
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solid-phase microextraction low temperature plasma mass spectrometry for the direct and rapid analysis of chemical warfare simulants in complex mixtures†
Morphy C.
Dumlao
,
Laura E.
Jeffress
,
J. Justin
Gooding
and
William A.
Donald
*
School of Chemistry, University of New South Wales, Sydney, New South Wales 2052, Australia. E-mail: w.donald@unsw.edu.au
First published on 14th March 2016
Abstract
Solid-phase microextraction (SPME) is directly integrated with low temperature plasma ionisation mass spectrometry to rapidly detect organophosphate chemical warfare agent simulants and their hydrolysis products in chemical mixtures, including urine. In this sampling and ionization method, the fibre serves: (i) to extract molecules from their native environment, and (ii) as the ionization electrode that is used to desorb and ionize molecules directly from the SPME surface. By use of a custom fabricated SPME fibre consisting of a stainless steel needle coated with a Linde Type A (LTA) zeolitic microporous material and low temperature plasma mass spectrometry, protonated dimethyl methylphosphonate (DMMP), diethyl ethylphosphonate (DEEP) and pinacolyl methylphosphonic acid (PinMPA) can be detected at less than 100 ppb directly in water and urine. Organophosphates were not readily detected by this approach using an uncoated needle in negative control experiments. The use of the LTA coating significantly outperformed the use of a high alumina Zeolite Socony Mobil-5 (ZSM-5) coating of comparable thickness that is significantly less polar than LTA. By conditioning the LTA probe by immersion in an aqueous CuSO4 solution, the ion abundance for protonated DMMP increased by more than 300% compared to that obtained without any conditioning. Sample recovery values were between 96 and 100% for each analyte. The detection of chemical warfare agent analogues and hydrolysis products required less than 2 min per sample. A key advantage of this sampling and ionization method is that analyte ions can be directly and rapidly sampled from chemical mixtures, such as urine and seawater, without sample preparation or chromatography for sensitive detection by mass spectrometry. This ion source should prove beneficial for portable mass spectrometry applications because relatively low detection limits can be obtained without the use of compressed gases, fluid pumps, and lasers. Moreover, the ion source is compact, can be powered with a 10 V battery, and is tolerant of complex mixtures.
Introduction
The Organization for the Prohibition of Chemical Weapons (OPCW) has mandated by the Chemical Weapon Convention to prohibit the development, production, stockpiling, transfer and use of chemical warfare agents.1 There are many classes of chemical warfare agents, including blister agents (mustard gas), choking agents (phosgene), blood agents (arsines), and nerve agents. The latter are synthetic organophosphate neurotoxins (e.g., Fig. 1, 1–3) that are highly potent, volatile, and colourless liquids which are challenging to detect until symptoms appear. For example, Sarin (or GB, 2) is amongst the most powerful inhibitors of the acetylcholinesterase enzyme. For humans, direct inhalation of Sarin at a dose of ca. 35 μg min L−1 can cause death in 1 to 10 min owing to asphyxiation resulting from the loss of muscle control that is implicated in the accumulation of acetylcholine upon inhibition of acetylcholinesterase.2 A recent example of the ongoing threat of chemical weapons occurred in the chemical attack on Ghouta during the ongoing Syrian Civil War.3 A United Nations report issued nearly a month later confirmed the use of GB as the neurotoxin responsible for the deaths of approximately 1000 civilians. The unknown cause of the symptoms displayed by the victims may have contributed to the confusion and panic in the aftermath of the attack. Thus, there is a need for reliable real-time methods for detecting chemical warfare agent exposure to assist first responders and authorities in rapidly implementing medical treatments and environmental containment measures.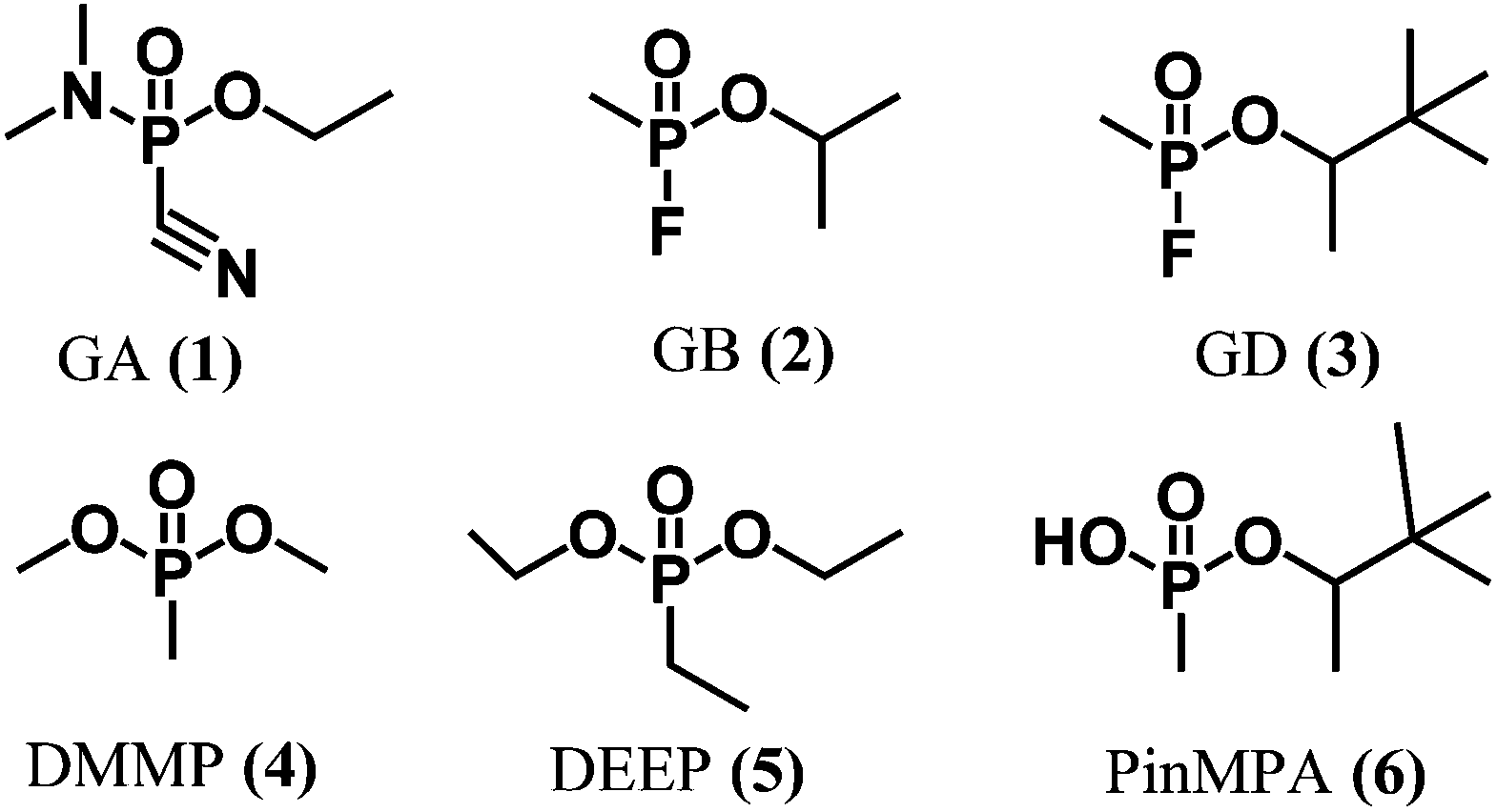 | ||
| Fig. 1 Chemical structures of chemical warfare agents (1–3), simulants (4–5), and a hydrolysis product (6). | ||
There are many different types of chemical sensors and instruments that are commercially available for the detection and identification of chemical warfare agents, including ion-mobility spectrometry,4 surface-acoustic wave devices,5 infrared spectroscopy,6 and colorimetry.6 The key limitations of many such devices include the high false positive rates and the challenge in detecting many different analytes nearly simultaneously from complex mixtures, such as bodily fluids. Thus, samples are generally analysed at OPCW-certified laboratories for more rigorous chemical analysis for confirming the presence and determining the concentrations of chemical warfare agents.
