
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Electropolymerized hydrophobic polyazulene as solid-contacts in potassium-selective electrodes
Ning
He
a,
Róbert E.
Gyurcsányi
*b and
Tom
Lindfors
 *a
*a
aÅbo Akademi University, Faculty of Science and Engineering, Johan Gadolin Process Chemistry Centre, Laboratory of Analytical Chemistry, Biskopsgatan 8, FIN-20500 Turku/Åbo, Finland. E-mail: Tom.Lindfors@abo.fi
bMTA-BME “Lendület” Chemical Nanosensors Research Group, Department of Inorganic and Analytical Chemistry, Budapest University of Technology and Economics, Szt. Gellért tér 4, 1111, Budapest, Hungary. E-mail: robertgy@mail.bme.hu
First published on 16th March 2016
Abstract
Electropolymerized hydrophobic polyazulene (PAz) based solid-state potassium ion-selective electrodes (SC-ISEs) have been characterized in terms of their suitability for clinical application. Polarization of the PAz solid contact before applying the plasticised poly(vinyl chloride) based K+-selective membrane was implemented as a convenient approach to address the general problem of the irreproducible standard potential (E0) of SC-ISEs. Using this method, the E0 reproducibility among different electrodes was, in the worst case, ±7.9 mV (n = 4). The effectiveness of the redox buffer-free approach presented here in stabilizing E0 is strengthened by the absence of light, oxygen and carbon dioxide sensitivity of the PAz SC-ISEs. No evidence was found for the formation of an aqueous layer for the PAz-based SC-ISEs. Thus the hydrophobic carbon structure of PAz having a water contact angle of 98 ± 11°, which is slightly higher than that for graphene, can apparently efficiently counteract the aqueous layer formation. In terms of the specific application, the PAz solid contact ISEs were found to show a remarkably good potential stability at their first contact with an aqueous sample. We also confirmed that the PAz-based SC-ISEs can be used for the accurate determination of the K+ concentration in serum solutions. Overall, the PAz solid contact shows significant advantages as compared to the state-of-the-art of electrically conducting polymer based SC-ISEs.
Introduction
Besides electrochemical glucose sensors, the ion-selective electrodes (ISEs) are the most employed electrochemical sensors in the routine analytical and clinical practice.1–4 In recent years, research interest has been redirected from the classical liquid contact ISEs toward solid-contact ion-selective electrodes (SC-ISEs). From a practical point of view, the main driving force for the SC-ISE development is its compatibility with mass production by standard microfabrication techniques5,6 and the opportunity for robust miniaturization.7 However, there are numerous additional benefits originating from these enabling technologies,8e.g., low cost and energy consumption, which make SC-ISEs appealing even for already established routine tasks that presently employ classical liquid contact ISEs. Additionally, SC-ISEs would meet the demands of emerging applications such as the use of ISEs as detectors for low-cost paper-based analytical platforms,9–11 wearable sensors,12,13 and bioassays.14,15 For widespread implementation, the SC-ISEs need to approach or surpass the potential stability of their liquid contact alternatives by establishing a thermodynamically well-defined interface between the ion conducting ion-selective membrane (ISM) and the electron conducting electrode substrate.16 Therefore, electrically conductive polymers (ECPs) possessing mixed electronic and ionic conductivity have become one of the most popular ion-to-electron transducer materials in SC-ISEs.17Over the years, different ECPs have been applied as the SC layer, such as polypyrrole,6,17 poly(3-octylthiophene) (POT),18 poly(3,4-ethylenedioxythiophene)19 and polyaniline (PANI).20 However, the commercialization of ECP-based SC-ISEs has been so far limited by the poor reproducibility of their standard potential and their unsatisfactory long term potential stability. These deficiencies may originate from (i) the substrate electrode,21 (ii) intrinsic equilibration processes of the deposited ECPs,22,23 and (iii) the sensitivity of ECPs to one or more environmental parameters, e.g., pH,20 O2, CO2, temperature and light,24 but most often from (iv) the accumulation of a water layer/pool at the buried interfaces of the SC-ISEs.25 Presently, the full prevention of the formation of a thin aqueous layer beneath the ISM caused by the water uptake of ISMs26–28 is one of the most challenging tasks in SC-ISE development.29,30 It was recently proven by the Fourier transform infrared attenuated total reflection (FTIR-ATR) technique that the water uptake of the ISMs is influenced by