
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
High desolvation temperature facilitates the ESI-source H/D exchange at non-labile sites of hydroxybenzoic acids and aromatic amino acids†
Alexander
Zherebker
a,
Yury
Kostyukevich
bde,
Alexey
Kononikhin
cde,
Vitaliy A.
Roznyatovsky
a,
Igor
Popov
ce,
Yuri K.
Grishin
a,
Irina V.
Perminova
*a and
Eugene
Nikolaev
*bcde
aLomonosov Moscow State University, Department of Chemistry, Leninskie Gory 1-3, 119991 Moscow, Russia. E-mail: iperm@org.chem.msu.ru
bSkolkovo Institute of Science and Technology, Novaya St., 100, Skolkovo 143025, Russian Federation. E-mail: ennikolaev@rambler.ru
cEmanuel Institute for Biochemical Physics Russian Academy of Sciences, Kosygina st. 4, 119334 Moscow, Russia
dInstitute for Energy Problems of Chemical Physics Russian Academy of Sciences, Leninskij pr. 38 k.2, 119334 Moscow, Russia
eMoscow Institute of Physics and Technology, 141700 Dolgoprudnyi, Moscow Region, Russia
First published on 10th March 2016
Abstract
Hydrogen/deuterium exchange coupled with high-resolution mass spectrometry has become a powerful analytical approach for structural investigations of complex organic matrices. Here we report the feasibility of the site-specific H/D exchange of non-labile hydrogens directly in the electrospray ionization (ESI) source, which was facilitated by an increase in the desolvation temperature from 200 °C up to 400 °C. We have found that the exchanges at non-labile sites were observed only for the model compounds capable of keto–enol tautomeric transformations (e.g., 2,3-, 2,4-dihydroxybenzoic acids, gallic acid, DOPA), and only when water was used as a solvent. We hypothesized that the detected additional exchanges were induced by the presence of hydroxyls in the sprayed water droplets generated in the negative ESI mode. It was indicative of the exchange reactions taking place in the sprayed droplets rather than in the gas phase. To support this hypothesis, the H/D exchange experiments were run in deuterated water under base-catalyzed conditions for three model compounds, which showed the most intensive exchanges in the MS experiments: DOPA, 2,4-DHB, and 5-acetylsalicylic acid. 2H NMR spectroscopy has confirmed keto–enolic transformations of the model compounds leading to the specific labeling of the corresponding non-labile sites. We believe that the proposed technique will be useful for structural investigations of natural complex mixtures (e.g. proteins, humic substances) using site-specific H/D exchange.
Introduction
Hydrogen/deuterium exchange (HDX) is an analytical approach, which is widely used for structural studies of proteins.1,2 The labile hydrogens attached to heteroatoms such as oxygen, nitrogen, and sulfur, can be easily replaced on deuterium by incubation with deuterated solvents such as D2O, MeOD, and others.3 Mass measurement of the target molecule is a method of choice to monitor HDX reactions.4–7 The use of soft ionization techniques, such as electrospray ionization (ESI), coupled with HDX allows for the identification of labile hydrogens in proteins2,8,9 and other polymers.10–12 That is why numerous studies were performed on H/D exchange in ESI,13–17 APPI,18,19 APCI,20 and others.21 The specific option of atmospheric pressure HDX mass-spectrometry is a facile conversion of folded to unfolded proteins during ionization, which can be achieved by heating the desolvation capillary.22 This conformational change gives rise to a number of exchanged labile hydrogens which are easily registered by using a mass-spectrometer and used for data interpretation. However, some studies on the gas phase H/D exchange at the atmospheric pressure reported deuterium enrichment exceeding the maximum amount of labile hydrogens present in the system.4,23,24 The authors related these extra exchanges to a contribution from C–H acidity, which can become profound under conditions of mass-spectrometric ionization, and warned of misinterpretation of these kinds of results. This phenomenon could be of particular importance for ESI: during ionization the solvent undergoes slight electrolysis, which, in the case of water, might lead to the generation of trace amounts of hydroxyls.25 The presence of a base might induce exchanges of non-labile hydrogens, which are typical for a base-catalyzed H/D exchange, e.g. due to keto–enol tautomerism.3To demonstrate the feasibility of the in-ESI H/D exchange at the C–H sites capable of keto–enol transformations, we have relied on our previous studies with respect to both acid/base catalyzed HDX at non-labile sites in aqueous solutions of the synthetic humic substances,26 and to in-ESI HDX at non-labile sites of 2-nitrophloroglucinol observed due to tautomeric transformations induced by heating of the desolvation capillary.27 The last finding allowed us to suggest that in the presence of a base in the sprayed solvent, heating of the capillary might facilitate conversion of the compounds capable of keto–enol tautomerism which would give rise to HDX at these specific non-labile sites. If this is the case, a simple and robust technique could be developed for conducting selective H/D exchange directly in the mass-spectrometer.