Mass spectrometry is highly sensitive, selective, and can be used to detect many different chemical warfare agents and their degradation products nearly simultaneously from many different types of samples, including complex chemical mixtures. For chemical warfare agent confirmation, such analyses are generally performed by use of gas-chromatography mass spectrometry (GC/MS)7,8 and liquid chromatography tandem mass spectrometry (LC/MS/MS).9 Based on standard GC/MS and LC/MS/MS10 protocols for chemical warfare agent detection in common bodily fluids such as urine, sample preparation and analysis requires several hours and the minimum detection thresholds are less than 1 ppm. Thus, there is a clear need to develop methods for rapidly detecting chemical warfare agents and their degradation products directly from complex mixtures in near real-time using mass spectrometry.
Ambient ionization mass spectrometry methods11 are useful for rapidly and directly forming ions from samples in their native in situ environment without sample preparation and chromatography. Ambient ionisation methods can be categorized as being based on plasma,12,13 spray,14 and laser desorption ionisation.15 A key challenge with all ionization techniques is matrix ion suppression, in which molecules from the sample matrix can compete for charge with the analyte molecules to significantly reduce the analytical performance of the mass spectrometer. Plasma based ion sources, such as direct analysis in real time,16 plasma-assisted desorption ionisation,17 low temperature plasma,18,19 active capillary ionization,20 and other novel ion sources are gaining importance for the detection of volatile molecules directly from complex mixtures partly because plasma sources: (i) do not require solvent pumps and lasers like spray and laser based ion sources; and (ii) plasma based ion sources are compact and are thus well suited for integration with portable mass spectrometers. More recently, active capillary plasma ionization is particularly attractive because the use of compressed gases in the plasma ion source is eliminated12 and the extent of internal energy deposited upon ion formation, transfer and detection is relatively low.21 Although plasma ion sources tend to be less susceptible to matrix ion suppression than spray and laser based ion sources,22 the analytical performance of such sources can be detrimentally affected by complex sample matrices, such as urine.
An important strategy to rapidly reduce matrix effects in chromatographic based methods is solid-phase microextraction (SPME),23 in which a sampling fibre that is on the order of micrometre thickness and contains a specific surface coating is used to extract analyte molecules from a chemical mixture prior to analysis. However, the use of SPME in combination with ambient mass spectrometry is limited. Here, our approach is to develop a portable and compact ionization source that directly integrates SPME and low temperature plasma mass spectrometry (Fig. 2). Custom integrated SPME fibres and ionization electrodes were fabricated by coating stainless steel needles with zeolitic microporous materials. Using this approach, G-series nerve agent simulants and common hydrolysis products can be rapidly extracted from complex mixtures, ionized and detected by mass spectrometry in two simple steps: (i) directly inserting the probe into the mixture for <1 min, and (ii) inserting the SPME probe and electrode into a low temperature plasma. This ion source has the advantages that chemical warfare agents can be rapidly sampled and detected directly from complex mixtures, including urine, in less than 2 minutes with low limits of detection (<100 ppb) that are well within OPCW proficiency guidelines (<10 ppm).24
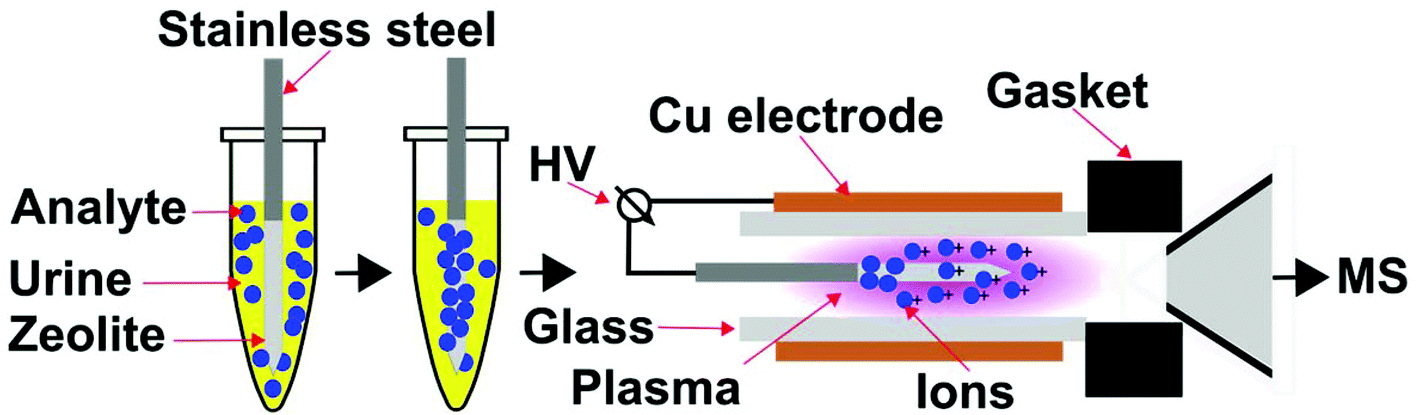 | ||
| Fig. 2 Schematic diagram of solid-phase microextraction low temperature plasma mass spectrometry. Upon sampling, the SPME fibre is inserted into a low temperature plasma ionisation chamber and serves as an ionization electrode. A high voltage (HV) alternating current is applied between the stainless steel electrode and the Cu electrode to initiate and maintain the dielectric barrier discharge low temperature ion plasma. Ambient gases are introduced continuously into the ion plasma owing to suction from the vacuum interface of the mass spectrometer. | ||
Experimental methods
Materials and reagents
The neurotoxin simulants and hydrolysis product were purchased from Alfa Aesar (Ward Hill, MA, USA) for dimethyl methylphosphonate (DMMP) (97%) and from Sigma Aldrich (St. Louis, MO, USA) for diethyl ethylphosphonate (DEEP) (>98%), and pinacolyl methylphosphonic acid (PinMPA) (97%). Reagent grade sodium hydroxide pellets, sodium aluminate, sodium silicate, silicic acid and 20% solution of tetrapropylammonium hydroxide (TPA-OH solution) were obtained from Sigma Aldrich. Sodium chloride and copper sulphate were obtained from Ajax Finechem (Sydney, Australia). All of these chemicals were used without further purification. Acupuncture needles (o.d. 0.30 mm, length 40 mm; Sensei) were purchased from Acuneeds (Melbourne, VIC, Australia). UltraLime soap was purchased from Septone (Dandenong South, VIC, Australia). Sea water (Bondi Beach, Sydney, Australia) and rain water (Kensington, NSW, Australia) matrices were collected and kept at 4 °C when not in use. Urine matrix, a synthetic urine negative control, was purchased from Cerilliant (Novachem, Collingwood, VIC, Australia) and also kept at 4 °C when not in use. Chemical warfare agent simulants and hydrolysis products were used in this study owing to safety and regulatory purposes.Surface immobilisation of zeolites and characterisation