the composition of the sample solution.31 Thus water accumulation is a complex process, in which a thin aqueous layer or water pools will be formed and behave as small electrolyte reservoirs with ill-defined ion activity. Owing to their extremely small volume, the ion composition of the aqueous layer can be changed even by minute transmembrane fluxes of the primary and interfering ions.25 This induces severe potential instability and hinders the wide applicability of the relevant SC-ISEs.25 Hence, it is crucial to minimize the water uptake of the ISM and the SC layer that could be achieved using highly hydrophobic SC materials, most optimally combined with a low diffusivity hydrophobic ISM membrane matrix.27,28,31
The dominance of ECPs was challenged lately by nanostructured carbonaceous materials, e.g. 3D ordered mesoporous carbon,32 carbon nanotubes,33 as well as graphene and reduced graphene oxide.34,35 These materials have a high surface area that, owing to their large capacitance, stabilizes very effectively the electrode potential of the relevant SC-ISEs. Importantly, they generally show no photovoltaic effects and are more inert than ECPs, which seems to offer essential advantages in terms of potential stability and avoiding the formation of an aqueous layer beneath the ISM.36,37 However, in many cases the handling and implementation of these carbonaceous materials in ISE fabrication is not as straightforward as that in the case of ECP-based SCs.38 Additionally, their inertness depends on the surface functionalities and/or surface adsorbed dispersing agents that should be minimized to avoid aqueous layer formation. This requirement is rather similar to many of the ECP solid contacts where the oxidation state of the polymer needs careful consideration as well as the removal of the excess of doping ions, which generally increases the hydrophilicity of the ECPs.39 Very recently, improved hydrophobicity was reported for the composite SC material of few-layered graphene and PANI in Ca2+-selective SC-ISEs.40 The synergistic effect between the ECP and the few-layer graphene results in improved redox capacitance. Therefore, we were interested in implementing an all hydrocarbon based ECP as a solid contact in conjunction with a lipophilic counter ion. Based on previous investigations,41 we expected polyazulene (PAz) to be especially beneficial as the SC layer in SC-ISEs owing to its high hydrophobicity in the semiconducting form (water contact angle: 98 ± 11°), which is slightly higher than that for graphene,42 and also due to its relatively high areal redox capacitance (ca. 45 mF cm−2; measured for the electrically conducting form).41 Therefore in this work, we studied the use of electrochemically synthesised PAz as the ion-to-electron transducer solid contact in potassium-selective SC-ISEs and tested it for potassium measurement in the clinically relevant range in artificial serum. This task is especially demanding as the reference range of potassium in serum or blood is 3.5–5.1 mM![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 43 and such a narrow range requires K+-selective SC-ISEs with extremely good potential stability.
43 and such a narrow range requires K+-selective SC-ISEs with extremely good potential stability.
Experimental
Chemicals
Azulene (99%), acetonitrile (ACN, 99.5%, anhydrous) and KCl were received from Aldrich and used without further purification. High molecular weight poly(vinyl chloride) (PVC), bis(2-ethylhexyl) sebacate (DOS), potassium tetrakis[3,5-bis(trifluoromethyl)phenyl]borate (KTFPB), potassium ionophore I (valinomycin), and tetrahydrofuran (THF), all of Selectophore grade, and tetrabutylammonium hexafluorophosphate (TBAPF6, 99%) were purchased from Fluka and used as received. The regioregular poly(3-octylthiophene-2,5-diyl) (POT; Mn = 34000) with an electrical conductivity of 10−6 S cm−1 was purchased from Sigma-Aldrich. Deionized water (ELGA, resistivity: 18.2 MΩ cm) was used to prepare the aqueous solutions. Thermo Scientific provided the three calibration solutions (Cal 1: 4.50 mM K+, 0.6 mM Mg2, 140 mM Na+, 1.25 mM Ca2+, 101.5 mM Cl−, pH = 7.40; Cal 2: 6.00 mM K+, 0.3 mM Mg2, 120 mM Na+, 0.75 mM Ca2+, 82 mM Cl−, pH = 7.00; Cal 3: 3.00 mM K+, 0.3 mM Mg2, 150 mM Na+, 1.75 mM Ca2+, 108 mM Cl−, pH = 7.80)44 and the human serum (Nortrol) which is used as control serum for in vitro validation of selected instruments and methods defined by Thermo Fisher Scientific.
Electropolymerization and polarization of azulene