The objective of this study was to demonstrate the feasibility of the in-ESI H/D exchange at the C–H sites capable of keto–enol transformations, which can be induced by an increase in the desolvation temperature as shown in Fig. 1.
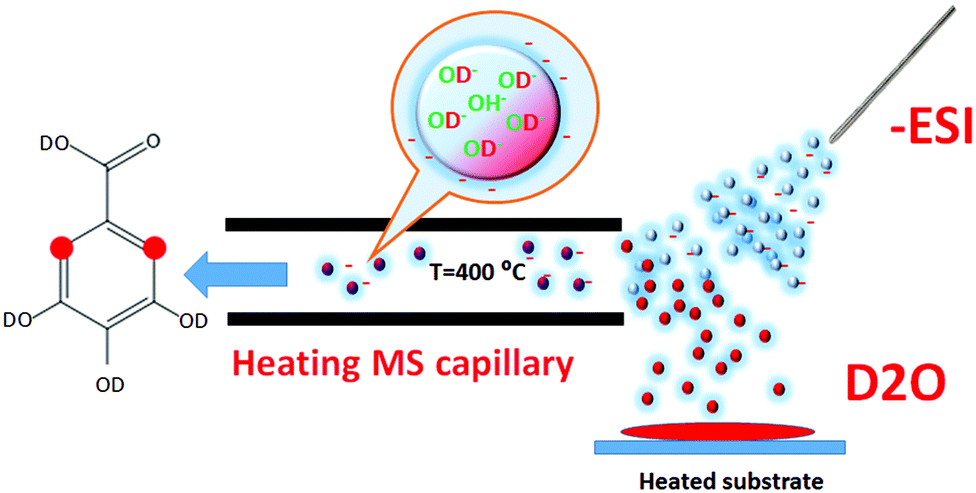 | ||
| Fig. 1 The design of the in-ESI source H/D exchange experiment. The charged droplets are produced by the ESI source; they pass through the D2O-saturated region, enter the heated capillary and evaporate producing the gas phase ions. During this transport the charged droplets interact with D2O containing trace amounts of hydroxyls, and exchange protium against deuterium. Red dots designate C–H sites capable of keto–enol tautomerism, which have undergone labeling. | ||
To avoid H/D back exchange, all in-ESI HDX experiments were run under a D2O-saturated atmosphere which was created by placing a D2O droplet right underneath the ESI cone as described previously.28,29 A model set of hydroxybenzoic acids and aromatic amino acids was used for this purpose, which possesses different non-labile sites capable of keto–enolic tautomerism. The obtained MS results were compared to the base-catalyzed HDX experiments in solution, which were quantified using 2H NMR spectroscopy.
Experimental
Materials
All reagents used in this study are commercially available. Methanol, ethyl acetate and ammonium hydroxide were of analytical grade. The enrichment of D2O (Merck) was 99.9%.All model compounds were purchased from Sigma-Aldrich with a purity of 99% and included: 2,5-dihydroxybenzoic acid, 2,3-dihydroxybenzoic acid, 2,4-dihydroxybenzoic acid, 3,4,5-trihydroxybenzoic (gallic) acid, tyrosine, 3,4-dihydroxyphenylalanine, 3,4-dihydroxyphenethylamine, 5-acetylsalicylic acid, and 2,2-diphenylacetic acid.
Sample preparation for FT ICR MS measurements
The samples were prepared in methanol and in its mixture with distilled water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v). All concentrations were 0.4 mg mL−1.
:1 v/v). All concentrations were 0.4 mg mL−1.