To fabricate integrated solid phase microextraction probes and ion emitter electrodes, zeolites were immobilised onto stainless steel acupuncture needles using a modified procedure reported by McDonnell et al.25 The stainless steel needles were cleaned by immersion in a soap solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, water:UltraLime) for 1 hour at 80 °C. The needles were rinsed with deionized water and dried in air. An autoclave (Amar Equipment, Mumbai, India) was fitted with a custom Teflon liner that can position up to 16 needles vertically. Needles were immersed ca. 1 cm vertically into a zeolite gel in the autoclave, which was sealed. Zeolite precursor gels were prepared for Linde Type A and high alumina ZSM-5.26 To prepare the Linde Type A precursor gel, deionized water, sodium hydroxide, sodium aluminate, and sodium silicate were mixed for 4 h until homogenized (3.165 Na2O:1 Al2O3:1.926 SiO2:128 H2O). To fabricate Linde Type A coated sampling probes, the precursor gel was heated in the autoclave for 24 h at 100 °C in the presence of the substrate stainless steel needles that were immersed in the gel solution. After synthesis, the probe needles and crystals were washed with deionized water until the pH of the deionized water was 9 pH units. The recovered samples and ion emitter probes were dried at 80 °C overnight.
:1 v/v, water:UltraLime) for 1 hour at 80 °C. The needles were rinsed with deionized water and dried in air. An autoclave (Amar Equipment, Mumbai, India) was fitted with a custom Teflon liner that can position up to 16 needles vertically. Needles were immersed ca. 1 cm vertically into a zeolite gel in the autoclave, which was sealed. Zeolite precursor gels were prepared for Linde Type A and high alumina ZSM-5.26 To prepare the Linde Type A precursor gel, deionized water, sodium hydroxide, sodium aluminate, and sodium silicate were mixed for 4 h until homogenized (3.165 Na2O:1 Al2O3:1.926 SiO2:128 H2O). To fabricate Linde Type A coated sampling probes, the precursor gel was heated in the autoclave for 24 h at 100 °C in the presence of the substrate stainless steel needles that were immersed in the gel solution. After synthesis, the probe needles and crystals were washed with deionized water until the pH of the deionized water was 9 pH units. The recovered samples and ion emitter probes were dried at 80 °C overnight.
For high alumina ZSM-5 synthesis, a seeding gel was prepared using deionized water, sodium hydroxide, TPA-OH, silicic acid and sodium aluminate (3.25 Na2O:1 Al2O3:30 SiO2:958 H2O). The resulting solution was mixed for one hour at ambient temperature and aged for 16 h at 100 °C. A separate synthesis gel was prepared by dissolving 8.8 g sodium hydroxide and 10.3 g sodium aluminate in 867.8 g water and mixed thoroughly. 113.1 g silicic acid was added to the separate synthesis gel while stirring and shaking vigorously for one hour at ambient temperature. The latter gel was added to 50 g of seeding gel and shaken for an additional hour. The final gel solution was heated for 40 h at 150 °C along with the stainless steel substrate needles that were vertically immersed in the solution. After synthesis, the needles and bulk samples were removed, washed with deionized water, and dried at 105 °C for 24 h.
To examine the morphology and estimate the thickness of surface coatings, a scanning electron microscope (SEM) with a secondary electron detector (S3400, Hitachi, Japan; operated at 20 kV) was used. The Si/Al ratio of the crystals, an important zeolitic characteristic, was measured by energy dispersive spectroscopy (EDS) SEM (QUANTAX, Bruker, Germany). X-ray photoelectron spectra were collected using an Escalab 250Xi X-ray photoelectron spectrometer (Thermo Scientific) with a mono-chromated Al K X-ray source. The data were obtained at room temperature and the operating pressure in the analysis chamber was below 2 × 10−9 mbar. The spot size was ca. 500 μm from the tip of the probe and the photoelectron take-off angle was 90°. The X-ray diffraction (XRD) patterns of zeolite powders were obtained using a PANalytical X'pert Multipurpose X-ray Diffraction System with Cu Kα monochromatic radiation (45 kV and 40 mA). The XRD patterns were identified and matched with the library using the PANalytical X'Pert HighScore Plus software (2015 version).
Low-temperature plasma ionization mass spectrometry
All mass spectrometry experiments used a custom low temperature plasma ionization source21 (Fig. 2) that was directly integrated with a fabricated solid-phase microextraction sample probe, which also served as an ionization electrode. The low temperature plasma source consisted of a quartz glass capillary (i.d. 1.5 mm, o.d. 1.8 mm, ca. 2.5 cm) that is surrounded by an outer Cu electrode (i.d. 1.9 mm, o.d. 2.5 mm, 1.5 cm). The inner electrode, a zeolite coated stainless steel solid-phase microextraction probe, was inserted on axis into the glass capillary. The glass capillary was positioned on axis with the inlet of mass spectrometer and sealed using a Swagelok fitting to ensure that the vacuum interface of the mass spectrometer delivers ambient gases to the plasma ion source by suction. That is, no compressed gases are required for this plasma ion source, which is based on the “active capillary” design of Zenobi and co-workers.12,20 The distance between the glass capillary and the capillary entrance to the mass spectrometer was 1 cm. The inner electrode was mounted on a 3D-positioning platform and the tip of the inner electrode was inserted 0.75 cm within the outer Cu electrode. To form a low temperature plasma, a 2 kV and 10 kHz square wave was applied between the inner stainless steel and Cu electrodes. The square wave was generated from a full bridge step-up transformer (6:1200) using 10 VDC input. To measure the temperature of the plasma (31.3 °C), the plasma was directed onto a probe thermometer (Fluke 62 Max, China). The thermometer was calibrated using an equilibrated ice-water bath (0 °C) and boiling water (100 °C; Milli-Q grade water). Mass spectra were obtained using a linear quadrupole ion trap mass spectrometer (LTQ XL, Thermo Scientific, San Jose, USA) that was operated in positive ion mode with a capillary temperature of 150 °C, capillary voltage of 13 to 25 V, and a tube lens voltage of 40 to 55 V. The humidity of the ambient air (55–57%) that maintains the continuous ion plasma was monitored regularly. For comparison of LTP-MS performance for different SPME surfaces and sampling matrices, mass spectrometry results were obtained on the same day. 10 mM stock solutions of organophosphate analytes in HPLC-grade methanol were stored at 4 °C when not in use. For experiments, stock solutions were diluted with deionized water (18 MΩ, Milli-Q, Merck Millipore, USA) or matrix solutions (e.g., urine). The diluted solutions were used immediately to minimise hydrolysis of the analytes.