Azulene was electrochemically polymerized on glassy carbon (GC) substrates incorporated into polyether ether ketone (PEEK) bodies. The GC/PEEK electrodes had an outer diameter of 6 mm with the GC surface area of 0.02 cm2 (Ø 1.6 mm). They were polished with 0.05 μm Al2O3 paste and rinsed with deionized water and ACN. The polymerization was performed in a three-electrode cell connected to an Autolab PGSTAT 30 potentiostat (equipped with the GPES software). A coiled Pt wire and a Ag/AgCl wire served as the counter and the pseudoreference electrode (calibrated vs. ferrocene/ferrocenium), respectively. All PAz films (Fig. 1a) were formed by cycling the potential between −0.6 V and 1.2 V (10 cycles, ν = 50 mV s−1) in ACN solutions containing 0.01 M azulene and 0.1 M TBAPF6. The TBAPF6 was dried under vacuum at 80 °C for 30 min prior to use. Prior to the ISM deposition, the PAz solid-contacts were polarized at 0.2 V for 5 min in a 0.1 M TBAPF6-ACN solution to improve the reproducibility of the SC-ISE by adjusting the oxidation state of the PAz films, which are both n- and p-dopable.45 The p-doping starts at ca. 0–0.1 V and the polymer backbone is therefore positively charged at E > 0.1 V. Three mL solutions purged with nitrogen gas for 5 min were used for both the electropolymerization and the polarization of the PAz films. After each step, the PAz films were rinsed with ACN and dried in air for 5 min.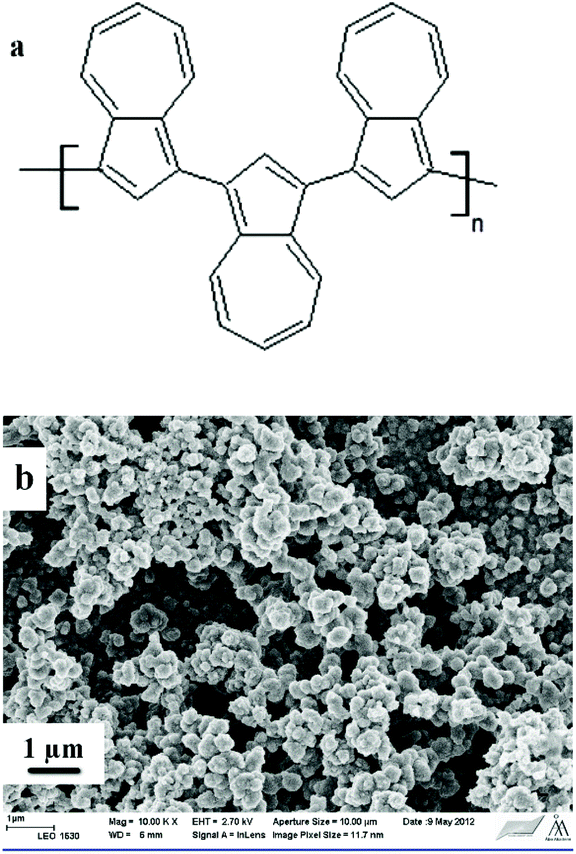 | ||
| Fig. 1 (a) The chemical structure of PAz. (b) SEM image of a PAz film electropolymerized on ITO glass, magnification: 10000×. | ||
Fabrication of the K+-selective SC-ISEs
A valinomycin based membrane cocktail solution was prepared by adding 100 mg of the membrane components (32.9% (w/w) PVC, 65.7% DOS, 1.0% valinomycin and 0.4% KTFPB) into 1 ml THF solution. After the electropolymerization and polarization of the PAz films, altogether 30 μl of the membrane cocktail was deposited onto electrosynthesised porous PAz films (Fig. 1b) to form ISMs with the thickness of 90–110 μm. The casting was made by applying successively three drops (3 × 10 μl) at ca. 1 min intervals to prevent sensor failure related to pinhole formation.6 The POT-based SC-ISEs were prepared by pipetting 2 × 0.5 μl of a POT solution (10 mg ml−1 in chloroform) on the GC substrate. After drying under an ambient atmosphere for 10 min, 30 μl (3 × 10 μl) of the K+-selective ISM cocktail (as described above but with 200 mg of the membrane components dissolved in 1 ml THF) was applied on top of the POT layer. For all SC-ISEs, the ISM covered the entire surface of the GC/PEEK electrode. The coated wire electrodes (CWEs) were prepared in a similar way, but the membrane cocktail was directly cast onto the GC/PEEK electrode surface. The electrodes were then allowed to dry overnight.Potentiometric measurements
The potentiometric response of the PAz-based SC-ISEs was measured with a 16-channel potentiometer (Lawson Labs, Inc., input impedance: 1015 Ω) at room temperature (23 ± 2 °C). The reference electrode used for all the potentiometric measurements was a double junction Ag/AgCl/3 M KCl//1 M LiOAc electrode. Four identical SC-ISEs were always used in the potentiometric measurements and their response characteristics were compared with four POT-based SC-ISEs or CWEs.The measurement of the initial potential stability upon making the PAz- and POT-based SC-ISEs come into contact, for the first time, with aqueous electrolyte solution was carried out in 10−3 M KCl without stirring for ca. 50 h. In the case where the PAz-based SC-ISEs and their CWE counterparts were evaluated in terms of their lower limit of detection (LOD), a special conditioning preceded the calibration, i.e., after measuring the initial potential stability, the electrodes were kept in 10−4 M KCl solution for 24 h and then placed in 10−9 M KCl solution for 4 h. During these last 4 hours, the KCl solution was replaced each hour by a fresh solution. Before the calibration of the electrodes, the electrodes were placed in 10−4 M KCl solution for ca. 10 min. The calibrations were done in KCl solutions from 10−3 to 10−9 M with the successive automatic dilution system (711 Liquino and Dosino 700, Metrohm, Herisau, Switzerland), by successively replacing 68.4% of the KCl solution in the cell with deionized water. The electrode potentials were measured in a Faraday's cage for 5 min at each concentration step by stirring the solution during the first minute. The potential (EMF) values were corrected for the liquid junction potentials and the activity coefficients were calculated according to the Debye–Hückel equation. The LOD was determined from the intersection of the two straight lines representing the linear part of the SC-ISE response and the low activity range where the electrode is not anymore responding to the changes in the K+-activity. The E0 values reported correspond to extrapolation of the linear potential response to aK+ = 1.
