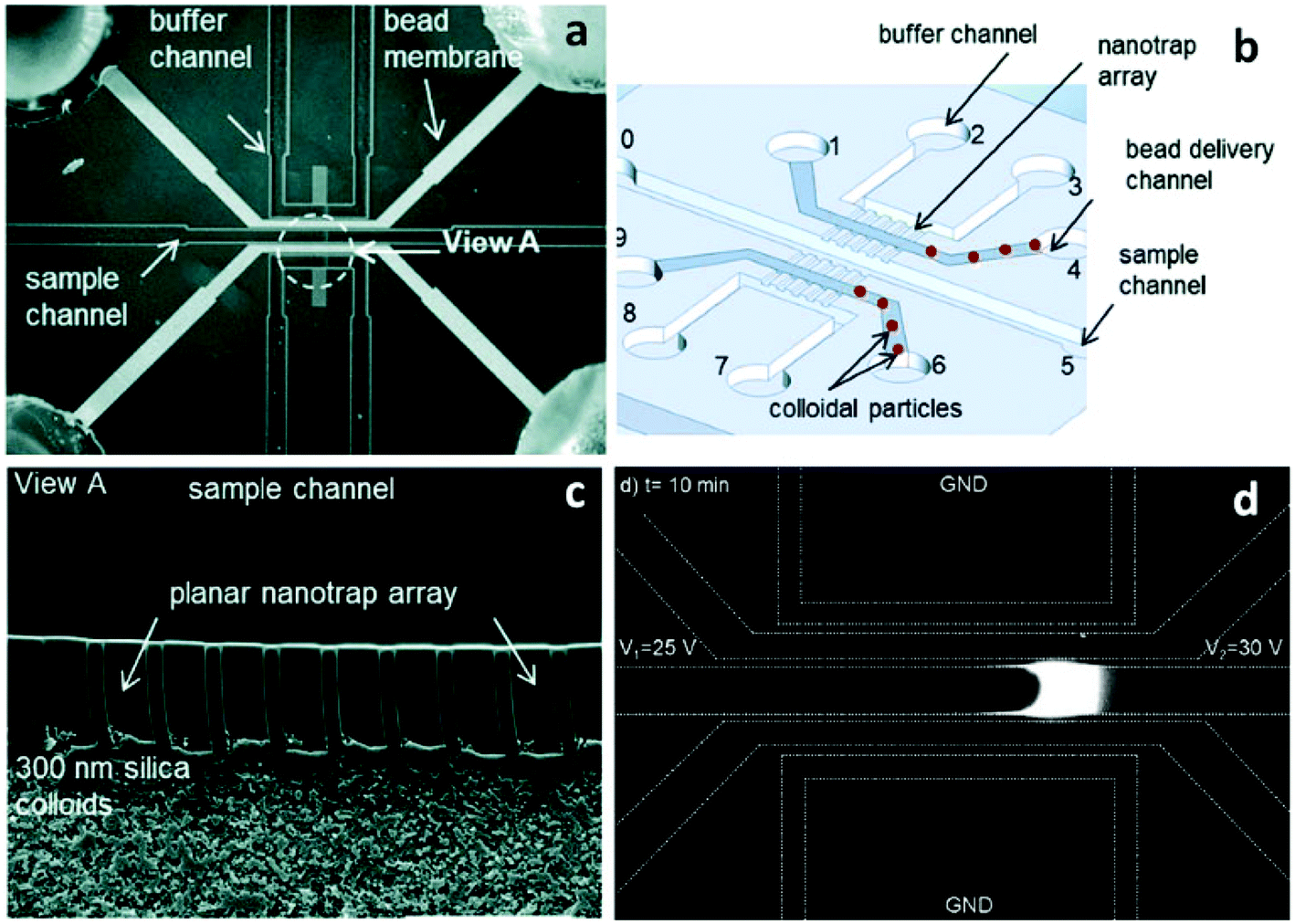 | ||
| Fig. 2 Schematic illustration of the microfluidic device made from self-assembled microbeads. (a) Micrograph of the device showing that the colloidal particles were assembled inside the upper and lower bead delivery channels (scale bar: 200 μm); (b) a schematic of the device design; (c) SEM image of the 300 nm silica particles trapped between the buffer and the sample channel (scale bar: 20 μm); (d) fluorescence micrograph showing 1700-fold DNA enrichment obtained using 300 nm silica beads. | ||
A 10 nM Cy5-tagged DNA 25-mer in 1 mM phosphate buffer solution was utilized as the target analyte for the ICP analysis. The increased zeta potential obtained through surface functionalization resulted in the initiation of ICP at low voltage (8 V–10 V) relative to uncoated beads (30 V). However, the high surface charge of polyelectrolyte-coated beads greatly propagated the ion depletion zone at higher voltage (e.g. 30 V), making the DNA plug unstable. This problem is often inevitable when applying a high voltage in ICP devices. However, it is possible to prevent the propagation of the depletion zone with an increase in the EOF (i.e. by channel surface modification) or by introduction of hydrodynamic flow. Pressure driven flow is preferred because running ICP in highly charged channels at high voltage may cause “bursting” of the ion depletion zone,8 which occurs when the EOF overcomes the repulsive force of the IDZ.
Another recently developed nanoporous junction for ICP employs single-wall carbon nanotube (SWNT) films.60 Carbon nanotubes (CNTs) are good candidates for membrane materials considering their high aspect ratio, electronic and mechanical properties.61 Specifically, the great potential of functionalization via various modifications makes CNTs widely used in applications such as electrochemistry, sensing and catalysis.62–64 With the aid of vacuum filtration and film transfer techniques, 60 nm-thick films of carboxyl-modified SWNTs were prepared and employed for ICP in a dual-channel microfluidic device. The formation of a limiting region in the i–v curve proved that ICP resulted from the permselectivity of the film. The 60 nm-thick SWNTs exhibited similar conductance to a 500 nm-thick Nafion membrane. The surface tunability and high conductance of SWNTs reported here suggest that this material has the potential to be a highly efficient ICP membrane.
3.3 Methods for controlling the location and stability of the concentrated analyte plug
Not only can flow rate be used to control ICP propagation at high voltage, it also the simplest means of achieving control over the location of the concentrated analyte plug. As a recent example, Choi et al. demonstrated enrichment of Alexa Fluor 488 in a Nafion-coated straight glass channel (inner diameter = 1.1 mm) under two sets of conditions. The architecture of Nafion65 allows the initiation of ICP via the overlap of EDLs. They first applied 30 μL min−1 flow toward the cathode with a DC voltage of 300 V, resulting in a mean fluorescence intensity of 93.66 When the flow rate was set to 40 μL min−1 at 400 V, the intensity increased to 243. A higher voltage shifted the concentrated plug away from the Nafion membrane, thus a higher flow rate was needed to overcome the enhanced electromigration and IDZ growth to fix the position of the concentrated plug.Once enrichment is achieved, it is advantageous to encapsulate and store the analyte for downstream analysis. To this end, Phan et al. demonstrated analyte sequestration using a continuous-flow droplet ICP microfluidic device. Their results underscore the balance of applied voltage and flow rate. The authors showed that the distance between the ICP boundary and the edge of the Nafion membrane varied linearly with the applied voltage.67 However, by increasing flow rate proportionally with voltage, the enriched band could be maintained at the droplet generator. Using this method, a maximum of 100-fold EF was obtained. The flow rate also controlled the size of the droplets formed. For instance, when the flow rate increased from 10 μL h−1 (30 V) to 30 μL h−1 (90 V), the droplet size changed from ∼240 mm to ∼370 mm. These results are significant because this technique could be interfaced with a suite of microfluidic tools for droplet manipulation.
Kwak et al. introduced a merged ion enrichment-depletion zone method to spatiotemporally define the preconcentrated plug in a way that is independent of ICP operating conditions.68 The key feature of this technique is two Nafion membranes positioned 100 μm apart on the bottom of a straight microchannel. When a voltage was applied, the anions that enriched at the boundary of the IDZ formed by the right-hand Nafion membrane were stabilized by the cations in the IEZ formed by the left-hand Nafion membrane, thus preventing expansion of the ICP boundary. A concentrated plug of a negatively charged fluorescent dye could be tuned to a specific region (100 μm × 50 μm × 10 μm) under a broad spectrum of testing conditions (voltage: 0.5 V to 100 V; ionic strength: 1 mM to 100 mM KCl; pH: 3.7 to 10.3; residence time: 0 s to 1200 s). This method addresses one of the greatest challenges encountered in ICP CFF and may render it more applicable to preconcentration in other microfluidic platforms.