For sampling, the solid-phase microextraction and low temperature plasma ionisation probes were immersed in 10 μL of a washing solution for one minute, and immersed in an aqueous 5% CuSO4 conditioning solution for one minute unless specified otherwise. The SPME probe was positioned in the low temperature plasma ion source and a high voltage was applied to ionize the molecules (see above; Fig. 2). At least three replicate runs were performed for each sample. A subsequent low temperature plasma mass spectrum was obtained after washing to ensure that there was essentially no carry over between measurements. Limits of detection (LOD) were obtained from linear regression calibration curves and eqn (1),
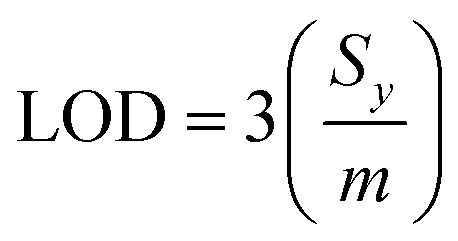 | (1) |
Results and discussion
Surface characterization
Heating stainless steel needles (300 μm in diameter) immersed vertically in a Linde Type A precursor gel enclosed in an autoclave at 100 °C for 24 h resulted in the immobilization of a 20.0 ± 0.2 μm layer of relatively evenly distributed material onto the stainless steel needles as measured by use of SEM (Fig. 3). Under these conditions, the thickness of the coating did not depend significantly as a function of needle depth (<5%) except at the tip of the needles (radial thickness of 30 ± 8 μm; 5 synthesis replicates). Well defined crystals with diameters of less than 5 μm were embedded in aggregates upon the surface (Fig. S1†). By use of energy dispersive SEM (Fig. S2†), the Si/Al ratio was measured to be 1.0 ± 0.3 (3 synthesis replicates), which is expected for Linde Type A zeolite materials (1.0).18,26 X-ray photoelectron spectroscopy of the surface coating resulted in peaks at 74.44, 102.52 and 1077.18 eV (Fig. S3†), which are near the expected values from the literature for Al 2p (73.7 eV), Si 2p (101.7 eV), and Na 1s (1072.0 eV),18,26 respectively. From the relative abundances, the ratio of Si/Al was measured to be 1.2 ± 0.6 (3 synthesis replicates), which is consistent with the expected ratio for Linde Type A18 and corroborates the energy dispersive SEM results (see above). X-ray powder diffraction spectroscopy of the filtered powder recovered from the autoclave resulted in 15 peaks between 0 and 40 degrees (2θ; Fig. S4†). The measured diffraction powder data matched the reference data with a scale factor of 0.9 out of 1.0 using the pattern profile fitting and pattern identification functions of the instrument software (PANalytical X'Pert HighScore Plus program). Overall, these data indicate that Linde Type A can be reproducibility immobilised onto stainless steel needles under these conditions.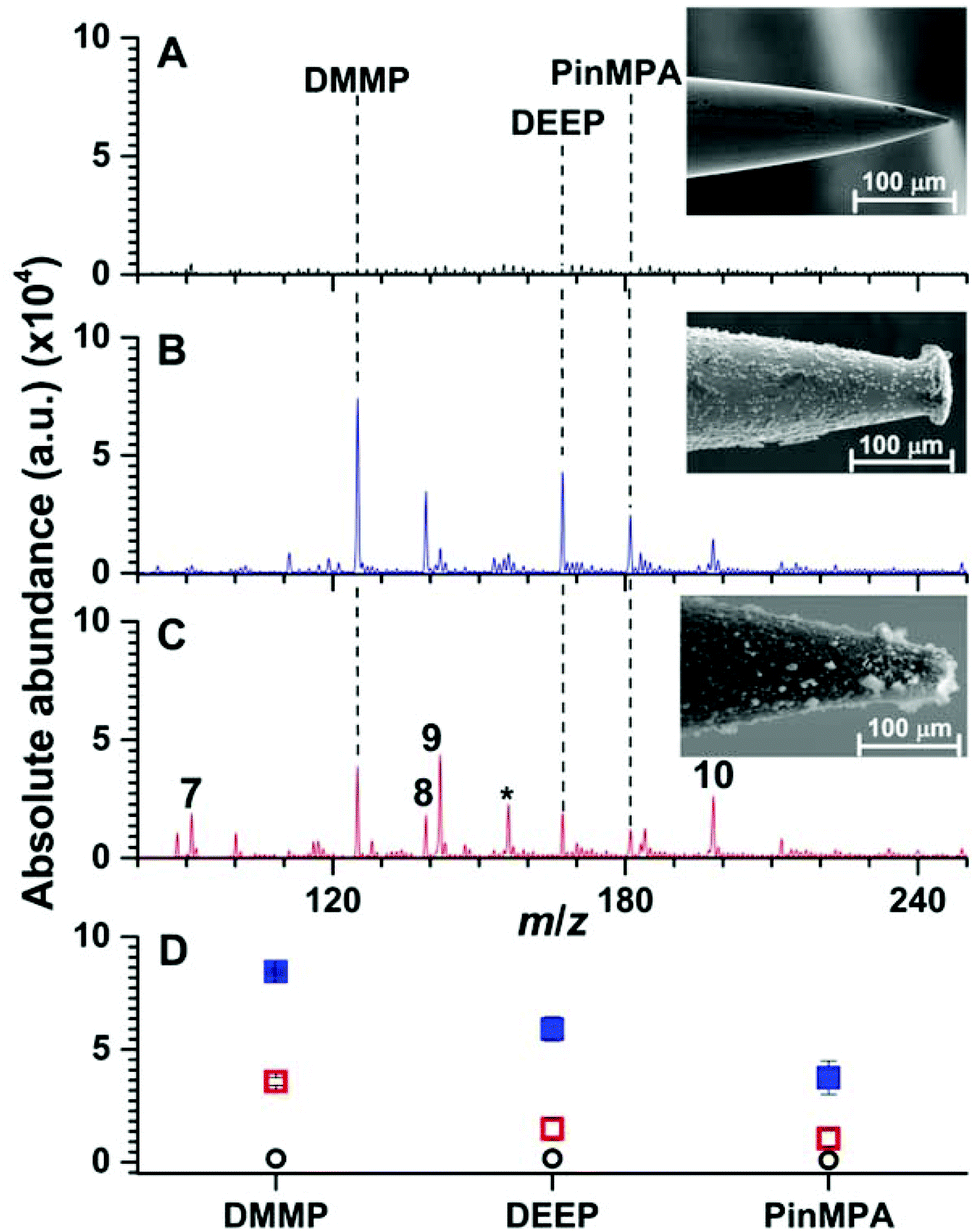 | ||
| Fig. 3 Representative solid phase micro extraction low temperature plasma mass spectra that were obtained by sampling an aqueous solution containing 100 μM of organophosphates 4, 5 and 6 using a (A) uncoated, (B) Linde Type A and (C) high alumina ZSM-5 probe. SEM images of each probe are inset. For the mass spectrum obtained by use of ZSM-5, the following additional peaks were assigned: (7) m/z 91, [H(H2O)5]+; (8) m/z 139, [DEEP-(CH2)2]+; (9) m/z 142, [DMMP,NH4]+; (10) m/z 199 [PinMPA, NH4]+ and (*) chemical noise. Ion abundances are in arbitrary units (a.u.). (D) The average and standard deviation of the ion abundances (5 replicates) for each analyte and surface coating (closed squares correspond to LTA, open squares correspond to ZSM-5, and open circles correspond to the uncoated probes). The solution sampling time was 1 min and the zeolites were conditioned by Cu2+-exchange (see Fig. S9 and S10†). | ||
In Fig. 3, an SEM image of a stainless steel needle that was coated using a high alumina ZSM-5 precursor gel is shown. The radial thickness of the coating is 22.1 ± 0.5 μm and is relatively uniform except at the tips of the coated needles (radial coating thickness of 17 ± 6 μm). Crystal prismatic aggregates of around 5 to 10 μm were formed on the surface (Fig. S5†). The Si/Al ratio of the surface coating were measured to be 13.6 ± 0.3 and 12.8 ± 0.4 by EDS-SEM (Fig. S6†) and XPS (Fig. S7†), respectively, which are near the expected ratio for high alumina ZSM-5 of 13.3.26 XRD (Fig. S8†) of the powder formed during synthesis resulted in more than 7 peaks that match the reference spectrum for high alumina ZSM-5 (score of 0.9 out of 1.0). These data indicate that high alumina ZSM-5 can be reproducibly immobilized onto stainless steel needles.
Solid phase microextraction low temperature plasma mass spectrometry