H/D exchange in deuterated solvents in solution
Deuterium (2H) NMR spectroscopy
2H NMR spectra (61.397 MHz) were acquired using the NMR spectrometer Agilent 400MR allowing the use of a lock channel for the observation of the deuterium–proton decoupled spectra and equipped with a 10 mm deuterium selective probe.The duration of the 90° pulse for 2H nuclei was 25 μs. The acquisition time of the free induction decay was at least 4 s and the relaxation delay between pulses was 1 s. The spectral width was 1100 Hz. Chemical shifts were measured with reference to the solvent (D2O −4.72 ppm, DMSO −2.47 ppm). Integral intensities of signals were determined by an iteration analysis of the total line shape taking into account the residual field inhomogeneity and phase distortions using the INTSPECT2 program.32
MS analysis
All experiments were performed using an LTQ FT Ultra (Thermo Electron Corp., Bremen, Germany) mass-spectrometer equipped with a 7 T superconducting magnet. Ions were generated by using an IonMax Electrospray ion source (Thermo Electron Corp., Bremen, Germany) in both negative and positive ESI modes. The temperature of the desolvating capillary varied from 200 °C to 400 °C. The length of the desolvating capillary was 105 mm and its inner diameter was 0.5 mm. The infusion rate of the sample was 1 μL min−1 and the needle voltage was 3 kV. Full-scan MS spectra (m/z 200–2000) were acquired in the FTICR with a resolution R = 400000 at m/z 400.
In-ESI source H/D exchange
The experimental setup for performing the H/D exchange was based on our previous developments described in ref. 22 and 28. The current design is shown in Fig. 1. The charged droplets are produced by using the ESI source; they pass through the heated capillary and evaporate producing the gas phase ions. During this transport, the ions interact with D2O vapors and exchange labile protium against deuterium. To create an atmosphere saturated with D2O vapor between the ESI needle and the MS entrance capillary, 400 μL D2O were placed on a copper plate positioned approximately 7 mm underneath the ESI needle. All experiments were run at normal and high desolvation temperatures, which accounted for 200 °C and 400 °C, respectively. A choice of the high desolvation temperature was based on preliminary HDX experiments on 2,4-dihydroxybenzoic acid (2,4-DHB), which were run at 300 °C, 400 °C, and 450 °C. The obtained results showed that a raise in the capillary temperature from 200 °C up to 400 °C was accompanied by a substantial growth in the intensity of the additional peak related to the non-labile HDX, whereas further heating up to 450 °C did not bring about any significant change in this peak intensity. The corresponding data are shown in Fig. S1 in the ESI.† As a result, all HDX experiments at the elevated desolvation temperature were run at 400 °C.Results and discussion
In-ESI source H/D exchange
To explore if an increase in the desolvation temperature may facilitate exchange of the non-labile protons, we have run H/D exchange reactions at normal and high desolvation temperatures (200 °C and 400 °C, respectively) for all model compounds shown in Table 1. The model set was composed of hydroxybenzoic acids and aromatic amino acids which can be considered as constituents of natural complex systems such as humic substances (HS), proteins, and others.33 To account for a strong −M effect of the carboxylic group on the electronic density of the ring, we included into a model set three dihydroxybenzoic acids (DHBs) and gallic acid as representatives of the aromatic compounds with the carboxyl group as a ring substituent (compounds from 1 to 4), whereas two amino acids were used as compounds with the carboxyl group in the side chain (5, 6). We also used an aromatic amine (compound 7) which does not possess any carboxyls, and 5-acetylsalicylic and 2,2-diphenylacetic acids (8 and 9, respectively) as containing α-carbons. The use of this model set was to allow us for the in-depth exploration of the relationship between the structure and reactivity governing the H/D exchange reactions. The formula designations and amounts of the H/D exchanges observed at the two desolvation temperatures for each compound are summarized in Table 1.| Neutral compound producing ion | No. of labile H atoms in anion or cationa | Maximum no. of H/D exchanges observed at 200 °C | Maximum no. of H/D exchanges observed at 400 °Cb |
|---|---|---|---|
|
a Cations in the positive-ion mode.
b Mixture of water–methanol (1:1 v/v) was used as a solvent.
|
|||
| 2,4-OH[C6H3]COOH (1) | 2 | 2 | 4 |
| 2,5-OH[C6H3]COOH (2) | 2 | 2 | 2 |
| 2,3-OH[C6H3]COOH (3) | 2 | 2 | 3 |
| 3,4,5-OH[C6H2]COOH (4) | 3 | 3 | 5 |
| 4-OH[C6H4]CH2CH(NH2)COOH (5) | 3 | 3 | 3 |
| 3,4-OH[C6H3]CH2CH(NH2)COOH (6) | 4 or 6a | 4 or 6a | 7 or 9a |
| 3,4-OH[C6H3]CH2CH2NH2 (7) | 5a | 5a | 8a |
| 2-OH,5-CH3CO[C6H3]COOH (8) | 1 | 1 | 4 |
| 2,2-C6H5[CH]COOH (9) | 0 | 0 | 1 |
For a desolvation temperature of 200 °C, the number of isotopic exchanges observed for all model compounds is equal to that theoretically expected from the number of labile H-atoms present in the anion or cation under study. However, a very different situation is observed when the desolvation temperature is set to 400 °C.