Recently, another ICP concentrator driven by capillary force was introduced to eliminate the undesirable instability of the concentrated plug.69 Instead of applying a voltage to drive ions into a permselective membrane, a negatively charged hydrogel was patterned70 and employed for continuous imbibition of cations by capillary force, thus initiating the formation of an IDZ. Once the IDZ formed, anions migrated away by electrostatic repulsion and enriched at a fixed position when a counter flow (e.g. pressure-driven flow) was applied. The propagation of the IDZ was found to be dependent on the electrolyte concentration – low buffer concentration resulted in a thick EDL, which in turn led to a high propagation velocity. Compared to electrically driven ICP, the EF was relatively low (100-fold in 60 s) due to the limited absorbing capability of the hydrogel and a lack of global electromigration. However, this ICP device is significant because it operates in the absence of an electrical power source, thereby expanding the potential utilization of ICP in POC applications. In addition, the simultaneous enrichment of cations and anions by patterning functionalized charged hydrogels71,72 in parallel along a microchannel can be expected.
3.4 Nafion-impregnated paper-based microfluidic devices
Several characteristics of paper-based microfluidic devices have motivated their recent surge in development: (i) they are low-cost, amenable to mass production, and easy to transport. A simple microfluidic paper-based analytical device (μPAD) is about $0.01 for the cost of the paper and patterning;73 (ii) typical μPAD materials, cellulose or cellulose–polymer blends, are compatible with various chemical/biochemical/medical applications;74 (iii) the devices can be disposed of by incineration safely after use.75 Therefore, since the pioneering study in 2007,76 μPADs have become a versatile and scalable platform for a broad spectrum of diagnostic applications.77–79 However, concentration enrichment in μPADs has been a challenge. The prevailing paper-wetting concentrators rely on capillary forces generated in dry portions of the paper matrix. Examples include sliding paper strips through a stationary reagent loading unit for performing enzyme-linked immunosorbent assays (ELISA),80 adding soluble sugar to create programmable flow delays,81 and actuating paper cantilevers to connect channels.82 Once a paper-based assay is fully wet, these approaches based on capillary forces cannot be utilized for the concentration and transport of analytes.The use of nanoporous membranes and ICP in μPADs opens up the avenue to develop paper-based concentrators which are independent of capillary forces. Of various nanoporous membrane materials, Nafion is most commonly used since it creates a hydrophobic barrier in paper devices while allowing selective cation transport through the hydrophilic nanopores.
Gong et al. have demonstrated the feasibility of concentration and transport of molecules using a Nafion membrane for ICP in a fully-wetted paper-based assay.11 Two classes of devices were developed – an external stamp-like silicone platform comprising the driving electrodes, reservoirs, and Nafion membranes separate from the paper, and an in-paper device patterned directly with Nafion (Fig. 3). For the external device, two ICP regions exist, one at each of the Nafion membranes in contact with the two ends of the paper fluidic channel. Depending on the polarity of the applied voltage, an enrichment zone forms at one end of the paper and depletion at the other. An enriched analyte band formed at the boundary of the depletion zone. This configuration is not unlike that introduced by Kwak et al. (vide supra). The silicone layer containing the embedded Nafion nanoporous membranes served to prevent direct mixing of samples with buffer solutions contained in the external reservoirs. For the in-paper devices, however, the nanoporous membranes were directly patterned into the paper, which served both to separate two wetted regions and to enable ICP-based enrichment. The enrichment of fluorescein achieved with the external-device became visible at 50 s, and reached saturation at 155 s with 40-fold enrichment; while the fluorescence intensity for the in-paper device became significant at 90 s, and the maximum value was 22-fold at 510 s. The transport efficiency (percent of analyte enriched) upon reversing the polarity was investigated for two cycles in both external and in-paper devices. In both devices, the transport efficiency dropped significantly with repeated cycles. The utilization of a glass coverslip resulted in a consistent transport efficiency of ∼90%, which indicated that the reduction of transport efficiency could be ascribed to evaporation.
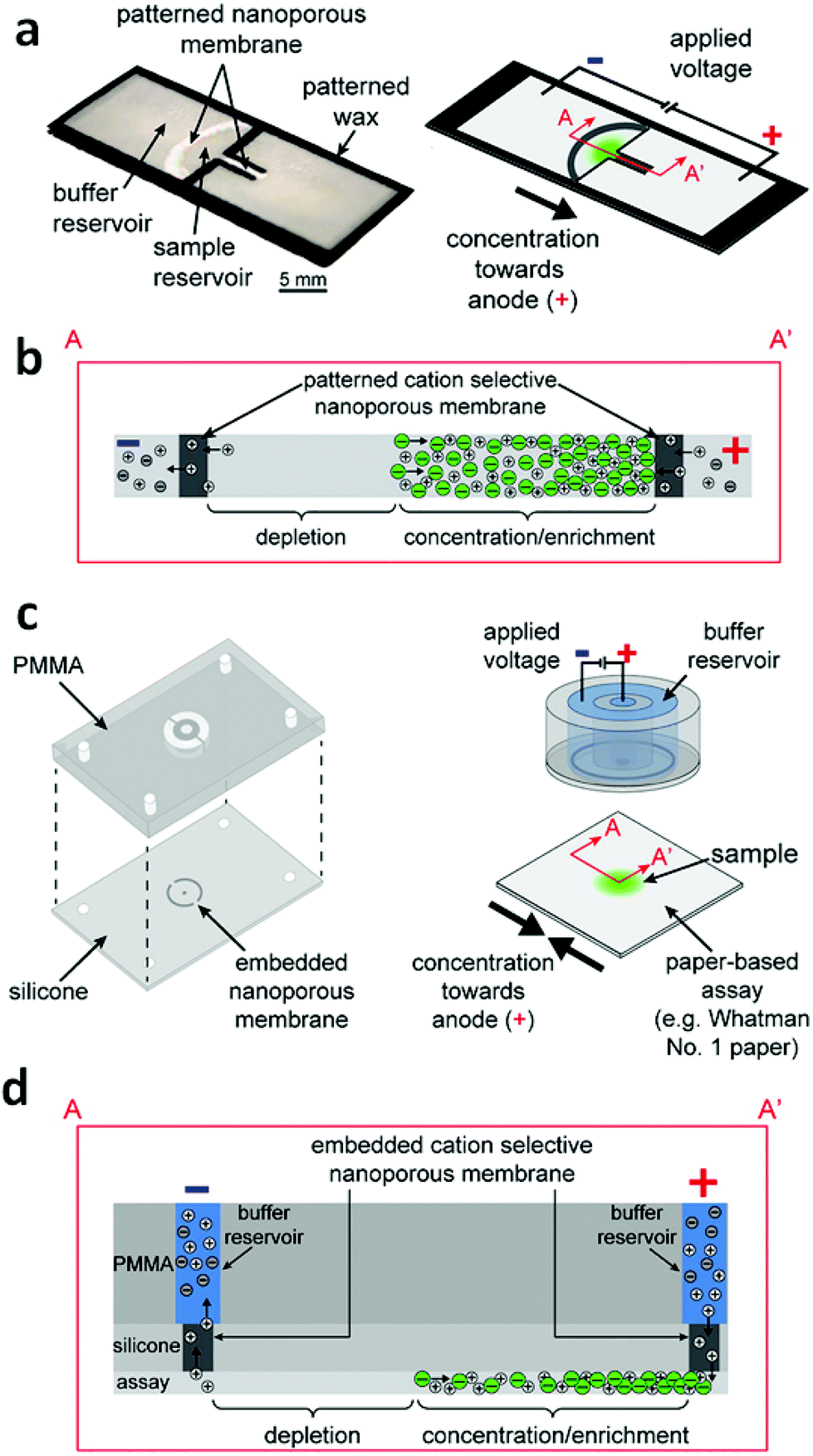 | ||
| Fig. 3 Design and operation of the external and in-paper devices for concentration. (a) Schematic of the in-paper transport configuration. (b) Cross-section of the in-paper devices illustrating ICP under an applied voltage. (c) Schematic of the external concentration device. (d) Cross-section of the devices and paper-based assays depicting ICP under an applied voltage. (Image reproduced from ref. 11 with permission. Copyright 2016, American Chemical Society.) | ||
Using the in-paper device, the authors achieved a limit of detection (LOD) for FITC-albumin of 2 pmol mL−1 (vs. 10 pmol mL−1 before concentration) and for bromocresol green dye ∼10 μM (vs. 40 μM). This improvement is significant because it extends the capabilities of low-cost paper-based assays and broadens the application of ICP beyond traditional device materials.