Representative SPME low temperature plasma mass spectra of aqueous solutions containing 100 μM of organophosphates 4, 5 and 6 (Fig. 1) that were obtained using an uncoated, Linde Type A, and high alumina ZSM-5 coated probe are shown in Fig. 3. Ions corresponding to protonated dimethyl methyl phosphonate (m/z 125), diethyl ethylphosphonate (m/z 167), and pinacolyl methylphosphonic acid (m/z 181) were detected with Linde Type A and high alumina ZSM-5 coated probes in abundances that were at least 1000% higher than that of the uncoated probe. The three protonated organophosphate ions were not detected by use of the Linde Type A and high alumina ZSM-5 coated probes when sampling water that did not contain the organophosphate analytes. These results indicate that zeolite coatings can be effective for extracting organophosphates from aqueous solution for detection by low temperature plasma mass spectrometry. The ion abundance values obtained by SPME low temperature plasma mass spectrometry and the use of Linde Type A coatings were over 100% higher than that obtained by use of ZSM-5 (Fig. 3). High alumina zeolites may outperform lower alumina zeolites in these experiments owing to the higher density of formal negative charges in the Linde Type A zeolitic scaffolds (1:1 Al:Si) compared to ZSM-5 (1:13.5 Al:Si); that is, Linde Type A is a significantly more polar microporous material than ZSM-5. The increased polarity of the Linde Type A scaffold may result in more favourable interactions with the polar organophosphate analytes, yielding more efficient extraction of the analytes from the solution than that for ZSM-5.
Interestingly, the use of ZSM-5 resulted in the formation of an ion series corresponding to the adduction of ammonium to the organophosphates, [M, NH4]+ (M = 4, 5 and 6), that were formed in approximately equal or slightly higher abundances than those for the protonated organophosphates (Fig. 3). Thus, the analyte signal is split into multiple detection channels, which should adversely affect the sensitivity and the detection limits under these conditions (ceteris paribus). In contrast, ammonium adducts were not detected in appreciable abundance by use of a Linde Type A zeolite surface coated probe (Fig. 3). Cu-exchanged ZSM-5 can be used as a catalyst in the reduction of N2 to NOX under plasma conditions at high temperatures.27 In addition, ammonia can be synthesised from N2 and a hydrogen source (e.g., H2 and CH4) in a dielectric barrier discharge reactor.28,29 In our low temperature plasma mass spectrometry experiments, ammonia may possibly originate from nitrogen reduction in an ambient dielectric barrier discharge using a hydrogen source other than H2 or CH4, such as water. Ammonium can also be formed in direct analysis in real time experiments, in which metastable atoms (e.g., N2, O2, H2O) are used to induce ambient ionisation,30 in significantly higher abundance than other plasma based ionisation sources.31 Recently, Bierstedt et al. reported that ammonium water clusters can be readily formed in a dielectric barrier discharge ionisation source operated at relatively high voltages (around 15 kV)32 compared to the lower voltages operated in our experiments (2 kV). For the latter conditions, the temperature of the ion plasma is near ambient temperature (31.3 °C) and benzylammonium thermometer ions with very low barriers to dissociation (≥105 kJ mol−1) can survive ionisation intact for detection by mass spectrometry.21
SPME conditioning and sampling time
Zeolite materials are comprised of a three dimensional scaffold of tetrahedral Al and Si atoms (AlO4 and SiO4 tetrahedra) that are linked together by oxygen atoms shared between adjacent tetrahedra building blocks (Fig. S1 and S5†).33 Although the SiO4 tetrahedra are uncharged, the AlO4 tetrahedra are formally negatively charged and thus are balanced by an extra-framework cation(s) that can be readily exchanged for other positively charged ions. The effect of conditioning the SPME probes by immersion in MilliQ-grade water, aqueous 5% NaCl, or aqueous 5% CuSO4 for 1 min on the performance of low temperature plasma mass spectrometry for the detection of dimethyl methyl phosphonate is shown in Fig. S9.† The solutions containing the ionic species of CuSO4 and NaCl for conditioning the SPME fibre resulted in an ion abundance of protonated dimethyl methyl phosphonate that was 311% and 250% higher, respectively, than that for MilliQ-grade water. Thus, a CuSO4 solution was used to condition the SPME probes for all subsequent experiments. It is hypothesized that exchanging the zeolite framework with Cu2+ may enhance the performance compared to Na+, perhaps due to the stronger Cu2+–phosphonate interactions relative to Na+–phosphonate interactions and thus, improving the extraction efficiency of phosphonates. These data are consistent with the use of Cu ions to enhance the preconcentration of phosphonates onto self-assembled monolayers featuring terminal carboxylate groups from aqueous solutions.34Low temperature plasma ionisation mass spectra obtained using a Linde Type A SPME probe that was immersed in aqueous solutions containing 100 μM of dimethyl methyl phosphonate for 1, 10 and 20 min, respectively, are shown in Fig. S10.† The average (and standard deviation) of the ion abundance for [DMMP + H]+ (5 replicates) increased by 205% as the immersion time increased from 1 to 20 min. Although higher sensitivity can be obtained by use of longer immersion times, a 1 min sampling time was used for all further experiments for rapid data acquisition.