Fig. 2 shows the mass-spectra of H/D series for the three DHBs used in our study. At 200 °C, the length of the isotopic exchange series is equal to the amount of labile H atoms (highlighted with blue color in the corresponding spectra and formulas). At 400 °C, the spectrum does not change for compound 2, but one and two additional peaks can be seen for compounds 1 and 3, respectively (highlighted with red color). Given the stability of the signals of deuterated analyte ions over the time demonstrated in our previous studies,13 we interpreted the observed impact of the capillary temperature on the H/D exchange of the DHBs as additional H/D exchanges occurring at the non-labile sites, which become reactive only under the conditions of keto–enol tautomerism (highlighted in red in molecular structures shown in Fig. 2). Those sites are 3,5 for 2,4-DHB (1), 3 for 2,5-DHB (2) and 5 for 2,3-DHB (3). The reason is the +M effect of the electron-donating OH-substituent and the neutral effect of the COOH-group on meta-positions in the aromatic ring. However, in the case of 2,5-DHB (2), additional exchanges at C–H sites were not detected at both the temperatures tested, whereas a significant labeling was observed for 2,3-DHB and 2,4-DHB at an elevated temperature.
 | ||
| Fig. 2 In-ESI source H/D exchange series of 2,4-DHB, 2,5-DHB, and 2,3-DHB at 200 °C (blue lines) and 400 °C (additional peaks are colored in red). Red dots in the structural formulas designate the feasible sites of deuteration in accordance with keto–enol tautomerism and mesomeric substituent effects in the aromatic ring. | ||
To explain these differences we have taken into account that the keto-form of phenol loses its proton to a base generated in the solvent in the ESI source25 to produce a carbanion intermediate, which undergoes HDX. In the absence of a base, the proton abstraction becomes a rate-limiting reaction34,35 and in the case of MS analysis it should impact the detected results. To increase the rate of H elimination, we used 10% NH3 solution in D2O that resulted in the appearance of an additional OH-anion in the reaction mixture, which is a strong base. We performed the H/D exchange of gallic acid in the presence and absence of NH3. The obtained mass-spectra are shown in Fig. 3. Gallic acid undergoes exchanges of all labile sites at 200 °C. An increase in the temperature leads to the incorporation of two additional deuterons, which is indicative of HDX at both C–H sites. An addition of NH3 to D2O significantly magnifies the enrichment of C–H sites with deuterium at an elevated temperature. We can assume that the addition of the base catalyzes the HDX reaction similar to base-catalyzed reactions in solution, and the proton elimination is the limiting stage.
 | ||
| Fig. 3 In-ESI source H/D exchange series of gallic acid (4) in the absence and presence of ammonia. The peaks corresponding to labile and backbone deuterium are highlighted with blue and red color, respectively. Red dots in the structural formulas designate the feasible sites for deuteration in accordance with keto–enol tautomerism. | ||
To evaluate an impact of the carboxyl substituent, which is a strong electron acceptor, on the H/D exchange in the aromatic ring, we conducted the labeling of tyrosine (5) and DOPA (6). The major structural difference of 5 and 6 is the presence of one and two hydroxyl groups, respectively. Fig. 4(a) shows 3 and 4 exchanges related to all labile protons in the case of tyrosine and DOPA, respectively. At the elevated desolvation temperature substantial differences can be seen for the HDX of tyrosine and DOPA: the former undergoes no additional exchanges, while the latter undergoes 3 additional exchanges at 400 °C.
 | ||
| Fig. 4 In-ESI source H/D exchange series at 400 °C of tyrosine (5) and DOPA (6) in the negative mode (a) and of DOPA (6) and dopamine (7) in the positive mode (b). The peaks corresponding to labile and backbone deuterium are colored in blue and red, respectively. Red dots in the structural formulas designate the feasible sites of deuteration in accordance with keto–enol tautomerism and mesomeric substituent effects in the aromatic ring. | ||
The observed phenomenon is in line with a similar trend for DHBs, while compounds with hydroxyl groups attached to adjacent carbon atoms underwent extended isotopic exchanges. At the same time, neither compound 2 nor 3 were labeled at all sites. This implies that the extent of deuteration is significantly influenced by the −M effect of the carboxyl group.