Before starting the selectivity measurements, the SC-ISEs and CWEs were equilibrated in a 10−4 M KCl solution for 4 h. The electrode potentials were measured first in 0.1 M chloride solutions of the interfering ions (Mg2+, Li+, Na+, NH4+, Ca2+, in that order) and finally in the 0.1 M KCl. The potentiometric aqueous layer tests were performed by recording the changes in the potential of three identically prepared SC-ISEs and CWEs in 0.1 M KCl, 0.1 M NaCl and then 0.1 M KCl for 4 h in each solution. The oxygen and carbon dioxide sensitivity of fully equilibrated PAz-based SC-ISEs was measured in a stirred 0.1 M KCl solution by purging it with pure O2, N2 or CO2 gas in the following measuring sequence: N2 (1 h)/O2 (1 h)/N2 (1 h)/CO2 (1 h)/N2 (1 h). The light sensitivity was measured in 100 ml of 0.1 M KCl solution by using a Leica CLS 150 XE cold light source equipped with a tungsten halogen lamp (Philips EKE 150 W, 21 V) directed through the electrolyte solution towards the SC-ISE surfaces. The ambient room light was provided by fluorescent lamps (Universal Thermo, 36 W/840). Before starting the test, the electrode potentials were monitored in the dark for ca. 4 h until the potentials had stabilized. The following measuring sequence was used during the light sensitivity test: ambient room light (30 min), cold light (30 min), ambient room light (30 min) and dark conditions (30 min).
For the measurement of potassium in human serum, three identical PAz-based SC-ISEs were fabricated. To determine the K+ concentration in the control serum, a three-point calibration was performed by using three different calibrator solutions with K+ concentrations of 3.0, 4.5, and 6.0 mM (±1%). Before these measurements, all the electrodes were conditioned overnight in 10−3 M KCl.
Polarization of the solid-contacts
Constant-current chronopotentiometry was performed to study the polarization of the PAz and POT solid contacts and their redox capacitances. The measurements were conducted using the Autolab PGSTAT 30 potentiostat in the three electrode setup with GC/PEEK, a GC rod and the double junction Ag/AgCl/3 M KCl//1 M LiOAc electrode serving as the working, counter and reference electrodes, respectively. The potentials of the PAz and POT-based K+-SC-ISEs were recorded in 0.1 M KCl while a constant current of +10−9 A was applied to the electrodes for 60 s followed by −10−9 A for another 60 s.Results and discussion
Potential stability of the SC-ISEs at their first sample contact
For the practical application of potassium electrodes either in a blood gas analyser or point of care devices, the time required for conditioning, i.e., the time required to achieve the desired potential stability, is essential. This is an often overlooked aspect and the potential stabilities of the SC-ISEs are generally reported after extensive conditioning, which does not guarantee their prompt use after fabrication or unpacking. Therefore, we investigated the potential stability at the first contact of the electrodes with an aqueous solution. In addition to the different equilibration processes, which include the water uptake and contingent formation of an aqueous layer, the PAz solid-contacts were additionally pre-polarized before the ISM deposition, which minimizes additional drifting. The pre-polarization was performed not only to adjust the oxidation state of the PAz polymer to obtain SC-ISEs with reproducible E0 values, but also to ensure a suitable hydrophobicity of the polymer film while keeping the potential in the thermodynamically stable region.The value of 0.2 V was chosen to fulfil these criteria as it was recently shown that the open circuit potential of neat PAz films polarized at 1.0 V slowly drifted to ca. 0.2 V upon their first contact with an aqueous solution (0.1 M CaCl2).41 Moreover, while PAz is only partially oxidized (i.e. electrically conductive) at this potential, it is still very hydrophobic with a water contact angle of 98 ± 11°.41 Therefore, we assumed that the PAz solid contact pre-polarized at this potential will exhibit minimal oxidation state change even in the worst case scenario of having an aqueous layer accumulated beneath the ISM as a result of the transmembrane flux of water.
We have compared the performance of the PAz solid contact with the commonly used chemically synthesised POT solid-contact due to their relatively similar hydrophobicity represented by the water contact angles of 98 ± 11° (PAz polarized at 0.2 V) and ca. 110° (POT).46Fig. 2 shows clearly the beneficial effect of pre-polarized PAz-based SCs on the potential stability of the respective SC-ISEs as compared with the commonly used POT-based SC-ISEs upon their first contact with an aqueous solution (10−3 M KCl). The average of the EMF values and their standard deviation were calculated for four identically prepared PAz- and POT-based SC-ISEs. After t = 2 min, 0.5 h, 10 h, 20 h, and 30 h in contact with the 10−3 M KCl solution, the standard deviations (SD) of the potentials of the PAz-based SC-ISEs were 3.1, 3.4, 2.6, 1.4, and 0.9 mV (n = 4), while they were 11.2, 12.7, 16.5, 18.1, and 17.4 mV (n = 4) for the POT-based SC-ISEs. Thus the potentials of the POT-based SC-ISEs show a divergence in time while the SDs of the PAz-based ISEs are in fact slightly decreasing so that they are almost 20 times smaller after 30 h than the SDs of their POT-based counterpart. The potential drifts calculated for PAz- and POT-based K+-selective SC-ISEs were 0.1 mV h−1 and 0.5 mV h−1, respectively, between t = 2 min and 49.5 h. However, between t = 10 h and 49.5 h the drift was only 28 μV h−1 for the PAz-based SC-ISE, unlike the POT-based SC-ISEs that still showed a potential drift of 0.5 mV h−1. These data show undeniably that the PAz-based SC-ISEs surpass the commonly used POT-based SC-ISEs (using chemically prepared POT) in their initial potential stability and the reproducibility of their standard potential. Moreover, the results presented here suggest that the PAz-based SC-ISEs may be used in the best case already after a conditioning (equilibration) time of only 2 min. However, the standard deviation of the PAz-based SC-ISEs is still too high to be used in calibration-free applications.