Reproducibility
To assess inter-probe reproducibility, the average ion abundances of protonated DMMP formed from aqueous solutions containing 100 μM DMMP are shown for three different Linde Type A SPME fibres and five sequential sampling cycles in Fig. S11.† Fibres were washed and conditioned by immersion in an aqueous 5% CuSO4 solution for 1 min prior to SPME sampling and low temperature plasma mass spectrometry. The ion abundances were the same or slightly decreased by ca. 10% over the course of at least five sequential sampling experiments. Inter-probe reproducibility was within ±15%. Energy dispersive SEM spectra of the Linde Type A coated probe immediately before use and after 30 sequential measurements are shown in Fig. S12.† The zeolite surfaces were Na+-exchanged by immersion in aqueous solutions of 5% NaCl immediately prior to imaging. For the zeolite surfaces, energy dispersive SEM peaks corresponding to zeolitic elements Na, Al, and Si, along with Cr and Fe from the stainless steel substrate were detected. The peaks corresponding to Na, Al and Si were not detected in the negative control spectrum of the uncoated needle (Fig. S12†). The abundances of the zeolite elemental peaks of the used SPME fibre is lower than that obtained before plasma use by between ca. 5 to 20% and the stainless steel elemental peaks increased in abundance by between 10 and 20%. Overall, these data indicate that the surface coatings are reasonably robust and can be used up to at least 30 times (Fig. S12†) with only a moderate decrease in performance.Rapid and direct analysis of chemical warfare agent simulants and hydrolysis products in complex mixtures
The effects of more complex chemical mixtures on the performance SPME low temperature plasma mass spectrometry were investigated. In Fig. 4, low temperature plasma mass spectra in which a Linde Type A SPME fibre was used to sample <1 μL from 10 μL of solutions containing 100 μM dimethyl methyl phosphonate spiked into water (pH = 7), rainwater (pH = 6), seawater (pH = 8) and synthetic urine (pH = 6) are shown. Peaks corresponding to [DMMP + H]+ and [DEEP + H]+ at m/z 125 and 167 were detected for each matrix with ion abundances that were more than a factor of 20 higher than the “unspiked” samples. A hydrolysis product, [PinMPA + H]+ (181 m/z), was detected at an abundance of more than 100% higher than the unspiked samples of different matrices. That is, the three organophosphate analytes were detected in comparable abundances across the different mixtures. Although the matrices reduce the ion abundances relative to water, these data indicate that SPME low temperature plasma mass spectrometry is relatively tolerant of complex mixtures and matrix ion suppression for these chemical warfare agent simulants and hydrolysis products. This ionisation approach may be relatively tolerant to these complex mixtures for at least two possible reasons: (i) the solid phase microextraction step can pre-concentrate the analyte onto the surface of the SPME probe prior to analysis, and (ii) non-volatile molecules are not sampled because ionization occurs after analyte vaporization in plasma-based ionisation methods.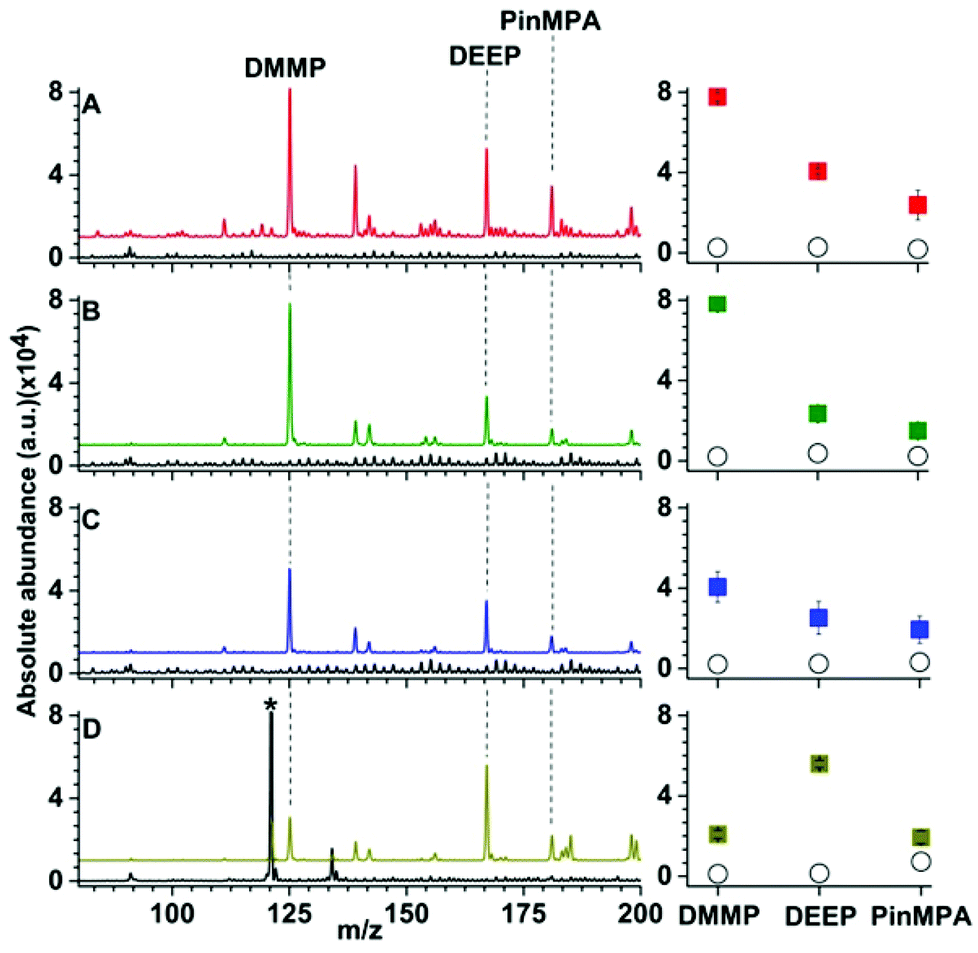 | ||
| Fig. 4 Representative Linde Type A solid-phase microextraction low temperature plasma mass spectra of solutions containing 100 μM of organophosphates 4, 5 and 6 in (A) water, (B) rain water (C) sea water from Bondi beach, and (D) synthetic urine. Ion abundances are in arbitrary units (a.u.). The corresponding mass spectra of the unspiked blank solutions for each matrix are shown in black. The average and standard deviation of the ion abundances (5 replicates) for each analyte for the corresponding mass spectra are shown in the right-most panels (closed squares correspond to LTA and open circles correspond to the uncoated probes). The peak labelled by “*” corresponds to [(urea)2 + H]+. | ||
Calibration curves obtained by SPME low temperature plasma mass spectrometry measurements for the simultaneous detection of organophosphates 4, 5 and 6 spiked in water and urine are shown in Fig. 5. For the calibration curves, each mass spectrum required <2 min of sampling and acquisition time and the three analytes were detected simultaneously. The R2 values of the linear calibration curves were at least 0.999 and the recovery values were 96% or higher for each of the three analytes in both water and urine (Table 1). The limits of detection values were at the low ppb level, which is well within the OPCW Proficiency Testing24 reporting limits.