To evaluate the influence of the ionization mode on the labeling reactions, we conducted the HDX experiments with DOPA (6) and dopamine (7) in the positive mode. The corresponding spectra are shown in Fig. 4(b). We observed 6 and 5 H/D exchanges for DOPA and dopamine, respectively, at 200 °C (Fig. 4b, blue lines), which equaled the amount of mobile protons, but at 400 °C, both compounds have undergone 3 more exchanges (Fig. 4b, red lines). At the same time the peak intensity of these ions was significantly smaller than in the case of the negative ESI mode. In the positive mode of ESI almost a full charge is provided by H3O+ ions, which possess a low catalytic activity for aromatic H/D exchange reactions.3 However, given that reactions in the charged droplets of electro-spray can occur at higher rates than in the bulk solution,36 we believe that in this case the H/D exchange proceeds through electrophilic substitution.
Collectively, the data obtained could be interpreted as a substantial contribution of keto–enol tautomerism to the H/D exchange at the non-labile sites at an elevated desolvation temperature. It is indicative of HDX reactions taking place in the charged droplets rather than in the gas phase. This looks feasible given the specific experimental conditions used in our study, where an atmosphere saturated with D2O vapor was created between the ESI needle and the MS entrance capillary. In this case, the capillary heating in the presence of a base might lead to a two-step dissociation of the DHBs bringing about the formation of phenoxide anions. This is facilitated by a substantial temperature dependence of the acidity of the hydroxyl group, which is not the case for the carboxylic group: phenols possess dpKa/dT values of about −0.1 to −0.2 units per 10 K.37 As a result, the ambident phenoxide anions form carbanions both at ortho- and para-positions.38 These positions may undergo HDX according to the relay mechanism proposed by Ghan and Enke39 for the gas phase HDX at the non-labile sites of aryl-compounds. This mechanism consists of the formation of a six-membered-ring intermediate between the localized negative charge site on the aromatic ring and D2O followed by the relocation of the charge site to the adjacent center.39 Still, another mechanism of skeletal HDX in the gas phase is possible: the intramolecular D-transfer proposed for anions produced by negative ESI.40,41 This mechanism is based on thermochemical calculations conducted by Tian et al.41 to explain additional HDX which was observed for the aromatic hydrogen atom located between two carboxylic groups. It was explained by the formation of hydrogen bonds with a deuterated solvent, which stabilize the aryl anion, followed by an intramolecular D transfer on the localized negative charge site.
It should be noted that both relay and D-transfer mechanisms alone (both of them are developed for the gas phase) did not explain the HDX results observed in our study. So, in accord with the D transfer mechanism we would expect 0 skeletal HDX for 2,3-DHB, 1 HDX for 2,4-DHB, and 1 significant HDX for 2,5-DHB (the latter contains a hydrogen atom located between carboxylic and hydroxyl groups). However, the experimental results contradict these suggestions: we observed 1 HDX for 2,3-DHB, 2 HDX for 2,4-DHB, and 0 HDX for 2,5 DHB. This could be explained by the occurrence of HDX reactions in the charged micro droplets rather than for gas-phase reactions into the vacuum zone of the mass-spectrometer. Indeed, if the observed HDX reactions would occur in the gas-phase, then the use of another non-deuterated solvent (e.g. methanol) would not impact HDX results. We conducted this experiment by dissolving model compounds in methanol, and none of them has undergone extradeuteration in this case. Moreover, it is known that the ionization of DHBs in methanol occurs via dissociation of phenolic OH which is more acidic than carboxyl under the conditions of ESI.42 In the case of gas phase HDX, this would lead to an intermediate favorable to the D transfer mechanism implementation, which was not observed in our experiments. The major difference is the composition of the spray droplets in the case of water as compared to methanol. Trace amounts of hydroxyls in the droplets which are generated in water are not produced in methanol.43 This highlights the particular importance of the presence of a base during the in-source H/D exchange and indicates that for our system the reactions occur in the charged micro droplets rather than in the gas phase as shown in Fig. 1.44,45
Taking the above considerations into account we propose the following HDX reactions at the non-labile sites of the DHBs which account for the contribution of keto–enol transformations taking place in the charged droplets at an elevated capillary temperature in the negative ESI mode (Fig. 3).