To decipher the influence of channel geometry on concentration performance, a separate study of fully-wet paper-based devices investigated convergent channels with different length and width.12 Note that the use of convergent channels could favor the aggregation of analytes (confinement effect), while narrow channels decrease the current and the net movement of ions which is not favorable for sample enrichment (current effect). The analysis of fluorescence intensity in different convergent channels demonstrated that a trade-off existed between the two counter-effects (confinement and current), and a convergent channel with a width of 1 mm (narrowed from 2 mm) yielded the best performance in this paper-based microfluidic device.
The ICP effect of the convergent channels on paper-based devices was also investigated by measuring the i–v curve. Similar to traditional PDMS/glass devices, three characteristic regions on this paper-based microfluidic device were observed. However, for the present paper-based devices, the overlimiting current increased only slightly with the voltage. This behavior can be attributed to the intricate structure of cellulose, which tends to decrease the electro-convective effect through hydrodynamic resistance.
It is also noteworthy that the use of one straight channel in a paper-based device greatly simplifies the fabrication process compared to the traditional two-channel ICP concentrators. Phan et al. further simplified the fabrication process by placing a thin Nafion membrane underneath the paper strip.83 Using such a device, an EF of 60-fold at 200 s was obtained, which was slightly higher than those obtained using manually patterned Nafion membranes (40-fold at 155 s for ref. 11, 20-fold at 130 s for ref. 12). The enhanced enrichment performance in the external Nafion device may be attributed to more uniform EOF throughout the device.
4 Simultaneous preconcentration and separation of analytes
4.1 Introduction
Separation of sample components is often the starting point of chemical or biochemical analysis. Separation conducted by ICP CFF not only allows continuous separation and simultaneous enrichment, but also creates the potential for LOC integration, rendering the isolation of targets from bulk solutions rapid and convenient. Separation resolution (SR), which is defined as the ratio of peak-to-peak distance between two adjacent bands to their average width, is commonly used to evaluate performance. Separation in an electric field is based on the differences among analytes in electrophoretic mobility (μ), which is equal to the charge of a molecule (Q) over friction (ζ). A limitation of electric field based separations is therefore cases for which μ is indistinguishable for two analytes. For example, the separation of DNA is often problematic since Q and ζ are both proportional to the number of monomers in the DNA chain (M) (i.e. μ = Q/ζ ∝ M/M).84 ICP CFF separation also has been limited by the difficulty of accessing enriched bands (e.g. via extraction or isolation). Recent advancements that are discussed in this subsection address these two limitations by integrating mobility shift strategies and a valve system with ICP CFF separation.4.2 Paper as a sieving matrix in ICP devices
Due to the indistinguishable μ of distinct DNA strands, a sieving matrix such as agarose gel is typically employed to force DNA to collide with fixed structures to a degree dependent upon strand length.85,86 The adaptation of ICP CFF to paper-based devices has raised the possibility that paper could serve as a suitable sieving matrix for DNA separation. It is reported that nitrocellulose papers with 0.2 μm, and 0.45 μm pore diameter correlate to 2% and 1% agarose gels, respectively.87 Thus, the paper-based ICP design promises to be a viable approach for the continuous separation and concentration of DNA.Recently, a paper-based ICP device using Nafion was prepared and used for the direct analysis of Hepatitis B and the assessment of human sperm DNA integrity (Fig. 4).13 Fluorescein isothiocyanate (FITC) and calcein were utilized as anionic fluorescent tracers for a model investigation of the performance of preconcentration and separation.
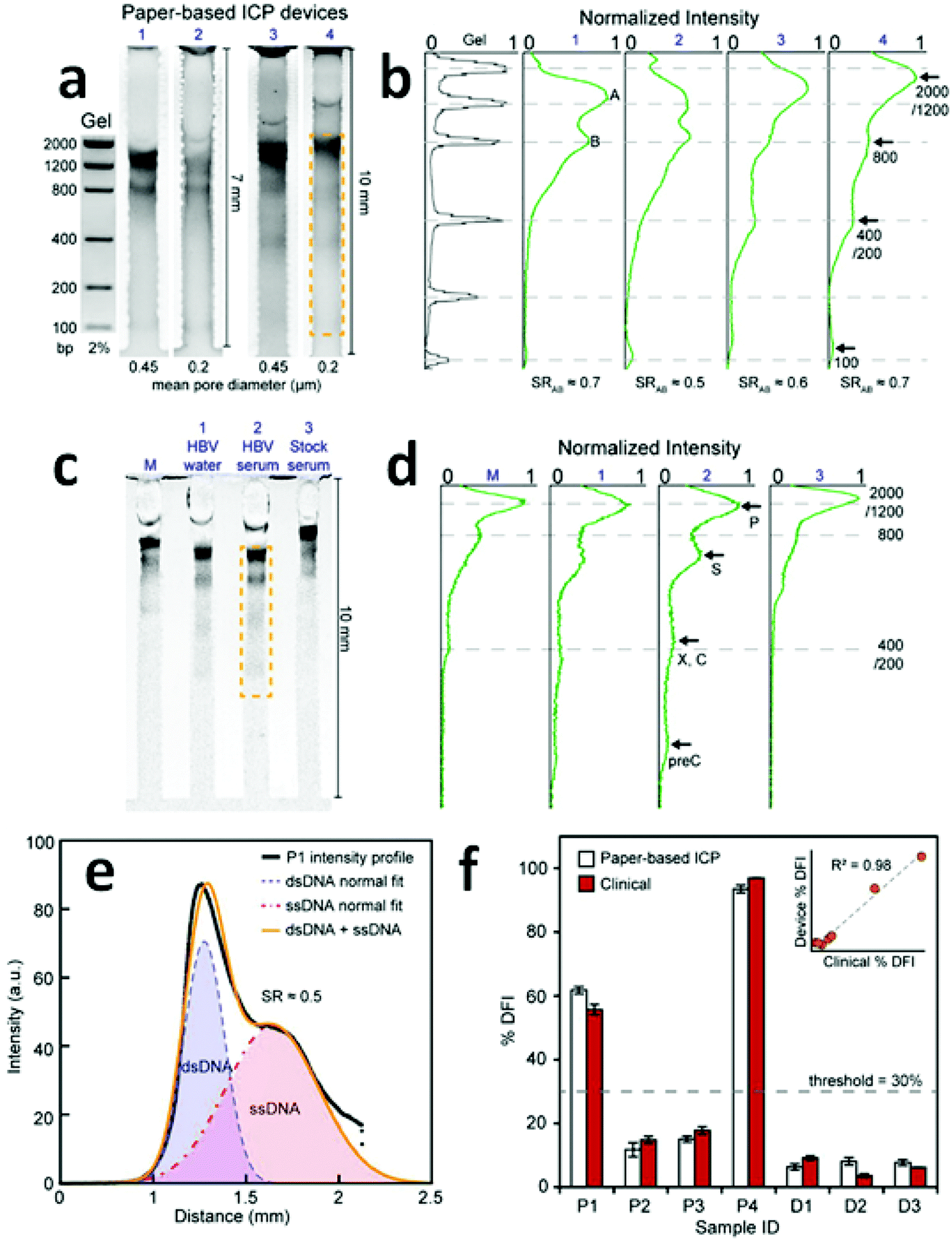 | ||
| Fig. 4 Results of direct DNA analysis using the paper-based ICP device. (a) Separation of a DNA standard containing six dsDNA fragments in gel and in four paper devices with varied length and pore size; (b) comparison of the band intensities obtained by the gel and the paper-based ICP devices in (a); (c) and (d) results obtained using the multichannel method for the analysis of (M) DNA standard, HBV fragments in (1) water and (2) serum, and (3) a stock serum sample; (e) intensity profile of the separation of ssDNA and dsDNA from a patient sperm sample; (f) %DFI testing results for patients and donors. 30% is used as the threshold to evaluate clinical outcome. (Image reproduced from ref. 13 with permission. Copyright 2016, American Chemical Society.) | ||
A multichannel device (shown in Fig. 4c) using the paper-based ICP design was then employed to simultaneously preconcentrate, separate and detect HBV DNA fragments (precore, core, surface, X, and polymerase). Though the device failed to distinctly identify the X and the core regions of the HBV genome, the capability to detect surface and precore regions allowed it to be used for HBV detection. Significantly, for hepatitis B testing, the low LOD (150 copies per mL) achieved without prior PCR amplification was comparable to that of commercial PCR systems (50–100 copies per mL). For the assessment of human sperm DNA integrity, the percent DNA fragmentation index (%DFI) obtained with the device strongly correlated to those detected by a flow cytometry-based sperm chromatin structure assay (SCSA) measurement (R2 = 0.98). These results indicate that this paper-based device may provide a platform for scalable fertility testing.