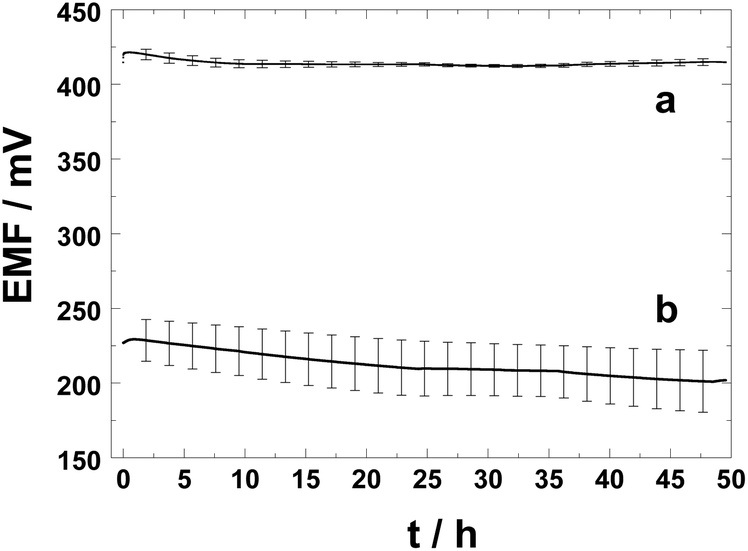 | ||
| Fig. 2 Potential response of (a) the PAz-based and (b) POT-based SC K+-selective electrodes (n = 4) at their first contact (after fabrication) with a 10−3 M KCl solution. | ||
Calibration of the K+-selective SC-ISEs
While the clinical application of SC K+-selective electrodes is not demanding, in terms of requiring very low LODs to fully explore the analytical performance characteristics of the PAz-based SC-ISEs, this aspect was also carefully investigated by using extensive conditioning at low K+ concentrations.47 The SC-ISEs were calibrated in 10−3 M to 10−9 M KCl solutions (high to low concentration) and the slopes of the electrode responses were calculated from the linear part (10−3–10−6 M) of the calibration plots. A representative potential trace for the K+-selective SC-ISEs is shown in Fig. 3a with the relevant calibration curves for 4 different electrodes plotted in Fig. 3b. The SC-ISEs had a close to Nernstian slope of 57.0 ± 0.3 mV per decade (r2 = 0.9999; mean residual standard deviation, sres = 1.4 ± 0.8) and a LOD of (7.2 ± 0.9) × 10−8 M. For comparison the CWEs (not shown) exhibited almost an order of magnitude higher LOD (2.8 ± 0.4) × 10−7 M with a slope of 56.6 ± 1.4 mV per decade (r2 = 0.9997). The SD of the E0 values for the K+-selective SC-ISEs was ± 7.9 mV, which is inferior to SC-ISEs based on implementing dedicated redox species loaded to the membrane to adjust the E0.48 However, the simplicity of the preparation procedure may easily compensate for this deficiency for most routine applications and as expected the benefits are obvious compared with CWEs showing SD exceeding 40 mV (not shown).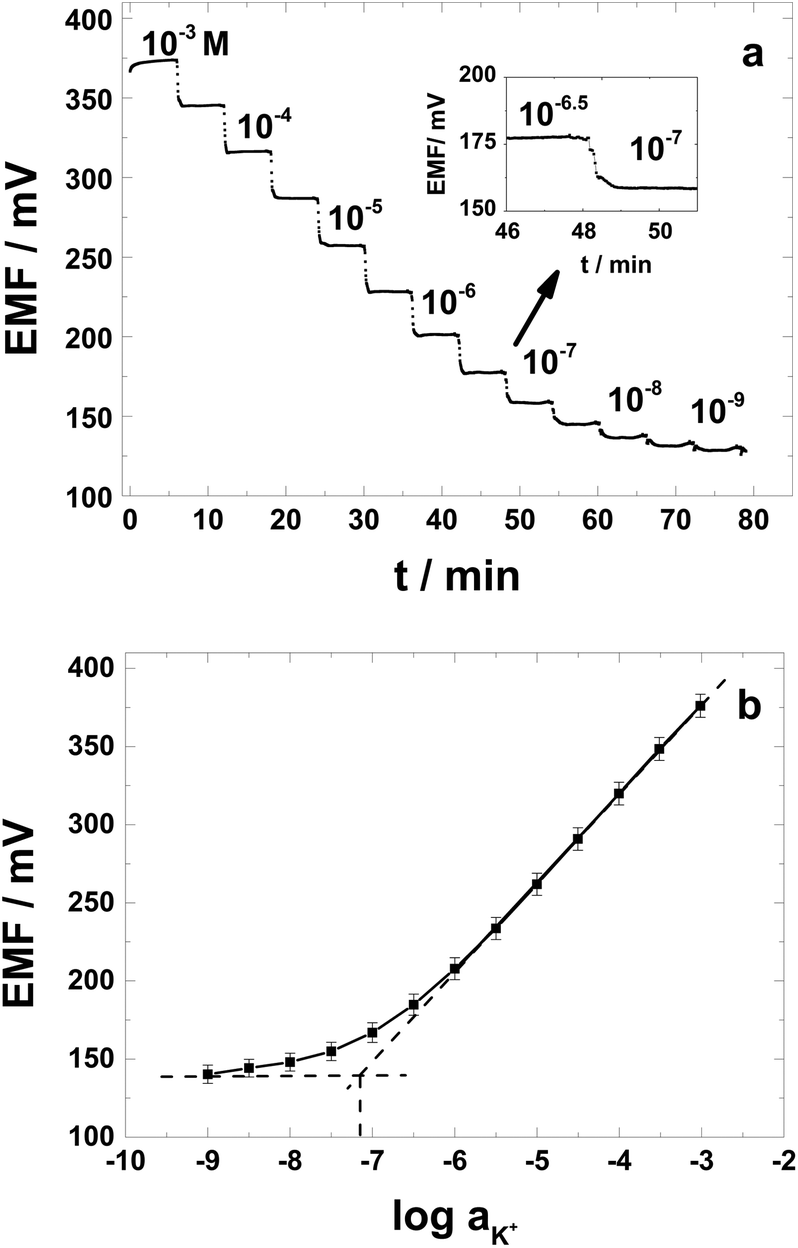 | ||
| Fig. 3 PAz-based SC-ISEs: (a) the potential trace curve measured when the K+ concentration was successively lowered and (b) the calibration graph (n = 4). Inset in (a): expansion of the selected concentration range. | ||
After conditioning in 10−4 M KCl solution, the potentiometric selectivity coefficients of the SC-ISEs were determined by the separate solution method (SSM)49 at the 0.1 M level using chloride salt solutions of Mg2+, Li+, Na+, NH4+ and Ca2+. The obtained potentiometric selectivity coefficients of K+ against Mg2+, Li+, Na+, NH4+ and Ca2+ were −4.0 ± 0.1, −5.0 ± 0.1, −4.3 ± 0.1, −4.3 ± 0.1 and −4.0 ± 0.1, respectively (n = 4). In general, the choice of the solid-contact should not influence the selectivity of the ISM and indeed these values were in relatively good agreement with the selectivity coefficients reported in the literature for the valinomycin-based electrode and fulfill the selectivity requirements for measurements in blood or serum.50