In the case of 2,4-DHB an intermediate carbanion possesses two resonance structures, while two other DHBs possess only one. This is because the structures with a formal negative charge adjacent to substituents are energetically unfavorable. Therefore, the tautomeric form of 2,4-DHB is relatively stabilized which results in a significant H/D exchange of both C–H protons at 400 °C. The feasible reaction pathway is shown in Fig. 5A. The high capillary temperature leads to phenol dissociation, causes keto–enol transformation of the phenoxide anion followed by deuteration of the carbanion by the D2O molecule oriented by carbonyl oxygen and proton elimination resulting in a stable aromatic structure formation in terms of substituent effects. The corresponding HDX pathways for 2,3-DHB and 2,5-DHB are shown in Fig. 5B and C, respectively. Despite 2,3- and 2,5-DHBs having similar substituent patterns, 2,3-DHB undergoes 3 H/D exchanges, whereas 2,5-DHB – only two. This is indicative of one additional H/D exchange in the case of 2,3-DHB. It might result from the stabilization of a short-living ketone by hydrogen bonds between two adjacent groups in accord with the relay mechanism facilitating an exchange at the non-labile site.46 A similar stabilization cannot be implemented in the case of 2,5-DHB which does not show exchanges at the non-labile sites.
 | ||
| Fig. 5 The proposed reactions of H/D exchange of the aryl protons at an elevated desolvation temperature (400 °C) in the charged microdroplets of the saturated atmosphere of D2O favoring keto–enolic transformations of the DHBs in the negative ionization mode: (A) 2,4-DHB, (B) 2,5-DHB, (C) 2,3-DHB. (The second non-labile proton of 2,4-DHB (A) is exchanged via a similar pathway which is realized for the first resonance structure as well.) | ||
To confirm the suggestion that keto–enol tautomerism is an essential part of skeletal HDX, we carried out the MS labeling reactions with 5-acetylsalicylic (8) and diphenylacetic (9) acids, which are characterized by inactive aromatic protons and active α-protons. We anticipated observing 3 extra H/D exchanges in the case of acetylsalicylic acid related to the CH3-group of a side chain, and only one exchange in the case of diphenylacetic acids. The corresponding results are shown in Fig. 6. The heating of the capillary to 400 °C gave rise to 3 and 1 additional H/D exchanges for compounds 8 and 9, respectively. These observations are in accordance with our suggestion about the significant contribution of tautomerism to H/D exchange under the conditions studied.
 | ||
| Fig. 6 In-ESI source H/D exchange series of (a) diphenylacetic acid (8) and (b) 5-acetylsalicylic acid (9). Red dots indicate α-carbons. | ||
Therefore, we concluded that additional H/D exchanges could occur via keto–enol tautomerism at the most reactive non-labile sites similar to the reactions in the solution phase HDX followed by the conventional relay mechanism leading to deuterated products.
H/D exchange in the liquid phase
To confirm that H/D exchange during ESI under the conditions studied occurs in the charged droplet rather than in the gas phase, our next goal was to validate sites capable of H/D exchange due to keto–enolic transformations in the liquid phase by running the same reactions in deuterated water and analyzing them with the use of 2H NMR spectroscopy. For this purpose, we used three model compounds, which showed the most intensive exchanges in the MS experiments: DOPA (6), 2,4-DHB (3), and 5-acetylsalicylic acid (8). The corresponding 2H NMR spectra are shown in Fig. 2S–4S in the ESI.† For DOPA (Fig. 2S†) we observed a large wide peak at 6.71 ppm corresponding to overlapped signals of all aromatic deuteriums and negligibly small signals assigned to α-deuterium at 3.76 ppm. The 2H NMR spectrum of 2,4-DHB (Fig. 3S†) shows a large peak at 6.13 ppm, which corresponds to the deuterium at positions 3 and 5, and a very small signal at 6.92 ppm related to the deuterium at position 6. The 2H NMR spectrum of the 5-acetylsalicylic acid shows a peak at 2.42 ppm (Fig. 4S†) assigned to α-protons which is in line with our expectations. ESI FT ICR MS spectra of the same labelled compounds obtained from a protic solvent are shown in Fig. 5S.† The lengths of exchanging series (3 for both DOPA and 5-acetylsalicylic acid) are consistent with the results of 2H NMR spectroscopy.2H NMR spectroscopy enables an evaluation of not only the number of exchanges, but also of the degree of deuteration. This is because NMR 2H–{1H} provides a high accuracy estimation of the integral intensity of signals even when measured at a natural abundance level of deuterium using precision methods of integration.32,47 In the case of DHB-2,4 the ratio of total deuterium integrals at 3 and 5 positions to the integral of deuterium at position 6 was 215. Further, the relative integral intensity of the methyl group of acetylsalicylic acid was 100% and there were no signals observed within an aromatic region. The calculated integral intensities are in agreement with the results of MS experiments, which show 2 and 3 additional exchanges, respectively.
Therefore, the analysis of base-catalyzed H/D exchange reactions in the liquid state indicates the site specificity, which proves the proposed selectivity of deuteration during the in-ESI source exchange.