Despite the capability of simultaneous preconcentration, separation and detection of DNA targets in a single operation, the low SR values achieved (0.5–0.7) indicate that there is still a need for data that aid in understanding the key factors impacting separation using paper-based ICP devices. One possible reason for the low SR values might be an additional dispersive mechanism (e.g. non-specific adsorption or inconsistent pore size) encountered in paper fibers. The shallow reservoirs, as defined by the paper thickness, may also hinder the supply of sample solutions from which to draw, thereby decreasing the enrichment of ions.
4.3 End-labeled free-solution electrophoresis (ELFSE) principle
To address the “free-draining” property of DNA, end-labeled free-solution electrophoresis (ELFSE), which breaks the charge-to-drag balance by labeling DNA with a large neutral molecule (“drag-tag”), is another way to achieve separation.88 Unlike size-based separation, no sieving matrix is needed, and fast separation can be expected. Song et al. demonstrated the versatility of the ELFSE approach for the separation of DNA via ICP CFF in a Nafion-based PDMS/glass microfluidic device.39 Two different classes of DNA (free biotinylated DNA (25 nM) and 3′ streptavidin labeled DNA (5 nM)) were separated at 60 V. At 120 s, the simultaneous preconcentration and separation of the two bands became clear as the free DNA exhibited higher electrophoretic mobility than the bound DNA. A maximum enrichment of 900-fold and 500-fold was reached at 240 s for the bound, and the free DNA, respectively. Increasing the voltage difference to 80 V resulted in 1150-fold enrichment for the bound DNA and a SR of 1.85. Together with the benefits accomplished by ELFSE, the Nafion-based ICP device demonstrated fast and effective separation and preconcentration of DNA. Note that while nitrocellulose paper acted as a sieving matrix for separation, the SR value (∼0.5) was much lower than that achieved by ELFSE. The introduction of the ELFSE principle in an ICP paper-based device can be anticipated to produce a fast, efficient and cost-effective separation approach.4.4 Combination of ELFSE and FICP
FICP driven by a bipolar electrode (BPE) embedded in a microfluidic system provides another way to enrich charged analytes via the balance of electromigration velocity against counter flow. It has been reported for such a system that the enrichment factor for a fluorescent anionic tracer reached 1.42 × 105-fold in about 33 min (71-fold per s).21 The ease of the fabrication process, also makes BPEs a convenient tool for the preconcentration of analytes. In this context, Song et al. have set forth the idea of integrating a BPE-based device with ELFSE for the simultaneous preconcentration and separation of DNA.89 Compared to the aforementioned study, which employed a Nafion-based device for ELFSE, a lower enrichment factor (600-fold and 500-fold) was obtained with longer time (300 s) when the same voltage (60 V) was applied, while the SR value was similar (1.49) since the same DNA and drag-tag were utilized.There are several possible ways to improve enrichment performance in BPE-based microfluidic devices. First, considering that a steeper electric field is favorable for enhancing enrichment process, an increase in buffer concentration can improve the EF.90 Second, a major limitation of BPE-based device is faradaic degradation of the BPE material or electrolysis of the solution to produce gas bubbles. These problems can be alleviated by applying a non-zero voltage in the buffer (auxiliary) channel, which enables one to maintain a low voltage drop across the BPE, while increasing the voltage drop across the focusing channel. Third, increasing the surface area-to-volume ratio of the BPE, for example by the utilization of a microchannel plate (MCP), takes the BPE concentration capacity to the next level: up to 175-fold per s was obtained for an MCP exceeding rates reported for planar BPEs.91 The authors claimed that each microcapillary tube in the MCP acted as a tiny BPE, resulting in a larger IDZ. It is worth noting that because of the reduction and oxidation reactions occurring individually at the cathodic and anodic poles (respectively), a single BPE can approximate both selective cation and anion transport, thus enabling the simultaneous preconcentration and separation of cationic and anionic analytes.24 These results, alongside those obtained through the systematic study of BPEs, such as was conducted by the Crooks group,19,21,24,40,41,92,93 indicate that this “faradaic ICP” may provide an opportunity to prepare new ICP concentrators with properties not attainable by current ion-selective membranes.
4.5 ICP with isotachophoresis to enhance SR
In parallel with the advancement of ICP devices for DNA separation, the development of single-cell analytical approaches has been rapidly expanding due to their ability to provide an accurate picture of the heterogeneity within a population of cells. This information is of particular significance in systems for which a minority of cells has a disproportionate influence on clinical outcomes such as in drug response and disease diagnosis. Kinases, enzymes that catalyze phosphorylation, are of importance in many cell signaling pathways and are an example of a key indicator of cell behavior. Kinase activity provides information about the kinetics and state of the signal transduction network. Recently, Cheow et al. have developed an ICP-enhanced mobility shift assay for real-time detection of kinase activities in single-cell lysate.18Their strategy leveraged the differing electrophoretic mobilities of a fluorescent substrate before and after phosphorylation. When only one kinase and its substrate were added, the reaction kinetics could be obtained by monitoring the ratio of the fluorescence intensities in enriched bands of the substrate and product with time. However, when more than one kinase was present, the small differences in electrophoretic mobility between multiple substrate/product pairs made it challenging to differentiate the bands. Moreover, there remained a need for data that could aid in evaluating the network relationship among various kinases. To improve the separation resolution, isotachophoresis (ITP)94–96 was combined with ICP CFF, using custom synthetic peptides as spacers between each substrate. The highly concentrated spacers altered the electric field, from a smoothly sloping gradient to a stair-like profile. Therefore, each substrate was focused on a more highly spatially confined portion of the gradient flanked by spacers bookending the substrate's mobility. With this approach, the activities of three kinases (PKA, AKt, and MK2) in a single cell were investigated.
This work provides a potential platform for in situ monitoring of kinase activities at a single-cell level. Multiple kinases were simultaneously detected using ICP CFF, and the separation resolution was greatly improved with the aid of ITP. Finally, this method was shown to be robust, allowing the addition of various inhibitors (off-target kinase, protease, and phosphatase) so that substrate specificity could be significantly enhanced. The authors expanded the use of ICP CFF and created a powerful tool for cell diagnosis and drug development. It has been separately reported that up to 128 samples can be analyzed in parallel in an ICP-based microfluidic chip.97 Therefore, the integration of high-throughput single-cell analysis tools with ICP CFF is anticipated.