Potentiometric aqueous layer test: oxygen, carbon dioxide and light sensitivity
As explained in the introduction, the formation of a thin aqueous layer or pools of aqueous solutions beneath the ISM in the SC-ISEs can counteract all the benefits of the SC.16,25 We used the potentiometric aqueous layer test to investigate whether the PAz-based SC can avoid the accumulation of such a layer.25 Thus the SC-ISEs were exposed successively to a relatively high concentration (0.1 M) of the primary ion (K+), then to the same concentration of the interfering ion (Na+) and finally to the primary ion.The lack of potential drifts for the K+-selective SC-ISEs in the aqueous layer test (Fig. 4) shows no indications of an aqueous layer/pool formation. In contrast, the CWE shows a drifting potential response pattern typical of the presence of an aqueous layer. This is very remarkable since for conventional plasticised PVC membranes, due to their high water uptake, it is very difficult to avoid the aqueous layer formation for most ECP-based solid contacts which are either in their semi-conducting or conducting form. Very importantly, upon placing back the SC-ISEs into the 0.1 KCl solution following exposure for 4 h to 0.1 M NaCl, the potential drift was less than 0.1 mV h−1, i.e., even after such large changes in concentration, the PAz-based SC-ISEs had a practically driftless response. It has been previously reported that no aqueous layer formation could be detected with solvent-cast POT-based Ca2+-selective SC-ISEs51 and therefore this electrode type was not included in the aqueous layer test in this work.
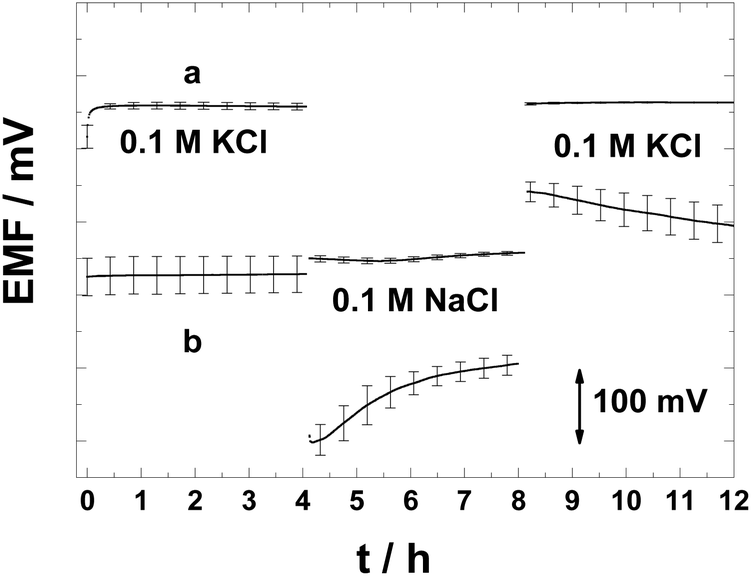 | ||
| Fig. 4 Potentiometric aqueous layer test for the K+-selective (a) PAz-based SC-ISEs and (b) CWEs. At t = 4 h, the primary ion (K+) was changed to the interfering ion (Na+) solution and replaced again with the primary ion solution at t = 8 h (n = 3). | ||
The sensitivities to O2 and CO2 of the polarized PAz-based SC-ISEs are shown in Fig. 5a. The potential increased only 1.0 mV during 1 h while the 0.1 M KCl solution was purged with pure oxygen. This minute change indicates that in practice, where such dramatic oxygen concentration changes are unlikely, the SC-ISEs are essentially insensitive to oxygen. The PAz-based SC-ISEs showed no significant changes in potential upon purging the KCl solution with pure CO2 for 1 h.