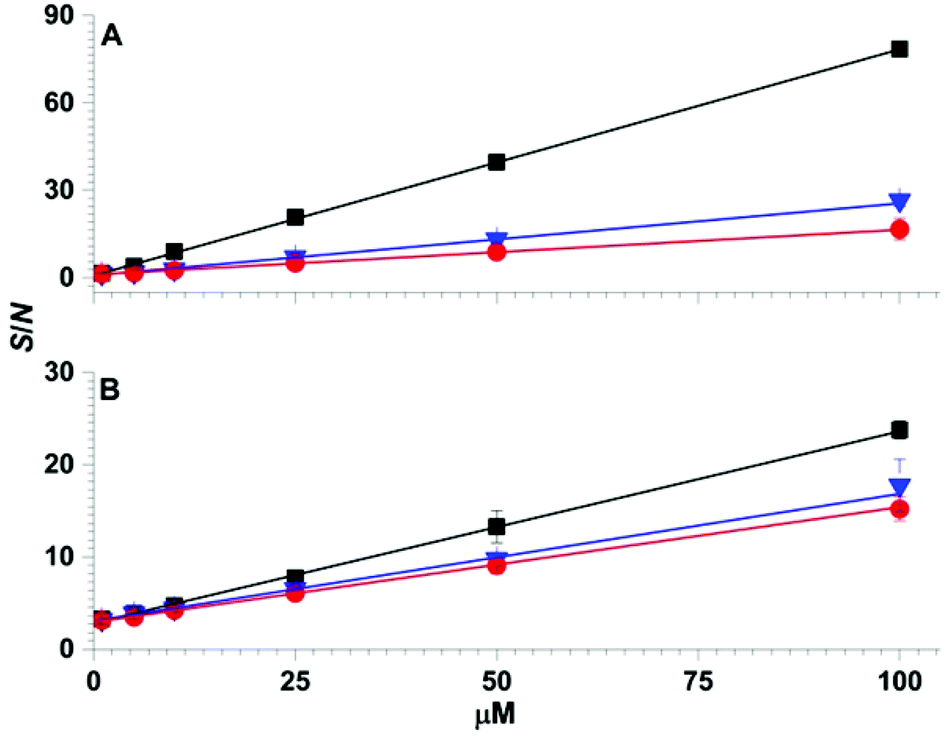 | ||
| Fig. 5 Solid-phase microextraction low temperature plasma mass spectrometry calibration curves obtained by sampling 1 μL of (A) aqueous solutions and (B) synthetic urine containing 1 to 100 μM of organophosphates 4 (squares), 5 (triangles) and 6 (circles). | ||
| Analyte | Water | Urine | ||||
|---|---|---|---|---|---|---|
| LOD (ppb) | R 2 | Recovery | LOD (ppb) | R 2 | Recovery | |
| DMMP | 24.46 ± 0.07 | 0.9998 | 98.0 ± 0.1 | 55.42 ± 0.08 | 0.9997 | 99.9 ± 0.9 |
| DEEP | 31.39 ± 0.13 | 0.9990 | 99.0 ± 0.1 | 27.33 ± 0.38 | 0.9978 | 101.0 ± 0.1 |
| PinMPA | 68.05 ± 0.14 | 0.9998 | 96.0 ± 0.1 | 98.89 ± 0.70 | 0.9999 | 99.9 ± 0.9 |
Conclusions
Coupling solid-phase microextraction to low temperature plasma mass spectrometry using a Linde Type A coated sampling fibre and ionisation electrode yields significant performance gains for the rapid and direct detection of organophosphate analytes directly from complex mixtures, such as urine, compared to the use of high alumina ZSM-5 coatings of approximately the same thicknesses (20 μM). The higher density of cation-exchange sites in Linde Type A relative to that for high alumina ZSM-5 should result in more sites for the coordination and preconcentration of organophosphate analytes in zeolitic microporous materials at metal cation centres. The use of Cu2+-exchanged high alumina ZSM-5 coatings in low temperature plasma mass spectrometry resulted in the formation of both protonated and ammonium adducted organophosphate complex ions, which are presumably an artefact of nitrogen fixation at near ambient temperature (31.3 °C). Ammonium adduction results in the analyte signal being split into an additional detection channel, which should reduce performance for the quantitation of organophosphates under these conditions and increase mass spectral complexity. In contrast, the use of Linde Type A surfaces resulted in the primary formation of protonated organophosphates and ammonium adducts of organophosphates were not detected in appreciable abundances under a wide range of conditions. Although the use of plasma ionisation and Cu2+-exchanged high alumina ZSM-5 may possibly provide a pathway for ammonia synthesis from N2 at ambient temperatures21 and pressures without the addition of H2 or CH428,29 or the application of voltages higher than 6 kV,32 future experiments are required to elucidate the mechanism of ammonium formation under such conditions.
Solid-phase microextraction low temperature plasma mass spectrometry has the advantages that organophosphate analytes can be detected with low ppb detection limits directly in urine, sampling and data acquisition takes less than 2 min, and multiple analytes can be detected nearly simultaneously in a single experiment. The solid-phase microextraction low temperature plasma ion source is compact (<2 cm3); does not involve the use of compressed gases, solvent pumps, and lasers; and can be powered by a 10 V battery. Thus, it is well suited for the direct integration with portable mass spectrometers. Because the temperature of the plasma is relatively low (31.3 °C) and substituted benzylammonium thermometer ions with low barriers to dissociation (≥105 kJ mol−1) can be formed intact with minimal fragmentation,21 this “gentle” sampling and ionisation method should be useful for directly detecting labile molecules such as explosives and other important analytes that contain weak chemical bonds. Moreover, the solid-phase microextraction sampling step could be readily automated using a fibre encased in a protective housing that is spring loaded for field sampling, storage, and transportation to a remote laboratory for analysis. In the future, it should be feasible to selectively extract different analyte classes from a broad range of matrices for rapid chemical analysis by the development of additional ion emitters that are coated with other solid-phase microextraction materials, such as polymers and hybrid microporous materials. It is anticipated that arrays of ion emitters with different coatings may be useful to extract and simultaneously analyse multiple different classes of analytes from complex mixtures rapidly and with high sensitivity without sample preparation or chromatography.