4.6 Continuous preconcentration, separation and collection using ICP and microvalves
Despite the demonstrated capabilities of ICP concentrators, the critical issue of how to achieve extraction of the concentrated plug for post-processing remains to be solved. Techniques that employ separately a two-phase droplet generator36 and pre-binding on-site reaction98 were reported to address this critical need. However, they suffer from the need for additional recovery and cleaning steps, respectively, that limit their utility for automation and commercialization. Moreover, for the extraction systems integrated in microfluidic devices, the following features are desired: (i) the concentrated plug needs to be well stabilized so that the subsequent extraction becomes possible; (ii) the undesirable dispersion of analytes should be eliminated as much as possible; (iii) multiple targets can be selectively preconcentrated and separated simultaneously; (iv) all targeted analytes can be collected; (v) all the processes can be continuously conducted.Recently, a pioneering study of a micro Total Analysis System (mTAS) using ICP has been reported (Fig. 5).17 First, the implementation of microchambers restricted the expansion of vortices and strong electrokinetic flow formed in the depletion zone, thus enabling the stabilization of sample plugs and increasing the enrichment of analytes.99,100 Second, the connection of microvalves to microchambers rendered it applicable for the collection of target analytes. Using this system, complete separation of two dyes (sulforhodamine B (SRB): z = −1; Alexa Fluro 488 (Alexa): z = −2) was achieved, with an SR of 1.75. Four microvalves were fabricated to collect the two dyes in individual microchambers. The authors indicate that depending on how the electric field is tuned, different dyes can be collected from the same microchamber. This study also demonstrated that continuous preconcentration, separation and collection can be achieved by sequential valve actuation. As shown in Fig. 5c, valve 1 was closed when running the enrichment step, while the concentrated plug was isolated in the microchamber 3 when microvalve 2 and microvalve 4 were shut. Subsequently, all the four valves were closed to squeeze the isolated plug into the rib-shape measurement window. After this valve sequence was repeated 9 times, the measurement window was filled with green Alexa dye at an EF of 100-fold.
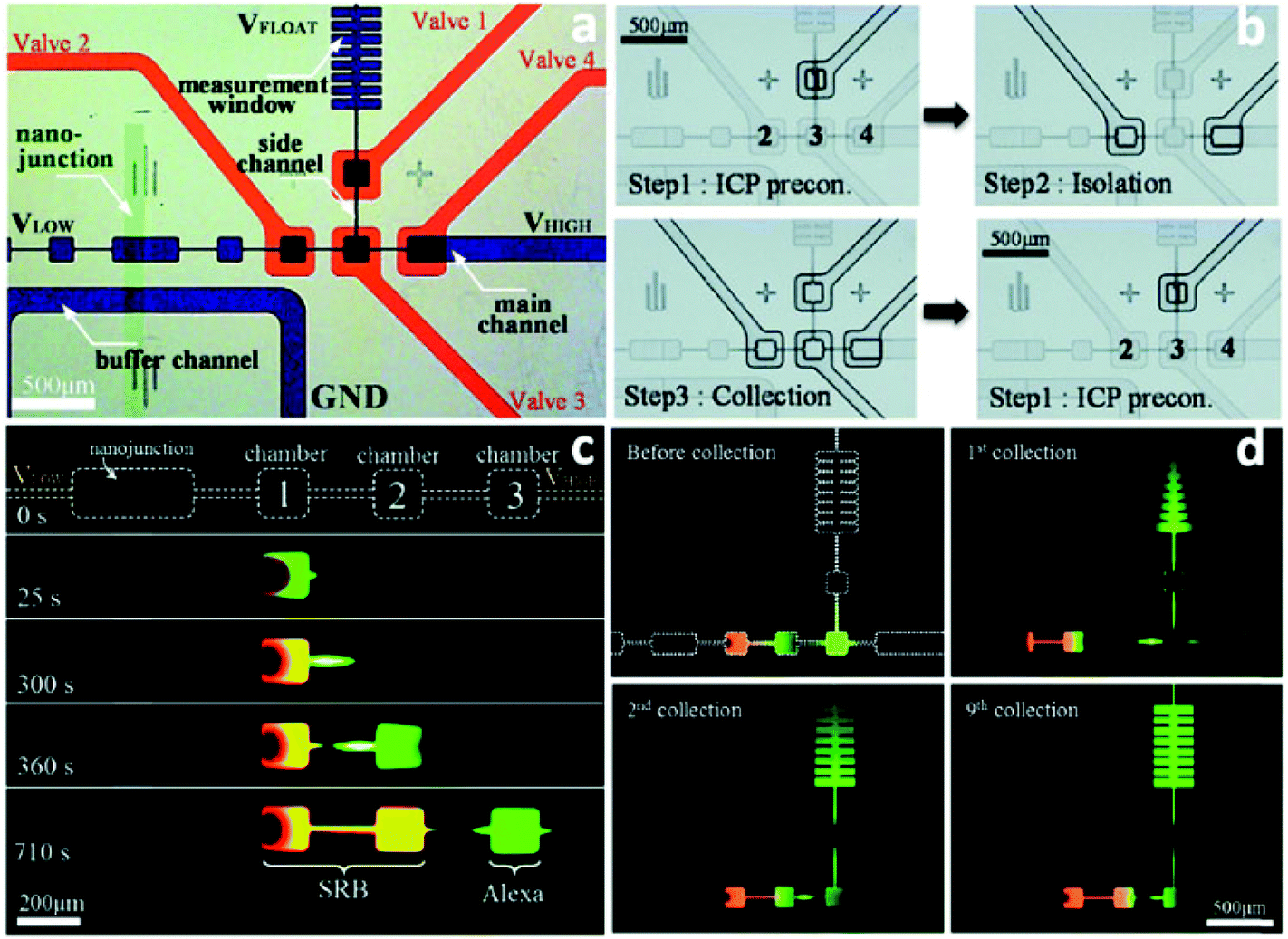 | ||
| Fig. 5 Schematic illustration of the mTAS using ICP and microvalves. (a) Microscopic view of the device (the microchannels in the ICP layer were indicated with blue and the microchannels in the valve layer were indicated red); (b) depiction of the three-step microvalve control; (c) time-lapse images of the simultaneous preconcentration and separation of SRB and Alexa; (d) snapshots of the repeated valve operations for collecting Alexa dye. | ||
This on-line collection strategy using microchambers and microvalves has demonstrated the versatility of ICP for the selective preconcentration, separation and collection of analytes. The robustness, diversity of charged materials available, and straightforwardness of the approach will facilitate the design of new ICP-based mTAS devices for a wide range of applications.
5 ICP-based desalination
Han and coworkers first recognized that the repulsion of all charged species from the IDZ could be exploited to accomplish desalination.14 Instead of focusing charged species along the IDZ boundary, these species are continuously diverted into a side channel (termed the brine stream) for removal. Using a Nafion membrane in a PDMS/glass device, the authors demonstrated approximately 99% rejection of salts with a 50:50 fluidic split and an inlet stream salinity approximating that of seawater (500 mM ionic strength, 45 mS cm−1). The freshwater output was 10 μL min−1.
More recently, Knust et al. demonstrated desalination by FICP at a carbon bipolar electrode (BPE) in a PDMS/glass device.15 The IDZ formed at the BPE anode via the oxidation of chloride ion to chlorine. This scheme resulted in 25% rejection of salts at an exceptionally high energy efficiency of 25 mW h L−1. The fresh water output was 0.04 μL min−1.
The obvious drawback of these microfluidic approaches is the miniscule freshwater output of an individual desalting junction. To produce sufficient drinking water for a household, thousands of parallel microfluidic devices would be required. MacDonald et al. recognized that the issue of throughput could be solved by increasing the size of the IDZ, thus allowing microchannels with larger cross-sectional area.16 The authors accomplished this goal by extruding the device design, increasing the interfacial area between the microchannel and a Nafion membrane. A cross-sectional view of the device is shown in Fig. 6. A device with a 200 μm × 2.0 mm inlet channel is capable of a freshwater production rate of 0.5 μL min−1 at 13.8 W h L−1 for an input salinity of 200 mM (ionic strength). Under separate conditions, the authors reported salt rejection as high as 95% and freshwater output at up to 20 μL min−1 for a single device.