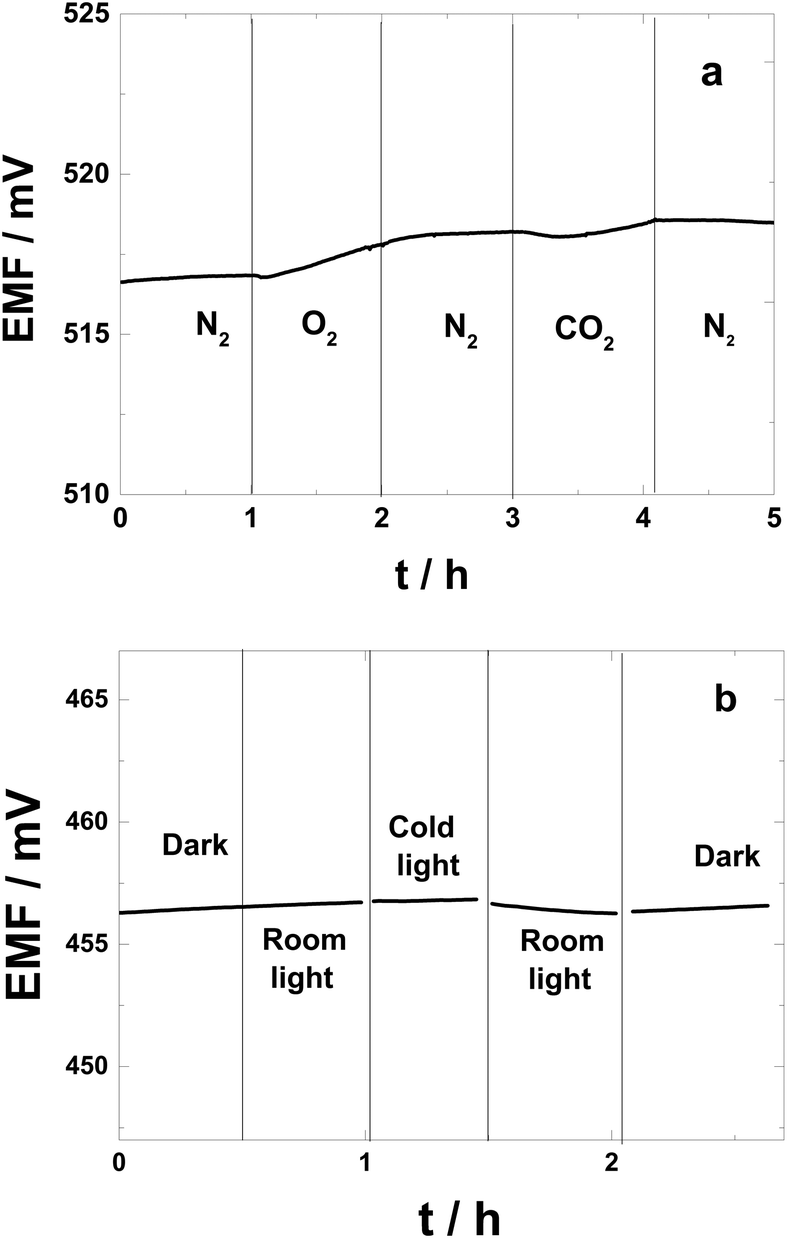 | ||
| Fig. 5 (a) The sensitivity to O2 and CO2 and (b) light of the PAz-based SC-ISE measured in 0.1 M KCl. | ||
To determine the light sensitivity of the SC-ISEs,24 the reference electrode was protected with an aluminum foil to eliminate the contribution of the photovoltaic response of the Ag/AgCl wire52 from the measured potential response. Assuming that the ISM components are photostable, the light sensitivity of the SC-ISEs can be ascribed to the redox reaction of the inner SC layer, i.e. changes in its oxidation state, which results in potential drifts. In the light sensitivity study, the changes in the potential of the PAz-based SC-ISEs were measured upon changing the illumination between dark conditions, ambient room light and intensive direct exposure to a cold light source as shown in Fig. 5b. The results show that the illumination conditions had very little effect on the potential of the SC-ISEs. The change in potential as a result of changing from dark to ambient room light conditions (and back) was practically undetectable. The most noticeable change in potential was recorded on exposure to cold light, but even in this case the associated potential decrease was less than 0.5 mV during 30 min. Thus the PAz solid-contact ISEs under normal measurement conditions should have no light sensitivity. This is a major advantage with respect to POT-based solid contact ISEs, one of the best performing ECP-based solid-contacts in terms of potential stability and aqueous layer elimination, which is extremely light sensitive (i.e., more than 300 mV when switching from dark to a cold light source).24
Constant-current chronopotentiometry
We have performed constant-current chronopotentiometric measurements to compare the polarizability of the PAz and POT solid contacts and to estimate the redox capacitance of the SCs, which is an important parameter for obtaining SC-ISEs with stable potentials.53 The chronopotentiograms in Fig. 6 show clearly that the POT solid contact is easily polarized when a constant current of ±1 nA was applied for 60 s causing the potential to increase by 48.2 mV (t = 2–60 s; Δt = 58 s). This corresponds to a low capacitance of 1.2 μF (59.7 μF cm−2; ΔE/Δt = I/C) that is close to the capacitance of the CWE.53 The low interfacial capacitance is due to the fact that pristine POT is practically non-conducting (ca. 10−6 S cm−1).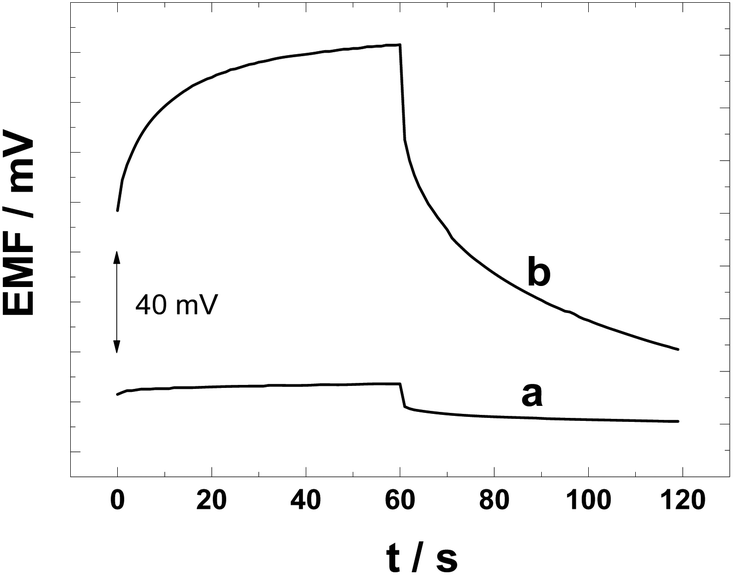 | ||
| Fig. 6 Chronopotentiograms measured in 0.1 M KCl for (a) the PAz-based and (b) POT-based K+-SC-ISEs. Applied current: +10−9 A for the first 60 s and −10−9 A for the next 60 s. The electrode potentials have been shifted along the y-axis in relation to each other. | ||