Conclusions
In this paper we demonstrate that the in-ESI source H/D exchange under atmospheric pressure leads to a site-specific exchange of non-labile protons at high desolvation temperatures. This might indicate that capillary heating facilitates keto–enol transformations taking place in the charged droplets followed by HDX at non-labile sites in accord with the relay mechanism. This allowed us to conclude that the negative ionization mode can be considered as an analogue of the base-catalyzed conditions in the liquid state and an increase in the desolvation temperature shifts equilibrium between tautomeric forms. Of importance is that a switch to the positive mode reduces the efficacy of H/D exchange. The use of 2H NMR spectroscopy allowed us to examine the selectivity of base-catalyzed labeling reactions qualitatively and quantitatively. The obtained results proved the crucial role of the compound structure in H/D exchange at non-labile sites and revealed an influence of the acceptor group (e.g. carboxyl) on reaction selectivity. We believe that the results obtained can be of use for structure elucidation using ESI-HDX mass-spectrometric experiments for proteins and natural organic matter. In addition, the observed phenomena could shed light on the driving force of the back exchange of labelled compounds caused by capillary heating during ESI MS analysis.Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Acknowledgements
This study was supported by the Russian Scientific Foundation grant no. 14-24-00114.References
- S. W. Englander, J. Am. Soc. Mass Spectrom., 2006, 17, 1481–1489 CrossRef CAS PubMed.
- T. E. Wales and J. R. Engen, Mass Spectrom. Rev., 2006, 25, 158–170 CrossRef CAS PubMed.
- J. Atzrodt, V. Derdau, T. Fey and J. Zimmermann, Angew. Chem., Int. Ed., 2007, 46, 7744–7765 CrossRef CAS PubMed.
- D. F. Hunt and S. K. Sethi, J. Am. Chem. Soc., 1980, 102, 6953–6963 CrossRef CAS.
- R. R. Squires, C. H. DePuy and V. M. Bierbaum, J. Am. Chem. Soc., 1981, 103 Search PubMed.
- J. C. Kleingeld and N. M. M. Nibbering, Tetrahedron, 1983, 39, 4193–4199 CrossRef CAS.
- J. J. Grabowski, C. H. DePuy and V. M. Bierbaum, J. Am. Chem. Soc., 1985, 107, 7384–7389 CrossRef CAS.
- F. W. McLafferty, Z. Guan, U. Haupts, T. D. Wood and N. L. Kelleher, J. Am. Chem. Soc., 1998, 120, 4732–4740 CrossRef CAS.
- A. Kharlamova, C. M. Fisher and S. A. McLuckey, J. Mass Spectrom., 2014, 49, 437–444 CrossRef CAS PubMed.
- K. B. Green-Church, P. A. Limbach, M. A. Freitas and A. G. Marshall, J. Am. Soc. Mass Spectrom., 2001, 12, 268–277 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov and E. Nikolaev, Anal. Chem., 2014, 86, 2595–2600 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov, N. Starodubtzevad, S. Pekov, E. Kukaev, M. Indeykina and E. Nikolaev, Eur. J. Mass Spectrom., 2015, 21, 59–63 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov and E. Nikolaev, Anal. Chem., 2013, 85, 5330–5334 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov and E. Nikolaev, J. Mass Spectrom., 2014, 49, 989–994 CrossRef CAS PubMed.
- M. E. Hemling, J. J. Conboy, M. F. Bean, M. Mentzer and S. A. Carr, J. Am. Soc. Mass Spectrom., 1994, 5, 434–442 CrossRef CAS PubMed.
- E. Gard, M. K. Green, J. Bregar and C. B. Lebrilla, J. Am. Soc. Mass Spectrom., 1994, 5, 623–631 CrossRef CAS PubMed.
- M. A. Freitas, C. L. Hendrickson, M. R. Emmett and A. G. Marshall, Int. J. Mass Spectrom., 1999, 185–187, 565–575 CrossRef.
- A. Ahmed and S. Kim, J. Am. Soc. Mass Spectrom., 2013, 24, 1900–1905 CrossRef CAS PubMed.
- Y. Cho, A. Ahmed and S. Kim, Anal. Chem., 2013, 85, 9758–9763 CrossRef CAS PubMed.
- N. W. Davies, J. A. Smith, P. P. Molesworth and J. J. Ross, Rapid Commun. Mass Spectrom., 2010, 24, 1105–1110 CrossRef CAS PubMed.