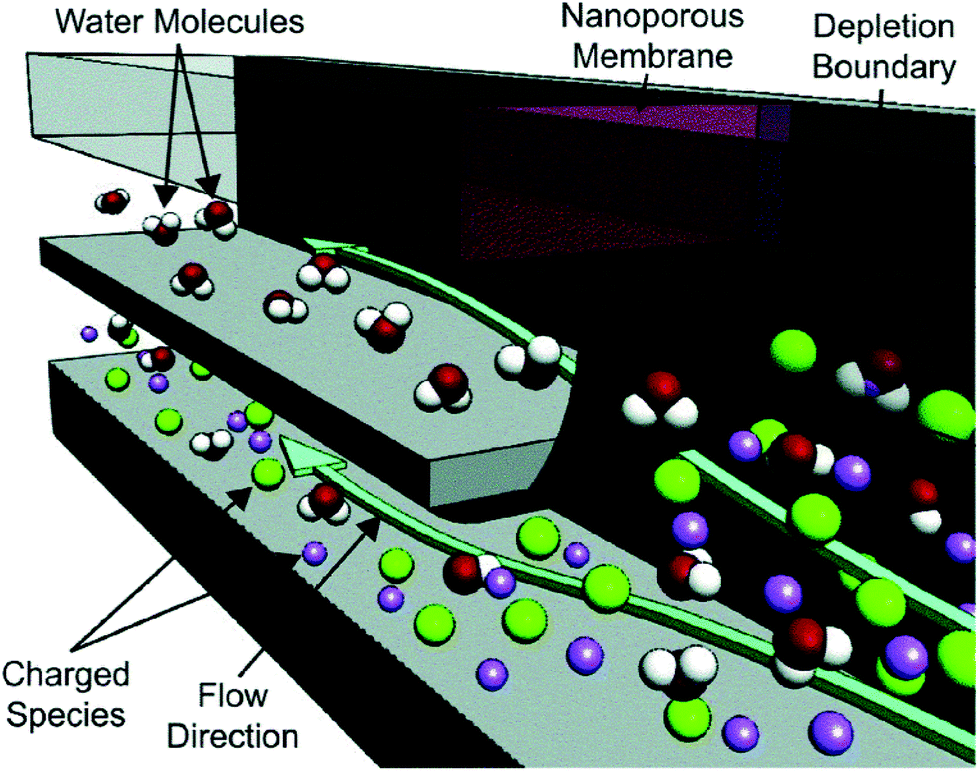 | ||
| Fig. 6 Depiction of the out-of-plane desalting device. The green plane indicates the boundary of the IDZ formed by the Nafion membrane at the intersection of the desalted (upper) and brine (lower) streams. By stacking these streams and the ground (auxiliary) channel vertically, the authors achieved greater functional density than can be obtained in a planar device. | ||
The full potential of these desalting devices can be realized by minimizing power consumption and maximizing throughput. For example, the driving voltage is reduced when applied as close to the desalting junction as possible, and a power-optimized device should have perfectly selective charge transport. Furthermore, the current across the nanofeature or BPE is linked to the degree of charge depletion and the size of the IDZ, therefore, impacting both salt rejection and throughput.
6 Dielectrophoresis of biological cells
The non-uniform electric field presented by ICP creates an opportunity to exert dielectrophoretic force on any polarizable particle. Dielectrophoresis (DEP) has been applied to trapping, transporting, sorting, and filtering particles ranging in size from proteins to biological cells.101 A non-uniform field exerts DEP force by simultaneously inducing a dipole in a dielectric material while exacting an electrostatic force differential on the two ends of that dipole. The dielectric properties of particles and the media in which they are suspended are influenced by time-dependent responses of molecular dipoles and ions to an applied field, and as a result the sign and magnitude of the dielectrophoretic force vary as a function of the electric field frequency. This property of DEP imparts selectivity, allowing different particles (such as cancer cells and white blood cells) to be distinguished.102 Therefore, DEP is often employed as an AC-field technique.The primary shortcoming of DEP is the short distance (tens of microns) over which electric field gradients, formed near electrodes or insulating barriers, typically exist. For this reason, recent advancements in DEP technology have been brought about by the development of 3D electrodes and insulating structures.103–106 Recently, Anand et al. utilized the extended electric field gradients generated by FICP to augment the reach of DEP.107 The authors employed an AC electric field with a DC offset to simultaneously drive both DEP and FICP. Given a sufficiently high frequency, the AC field has a minimal impact on the rate of ion depletion and enrichment at the BPE, thus allowing independent tuning of DEP force. The AC frequency (1.8 kHz) and low buffer conductivity employed led to negative DEP (nDEP), which is the repulsion of cells from high electric field regions. This strategy resulted in an AC-field-strength dependent (1) repulsion of cells from an IDZ and (2) attraction to an IEZ each formed at either the BPE anode or cathode. Most importantly, DEP force was demonstrated greater than 450 μm away from the BPE, a ten-fold extension of DEP reach versus standard approaches. This initial study opens the door to many possible extensions of this combined technique in terms of particle size, DEP mode (positive or negative), and ICP/FICP device design. Intriguingly, in a recently reported device107 intended for positive DEP (pDEP) capture of fluorescently tagged proteins in nanoconstrictions, an unusual response was observed and attributed by the authors to a combination of pDEP and ICP in the constrictions.108 This result implies that the combined technique is applicable over a wide range of particle sizes.
7 Conclusions
In conclusion, capabilities of ICP- and FICP-based techniques are rapidly expanding. Over the past three years, continued research has led to (1) increased throughput for enrichment, separation, and desalting schemes, (2) high resolution separation and focusing, especially through combined techniques (e.g. ICP with ITP and ELSFE), (3) extension into new materials for both the device (e.g. paper microfluidics) and the nanofeatures used for selective charge transport (e.g. silica beads and SWNTs), (4) schemes for recovering analyte bands (e.g. droplets and valves), and (5) leveraging extended electric field gradients for DEP of cells, to name a few. Although the scope of this review did not include advancements in theoretical computation, such studies are essential for progress in this field. For example, the combination of techniques such as DEP with ICP is expected to yield a complex interplay of electromigration, DEP, and both DC and AC driven fluid flow, and questions such as how an AC field impacts propogation of an IDZ remain unanswered. Further, a clearer understanding of the influence of paper fibers and their surface chemistry on EOF and analyte migration will aid in the development of higher performance paper-based ICP devices. Looking ahead, several of the remaining shortcomings of ICP-based techniques can be overcome by synthesizing ideas generated over the past three years. For example, one of the most scalable device architectures presented is that which used silica beads to achieve tunable surface chemistry. If leveraged for desalting, this scheme could lead to unprecedented fresh water output, and with highly charged bead surfaces, could do so at very low power. Further, the droplet generation scheme, if combined with droplet mass spectrometry, could produce an exquisitely sensitive and highly selective analytical tool. Clearly, ICP-based techniques are expected to enjoy continued growth and development.References
- R. M. Penner and C. R. Martin, J. Electrochem. Soc., 1985, 132, 514–515 CrossRef CAS.
- Z. Siwy, E. Heins, C. C. Harrell, P. Kohli and C. R. Martin, J. Am. Chem. Soc., 2004, 126, 10850–10851 CrossRef CAS PubMed.
- F. C. Leinweber and U. Tallarek, Langmuir, 2004, 20, 11637–11648 CrossRef CAS PubMed.
- T. C. Kuo, D. M. Cannon Jr., Y. Chen, J. J. Tulock, M. A. Shannon, J. V. Sweedler and P. W. Bohn, Anal. Chem., 2003, 75, 1861–1867 CrossRef CAS PubMed.
- J. Dai, T. Ito, L. Sun and R. M. Crooks, J. Am. Chem. Soc., 2003, 125, 13026–13027 CrossRef CAS PubMed.
- R. Dhopeshwarkar, R. M. Crooks, D. Hlushkou and U. Tallarek, Anal. Chem., 2008, 80, 1039–1048 CrossRef CAS PubMed.
- D. Hlushkou, R. Dhopeshwarkar, R. M. Crooks and U. Tallarek, Lab Chip, 2008, 8, 1153–1162 RSC.
- S. J. Kim, L. D. Li and J. Han, Langmuir, 2009, 25, 7759–7765 CrossRef CAS PubMed.
- T. A. Zangle, A. Mani and J. G. Santiago, Chem. Soc. Rev., 2010, 39, 1014–1035 RSC.
- S. J. Kim, Y.-A. Song and J. Han, Chem. Soc. Rev., 2010, 39, 912–922 RSC.