Acknowledgements
This work was funded by the Organization for the Prohibition of Chemical Weapons in The Hague, Netherlands (L/ICA/ICB/194482/15). MCD thanks UNSW for Postgraduate Research Scholarship. WAD thanks the Australian Research Council for a Discovery Early Career Researcher Award (DE130100424). JJG thanks the Australian Research Council for an Australian Laureate Fellowship (FL150100060).References
- Convention on the Prohibition of the Development, Production, Stockpiling and Use of chemical Weapons and on their Destruction, Technical Secretariat of The Organisation for the Prohibition of Chemical Weapons, The Hague, The Netherlands, 2005 Search PubMed.
- U. Ivarsson, H. Nilsson and S. J. Santesson, A FOA briefing book on biological weapons: threat, effects, and protection, National Defense Research Establishement, Umea, Sweden, 1992 Search PubMed.
- United Nations Security Council Resolution 2118 series 2013, UN Secuity Council, New York, USA, 2013 Search PubMed.
- M. A. Mäkinen, O. A. Anttalainen and M. E. T. Sillanpää, Anal. Chem., 2010, 82, 9594–9600 CrossRef PubMed.
- D. Matatagui, J. Martí, M. J. Fernández, J. L. Fontecha, J. Gutiérrez, I. Gràcia, C. Cané and M. C. Horrillo, Sens. Actuators, B, 2011, 154, 199–205 CrossRef CAS.
- R. Sferopoulos, A Review of Chemical Warfare Agent (CWA) Detector Technologies and Commercial-Off-The-Shelf Items, Defense Science and Technology Organization (DSTO), Department of Defense, Australia, 2009 Search PubMed.
- H. Nagashima, T. Kondo, T. Nagoya, T. Ikeda, N. Kurimata, S. Unoke and Y. Seto, J. Chromatogr., A, 2015, 1406, 279–290 CrossRef CAS PubMed.
- O. Terzic, H. Gregg and P. de Voogt, TrAC, Trends Anal. Chem., 2015, 65, 151–166 CrossRef CAS.
- R. M. Black and R. W. Read, J. Chromatogr., A, 1998, 794, 233–244 CrossRef CAS.
- I. Rodin, A. Braun, A. Stavrianidi, T. Baygildiev, O. Shpigun, D. Oreshkin and I. Rybalchenko, J. Anal. Toxicol., 2015, 39, 69–74 CrossRef PubMed.
- M. E. Monge, G. A. Harris, P. Dwivedi and F. M. Fernández, Chem. Rev., 2013, 113, 2269–2308 CrossRef CAS PubMed.
- M. M. Nudnova, L. Zhu and R. Zenobi, Rapid Commun. Mass Spectrom., 2012, 26, 1447–1452 CrossRef CAS PubMed.
- Z. Takáts, J. M. Wiseman, B. Gologan and R. G. Cooks, Science, 2004, 306, 471–473 CrossRef PubMed.
- Z. Takáts, J. M. Wiseman and R. G. Cooks, J. Mass Spectrom., 2005, 40, 1261–1275 CrossRef PubMed.
- V. V. Laiko, M. A. Baldwin and A. L. Burlingame, Anal. Chem., 2000, 72, 652–657 CrossRef CAS PubMed.
- J. Gross, Anal. Bioanal. Chem., 2014, 406, 63–80 CrossRef CAS PubMed.
- A. Bowfield, D. A. Barrett, M. R. Alexander, C. A. Ortori, F. M. Rutten, T. L. Salter, I. S. Gilmore and J. W. Bradley, Rev. Sci. Instrum., 2012, 83, 063503 CrossRef CAS PubMed.
- Y. Okamoto, M. Ogawa, A. Maezawa and T. Imanaka, J. Catal., 1988, 112, 427–436 CrossRef CAS.
- Y. Liu, X. Ma, Z. Lin, M. He, G. Han, C. Yang, Z. Xing, S. Zhang and X. Zhang, Angew. Chem., Int. Ed., 2010, 49, 4435–4437 CrossRef CAS PubMed.
- M. Dumlao, P. M.-L. Sinues, M. Nudnova and R. Zenobi, Anal. Methods, 2014, 6, 3604–3609 RSC.
- E. R. Stephens, M. Dumlao, D. Xiao, D. Zhang and W. A. Donald, J. Am. Soc. Mass Spectrom., 2015, 26, 2081–2084 CrossRef CAS PubMed.
- D. Snyder, C. Pulliam, Z. Ouyang and R. G. Cooks, Anal. Chem., 2016, 88, 2–29 CrossRef PubMed.
- É. A. Souza-Silva, R. Jiang, A. Rodríguez-Lafuente, E. Gionfriddo and J. Pawliszyn, TrAC, Trends Anal. Chem., 2015, 71, 224–235 CrossRef.
- J. Hendrikse, in Chemical Weapons Convention Chemicals Analysis, John Wiley & Sons, Ltd, 2006, ch. 3, pp. 89–132, DOI:10.1002/0470012285.ch6.
- A. M. P. McDonnell, D. Beving, A. Wang, W. Chen and Y. Yan, Adv. Funct. Mater., 2005, 15, 336–340 CrossRef CAS.
- International Zeolite Association, http://www.iza-online.org/synthesis/default.htm, (accessed September 21, 2015).
- B. S. Patil, Q. Wang, V. Hessel and J. Lang, Catal. Today, 2015, 256(Part 1), 49–66 CrossRef CAS.
- B. Mindong, Z. Zhitao, B. Hong Koo, B. Mindi and N. Wang, IEEE Trans. Plasma Sci., 2003, 31, 1285–1291 CrossRef.
- T. Mizushima, K. Matsumoto, J.-i. Sugoh, H. Ohkita and N. Kakuta, Appl. Catal., A, 2004, 265, 53–59 CrossRef CAS.
- J. M. Nilles, T. R. Connell and H. D. Durst, Anal. Chem., 2009, 81, 6744–6749 CrossRef CAS PubMed.
- A. Albert, J. T. Shelley and C. Engelhard, Anal. Bioanal. Chem., 2014, 406, 6111–6127 CrossRef CAS PubMed.
- A. Bierstedt, U. Panne, K. Rurack and J. Riedel, J. Anal. At. Spectrom., 2015, 30, 2496–2506 RSC.
- I. I. Ivanova and E. E. Knyazeva, Chem. Soc. Rev., 2013, 42, 3671–3688 RSC.
- G. G. Guilbault, J. Affolter, Y. Tomita and E. S. Kolesar, Anal. Chem., 1981, 53, 2057–2060 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6an00178e |
| This journal is © The Royal Society of Chemistry 2016 |