- A. B. Attygalle, R. Gangam and J. Pavlov, Anal. Chem., 2014, 86, 928–935 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov, A. Spasskiy and E. Nikolaev, J. Mass Spectrom., 2015, 50, 49–55 CrossRef CAS PubMed.
- D. R. Reed and S. R. Kass, J. Am. Soc. Mass Spectrom., 2001, 12, 1163–1168 CrossRef CAS PubMed.
- M. M. Siegel, Anal. Chem., 1988, 60, 2090–2095 CrossRef CAS.
- Electrospray and MALDI Mass Spectrometry, ed. R. B. Cole, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2010 Search PubMed.
- A. Y. Zherebker, D. Airapetyan, A. I. Konstantinov, Y. I. Kostyukevich, A. S. Kononikhin, I. A. Popov, K. V. Zaitsev, E. N. Nikolaev and I. V. Perminova, Analyst, 2015, 140, 4708–4719 RSC.
- Y. Kostyukevich, A. Kononikhin, I. Popov, N. Starodubtseva, E. Kukaev and E. Nikotaev, Eur. J. Mass Spectrom., 2014, 20, 345–349 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, I. Popov, O. Kharybin, I. Perminova, A. Konstantinov and E. Nikolaev, Anal. Chem., 2013, 85, 11007–11013 CrossRef CAS PubMed.
- Y. Kostyukevich, A. Kononikhin, A. Zherebker, I. Popov, I. Perminova and E. Nikolaev, Anal. Bioanal. Chem., 2014, 406, 6655–6664 CrossRef CAS PubMed.
- B. Lindström, B. Sjöquist and E. Anggard, J. Labelled Compd. Radiopharm., 1974, 10, 187–194 CrossRef.
- L. D. Saraswat, J. M. Kenny, S. K. Davis and J. B. Justice, J. Labelled Compd. Radiopharm., 1981, 18, 1507–1516 CrossRef CAS.
- V. A. Roznyatovsky, S. M. Gerdov, Y. K. Grishin, D. N. Laikov and Y. A. Ustynyuk, Russ. Chem. Bull., 2003, 52, 552–556 CrossRef CAS.
- E. A. Ghabbour and G. Davies and R. S. of C. G. Britain, Humic Substances: Structures, Models and Functions, Royal Society of Chemistry, 2001 Search PubMed.
- G. A. Olah, Acc. Chem. Res., 1971, 4, 240–248 CrossRef CAS.
- O. A. Reutov, Bull. Acad. Sci. USSR Div. Chem. Sci., 1980, 29, 1461–1478 CrossRef.
- T. Müller, A. Badu-Tawiah and R. G. Cooks, Angew. Chem., Int. Ed., 2012, 51, 11832–11835 CrossRef PubMed.
- J. C. Reijenga, L. G. Gagliardi and E. Kenndler, J. Chromatogr., A, 2007, 1155, 142–145 CrossRef CAS PubMed.
- W. B. Wheatley, L. C. Cheney and S. B. Binkley, J. Am. Chem. Soc., 1949, 71, 3795–3797 CrossRef CAS.
- S. Ghan and C. G. Enke, J. Am. Soc. Mass Spectrom., 1994, 5, 282–291 CrossRef CAS PubMed.
- J. E. Chipuk and J. S. Brodbelt, Int. J. Mass Spectrom., 2007, 267, 98–108 CrossRef CAS.
- Z. Tian, D. R. Reed and S. R. Kass, Int. J. Mass Spectrom., 2015, 377, 130–138 CrossRef CAS.
- D. Schröder, M. Buděšínský and J. Roithová, J. Am. Chem. Soc., 2012, 134, 15897–15905 CrossRef PubMed.
- S. Banerjee and S. Mazumdar, Int. J. Anal. Chem., 2012, 2012, ID282574 Search PubMed.
- A. P. Bruins, J. Chromatogr., A, 1998, 794, 345–357 CrossRef CAS.
- P. Kebarle and L. Tang, Anal. Chem., 1993, 65, 972A–986A CAS.
- V. F. Traven, V. V. Negrebetsky, L. I. Vorobjeva and E. A. Carberry, Can. J. Chem., 1997, 75, 377–383 CrossRef CAS.
- V. Silvestre, S. Goupry, M. Trierweiler, R. Robins and S. Akoka, Anal. Chem., 2001, 73, 1862–1868 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: FT ICR MS spectra of labeled DHB-2,4 at different capillary temperatures, 2H NMR spectra and FT ICR MS of labeled compounds 3, 6 and 8. See DOI: 10.1039/c5an02676h |
| This journal is © The Royal Society of Chemistry 2016 |