- M. M. Gong, P. Zhang, B. D. MacDonald and D. Sinton, Anal. Chem., 2014, 86, 8090–8097 CrossRef CAS PubMed.
- R.-J. Yang, H.-H. Pu and H.-L. Wang, Biomicrofluidics, 2015, 9, 014122 Search PubMed.
- M. M. Gong, R. Nosrati, M. C. San Gabriel, A. Zini and D. Sinton, J. Am. Chem. Soc., 2015, 137, 13913–13919 CrossRef CAS PubMed.
- S. J. Kim, S. H. Ko, K. H. Kang and J. Han, Nat. Nanotechnol., 2010, 5, 297–301 CrossRef CAS PubMed.
- K. N. Knust, D. Hlushkou, R. K. Anand, U. Tallarek and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 8107–8110 CrossRef CAS PubMed.
- B. D. MacDonald, M. M. Gong, P. Zhang and D. Sinton, Lab Chip, 2014, 14, 681–685 RSC.
- J. Choi, K. Huh, D. J. Moon, H. Lee, S. Y. Son, K. Kim, H. C. Kim, J. H. Chae, G. Y. Sung, H. Y. Kim, J. W. Hong and S. J. Kim, RSC Adv., 2015, 5, 66178–66184 RSC.
- L. F. Cheow, A. Sarkar, S. Kolitz, D. Lauffenburger and J. Han, Anal. Chem., 2014, 86, 7455–7462 CrossRef CAS PubMed.
- R. K. Perdue, D. R. Laws, D. Hlushkou, U. Tallarek and R. M. Crooks, Anal. Chem., 2009, 81, 10149–10155 CrossRef CAS PubMed.
- M. Kim, M. Jia and T. Kim, Analyst, 2013, 138, 1370–1378 RSC.
- R. K. Anand, E. Sheridan, K. N. Knust and R. M. Crooks, Anal. Chem., 2011, 83, 2351–2358 CrossRef CAS PubMed.
- S. E. Fosdick, K. N. Knust, K. Scida and R. M. Crooks, Angew. Chem., Int. Ed., 2013, 52, 10438–10456 CrossRef CAS PubMed.
- E. Sheridan, D. Hlushkou, K. N. Knust, U. Tallarek and R. M. Crooks, Anal. Chem., 2012, 84, 7393–7399 CrossRef CAS PubMed.
- K. N. Knust, E. Sheridan, R. K. Anand and R. M. Crooks, Lab Chip, 2012, 12, 4107–4114 RSC.
- Y. C. Wang, A. L. Stevens and J. Han, Anal. Chem., 2005, 77, 4293–4299 CrossRef CAS PubMed.
- M. Shen, H. Yang, V. Sivagnanam and M. A. Gijs, Anal. Chem., 2010, 82, 9989–9997 CrossRef CAS PubMed.
- H. Yang, M. Shen, V. Sivagnanam and M. Gijs, Solid-State Sensors, Actuators and Microsystems Conference (TRANSDUCERS), 2011 16th International, 2011, 238–241.
- D. W. Zhang, H. Q. Zhang, L. Tian, L. Wang, F. Fang, K. Liu and Z. Y. Wu, Microfluid. Nanofluid., 2013, 14, 69–76 CrossRef CAS.
- B. Kim, J. Heo, H. J. Kwon, S. J. Cho, J. Han, S. J. Kim and G. Lim, ACS Nano, 2013, 7, 740–747 CrossRef CAS PubMed.
- J. Mai, H. Miller and A. V. Hatch, ACS Nano, 2012, 6, 10206–10215 CrossRef CAS PubMed.
- L. H. Yeh, M. Zhang, S. Qian, J. P. Hsu and S. Tseng, J. Phys. Chem. C, 2012, 116, 8672–8677 CAS.
- J. H. Lee, B. D. Cosgrove, D. A. Lauffenburger and J. Han, J. Am. Chem. Soc., 2009, 131, 10340–10341 CrossRef CAS PubMed.
- L. F. Cheow and J. Han, Anal. Chem., 2011, 83, 7086–7093 CrossRef CAS PubMed.
- S. H. Ko, Y. A. Song, S. J. Kim, M. Kim, J. Han and K. H. Kang, Lab Chip, 2012, 12, 4472–4482 RSC.
- P. Dextras, K. R. Payer, T. P. Burg, W. J. Shen, Y. C. Wang, J. Y. Han and S. R. Manalis, J. Microelectromech. Syst., 2011, 20, 221–230 CrossRef CAS.
- C. H. Chen, A. Sarkar, Y. A. Song, M. A. Miller, S. J. Kim, L. G. Griffith, D. A. Lauffenburger and J. Han, J. Am. Chem. Soc., 2011, 133, 10368–10371 CrossRef CAS PubMed.
- S. Zheng, H. Lin, J. Q. Liu, M. Balic, R. Datar, R. J. Cote and Y. C. Tai, J. Chromatogr., A, 2007, 1162, 154–161 CrossRef CAS PubMed.
- A. C. Roque, C. S. Silva and M. A. Taipa, J. Chromatogr., A, 2007, 1160, 44–55 CrossRef CAS PubMed.
- H. Song, Y. Wang, C. Garson and K. Pant, Microfluid. Nanofluid., 2014, 17, 693–699 CrossRef CAS PubMed.
- D. R. Laws, D. Hlushkou, R. K. Perdue, U. Tallarek and R. M. Crooks, Anal. Chem., 2009, 81, 8923–8929 CrossRef CAS PubMed.
- F. Mavré, R. K. Anand, D. R. Laws, K.-F. Chow, B.-Y. Chang, J. A. Crooks and R. M. Crooks, Anal. Chem., 2010, 82, 8766–8774 CrossRef PubMed.
- P. Mao and J. Han, Lab Chip, 2009, 9, 586–591 RSC.
- A. Balducci, P. Mao, J. Y. Han and P. S. Doyle, Macromolecules, 2006, 39, 6273–6281 CrossRef CAS.
- M. Yamada, P. Mao, J. Fu and J. Han, Anal. Chem., 2009, 81, 7067–7074 CrossRef CAS PubMed.
- D. Huh, K. L. Mills, X. Zhu, M. A. Burns, M. D. Thouless and S. Takayama, Nat. Mater., 2007, 6, 424–428 CrossRef CAS PubMed.
- S. Chung, J. H. Lee, M.-W. Moon, J. Han and R. D. Kamm, Adv. Mater., 2008, 20, 3011–3016 CrossRef CAS.
- S. J. Kim and J. Han, Anal. Chem., 2008, 80, 3507–3511 CrossRef CAS PubMed.
- S. M. Park, Y. S. Huh, H. G. Craighead and D. Erickson, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15549–15554 CrossRef CAS PubMed.
- J. Heo, H. J. Kwon, H. Jeon, B. Kim, S. J. Kim and G. Lim, Nanoscale, 2014, 6, 9681–9688 RSC.
- Z. Slouka, S. Senapati and H. C. Chang, Annu. Rev. Anal. Chem., 2014, 7, 317–335 CrossRef CAS PubMed.
- H. C. Chang, G. Yossifon and E. A. Demekhin, Annu. Rev. Fluid. Mech., 2012, 44, 401–426 CrossRef.
- G. Yossifon and H. C. Chang, Phys. Rev. Lett., 2008, 101, 254501 CrossRef PubMed.
- A. Mani, T. A. Zangle and J. G. Santiago, Langmuir, 2009, 25, 3898–3908 CrossRef CAS PubMed.
- T. A. Zangle, A. Mani and J. G. Santiago, Langmuir, 2009, 25, 3909–3916 CrossRef CAS PubMed.
- A. Syed, L. Mangano, P. Mao, J. Han and Y.-A. Song, Lab Chip, 2014, 14, 4455–4460 RSC.
- Y. Zeng and D. J. Harrison, Anal. Chem., 2007, 79, 2289–2295 CrossRef CAS PubMed.
- O. Schepelina and I. Zharov, Langmuir, 2007, 23, 12704–12709 CrossRef CAS PubMed.
- J. J. Smith and I. Zharov, Langmuir, 2008, 24, 2650–2654 CrossRef CAS PubMed.
- O. Schepelina and I. Zharov, Langmuir, 2008, 24, 14188–14194 CrossRef CAS PubMed.
- B. Kim, J. Heo, H. J. Kwon, I. Kim, T. An and G. Lim, Jpn. J. Appl. Phys., 2015, 54, 035102 CrossRef.
- E. T. Thostenson, Z. F. Ren and T. W. Chou, Compos. Sci. Technol., 2001, 61, 1899–1912 CrossRef CAS.
- D. Vairavapandian, P. Vichchulada and M. D. Lay, Anal. Chim. Acta, 2008, 626, 119–129 CrossRef CAS PubMed.
- J. J. Gooding, Electrochim. Acta, 2005, 50, 3049–3060 CrossRef CAS.
- K. Gong, Y. Yan, M. Zhang, L. Su, S. Xiong and L. Mao, Anal. Sci., 2005, 21, 1383–1393 CrossRef CAS PubMed.
- K. A. Mauritz and R. B. Moore, Chem. Rev., 2004, 104, 4535–4585 CrossRef CAS PubMed.
- D. Choi, A. Choi and D. S. Kim, Int. J. Precis. Eng. Man., 2015, 16, 1467–1471 CrossRef.
- D. T. Phan, Y. Chun and N. T. Nguyen, RSC Adv., 2015, 5, 44336–44341 RSC.
- R. Kwak, J. Y. Kang and T. S. Kim, Anal. Chem., 2016, 88, 988–996 CrossRef PubMed.
- Y. Oh, H. Lee, S. Y. Son, S. J. Kim and P. Kim, Biomicrofluidics, 2016, 10, 014102 CrossRef PubMed.
- Y. N. Xia, E. Kim and G. M. Whitesides, Chem. Mater., 1996, 8, 1558–1567 CrossRef CAS.
- N. Sahiner, M. Singh, D. De Kee, V. T. John and G. L. McPherson, Polymer, 2006, 47, 1124–1131 CrossRef CAS.
- J. R. Saunders, S. Abu-Salih, T. Khaleque, S. Hanula and W. Moussa, J. Comput. Theor. Nanosci., 2008, 5, 1942–1960 CrossRef CAS.
- A. W. Martinez, S. T. Phillips, G. M. Whitesides and E. Carrilho, Anal. Chem., 2010, 82, 3–10 CrossRef CAS PubMed.
- X. Li, D. R. Ballerini and W. Shen, Biomicrofluidics, 2012, 6, 011301 CrossRef PubMed.
- A. K. Yetisen, M. S. Akram and C. R. Lowe, Lab Chip, 2013, 13, 2210–2251 RSC.
- A. W. Martinez, S. T. Phillips, M. J. Butte and G. M. Whitesides, Angew. Chem., Int. Ed., 2007, 46, 1318–1320 CrossRef CAS PubMed.
- A. W. Martinez, S. T. Phillips, E. Carrilho, S. W. Thomas 3rd, H. Sindi and G. M. Whitesides, Anal. Chem., 2008, 80, 3699–3707 CrossRef CAS PubMed.
- C. Renault, M. J. Anderson and R. M. Crooks, J. Am. Chem. Soc., 2014, 136, 4616–4623 CrossRef CAS PubMed.
- D. M. Cate, J. A. Adkins, J. Mettakoonpitak and C. S. Henry, Anal. Chem., 2015, 87, 19–41 CrossRef CAS PubMed.
- X. Liu, C. Cheng, A. Martinez, K. Mirica, X. Li, S. Phillips, M. Mascarenas and G. Whitesides, Micro Electro Mechanical Systems (MEMS), 2011 IEEE 24th International Conference, 2011, 75–78.
- B. Lutz, T. Liang, E. Fu, S. Ramachandran, P. Kauffman and P. Yager, Lab Chip, 2013, 13, 2840–2847 RSC.
- X. Li, P. Zwanenburg and X. Liu, Lab Chip, 2013, 13, 2609–2614 RSC.
- D. T. Phan, S. A. M. Shaegh, C. Yang and N. T. Nguyen, Sens. Actuators, B, 2016, 222, 735–740 CrossRef CAS.
- R. J. Meagher, J. I. Won, L. C. McCormick, S. Nedelcu, M. M. Bertrand, J. L. Bertram, G. Drouin, A. E. Barron and G. W. Slater, Electrophoresis, 2005, 26, 331–350 CrossRef CAS PubMed.
- E. M. Southern, J. Mol. Biol., 1975, 98, 503–517 CrossRef CAS PubMed.
- G. F. Carle and M. V. Olson, Nucleic Acids Res., 1984, 12, 5647–5664 CrossRef CAS PubMed.
- J. Narayanan, J.-Y. Xiong and X.-Y. Liu, J. Phys.: Conf. Ser., 2006, 28, 83–86 CrossRef CAS.
- R. J. Meagher, J. I. Won, J. A. Coyne, J. Lin and A. E. Barron, Anal. Chem., 2008, 80, 2842–2848 CrossRef CAS PubMed.
- H. Song, Y. Wang, C. Garson and K. Pant, Anal. Methods, 2015, 7, 1273–1279 RSC.
- R. K. Anand, E. Sheridan, D. Hlushkou, U. Tallarek and R. M. Crooks, Lab Chip, 2011, 11, 518–527 RSC.
- Z. Cao and L. Yobas, Electrophoresis, 2013, 34, 1991–1997 CrossRef CAS PubMed.
- I. Dumitrescu, R. K. Anand, S. E. Fosdick and R. M. Crooks, J. Am. Chem. Soc., 2011, 133, 4687–4689 CrossRef CAS PubMed.
- K. Scida, E. Sheridan and R. M. Crooks, Lab Chip, 2013, 13, 2292–2299 RSC.
- M. Bercovici, G. V. Kaigala and J. G. Santiago, Anal. Chem., 2010, 82, 2134–2138 CrossRef CAS PubMed.
- T. K. Khurana and J. G. Santiago, Anal. Chem., 2008, 80, 6300–6307 CrossRef CAS PubMed.
- J. Quist, K. G. Janssen, P. Vulto, T. Hankemeier and H. J. van der Linden, Anal. Chem., 2011, 83, 7910–7915 CrossRef CAS PubMed.
- S. H. Ko, S. J. Kim, L. F. Cheow, L. D. Li, K. H. Kang and J. Han, Lab Chip, 2011, 11, 1351–1358 RSC.
- Y. C. Wang and J. Han, Lab Chip, 2008, 8, 392–394 RSC.
- Q. Shi, L. Qin, W. Wei, F. Geng, R. Fan, Y. S. Shin, D. Guo, L. Hood, P. S. Mischel and J. R. Heath, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 419–424 CrossRef CAS PubMed.
- K. Eyer, P. Kuhn, C. Hanke and P. S. Dittrich, Lab Chip, 2012, 12, 765–772 RSC.
- C. Qian, H. Huang, L. Chen, X. Li, Z. Ge, T. Chen, Z. Yang and L. Sun, Int. J. Mol. Sci., 2014, 15, 18281–18309 CrossRef CAS PubMed.
- S. Shim, K. Stemke-Hale, J. Noshari, F. F. Becker and P. R. Gascoyne, Biomicrofluidics, 2013, 7, 11808 CrossRef PubMed.
- R. Martinez-Duarte, Electrophoresis, 2012, 33, 3110–3132 CrossRef CAS PubMed.
- R. Martinez-Duarte, P. Renaud and M. J. Madou, Electrophoresis, 2011, 32, 2385–2392 CAS.
- Y. Jia, Y. Ren and H. Jiang, Electrophoresis, 2015, 36, 1744–1753 CrossRef CAS PubMed.
- S. Zeinali, B. Cetin, S. N. Oliaei and Y. Karpat, Electrophoresis, 2015, 36, 1432–1442 CrossRef CAS PubMed.
- R. K. Anand, E. S. Johnson and D. T. Chiu, J. Am. Chem. Soc., 2015, 137, 776–783 CrossRef CAS PubMed.
- A. Nakano, F. Camacho-Alanis and A. Ros, Analyst, 2015, 140, 860–868 RSC.
| This journal is © The Royal Society of Chemistry 2016 |