In comparison to the POT-based SC-ISEs, the potential of the PAz-based SC-ISE increased only 2.7 mV (t = 2–60 s) upon the constant-current polarization (Fig. 6). This shows that the PAz-based solid-contact having a redox capacitance of 21.2 μF (1.05 mF cm−2) has much better resistance against polarization than the POT counterpart. It should be noted that the redox capacitance of PAz in this work is much lower than that reported earlier (45 mF cm−2) due to the semi-conducting nature of the PAz film that has been polarized at 0.2 V.
Measurement of potassium in human serum
To test the adequacy of the PAz-based SC K+-ISEs for potassium assay in the clinically relevant range, calibrations were performed using the same standard solutions as used for calibration of commercial clinical analyzers that mimicks the ion composition of the serum. As shown in Fig. 7a, staircase type potential responses were generated upon changing solutions, i.e., the three calibrator solutions and the serum. The potential stability in the standard solutions (Cal 1–Cal 3) expressed as the standard deviation of the potential values was less than 0.1 mV except for the calibration standard that was first measured (3 mM K+; i.e. Cal 3 with serum-like high ionic strength), where we registered a standard deviation of 0.12 mV. The higher standard deviation is most likely due to ionic strength effects when placing the SC-ISEs directly from the low ionic strength conditioning solution (1 mM KCl) to the first calibration solution with a much higher ionic strength. In the human serum (Nortrol) the short time stability slightly worsened as compared with the calibration solutions, i.e., 0.19 mV. The slope of three identically prepared PAz-based SC-ISEs was close to Nernstian (58.6 ± 0.6 mV per decade) (Fig. 7b) with an average E0 of 536.9 ± 7 mV. The potassium concentration in the serum determined with the three SC-ISEs was 3.93 mM with a remarkably low standard deviation of only 0.024 mM. Compared with the nominal concentration of K+ in the Nortrol solution, 3.8 ± 0.08 mM, this represents an error of 0.13 mM. Thus the PAz-based K+-selective SC-ISEs seem to fulfil the general requirements for clinical use. Since, in this preliminary study, we used a conventional reference electrode and routine open cell laboratory conditions, we consider this result to be especially promising for the application of PAz electrodes in clinical potassium analyzers with dedicated thermostated flow-through setups and built-in calibration routines.54 The slight discrepancy may have an essential contribution from the reference electrode potential, which might be affected in the high protein content serum solution.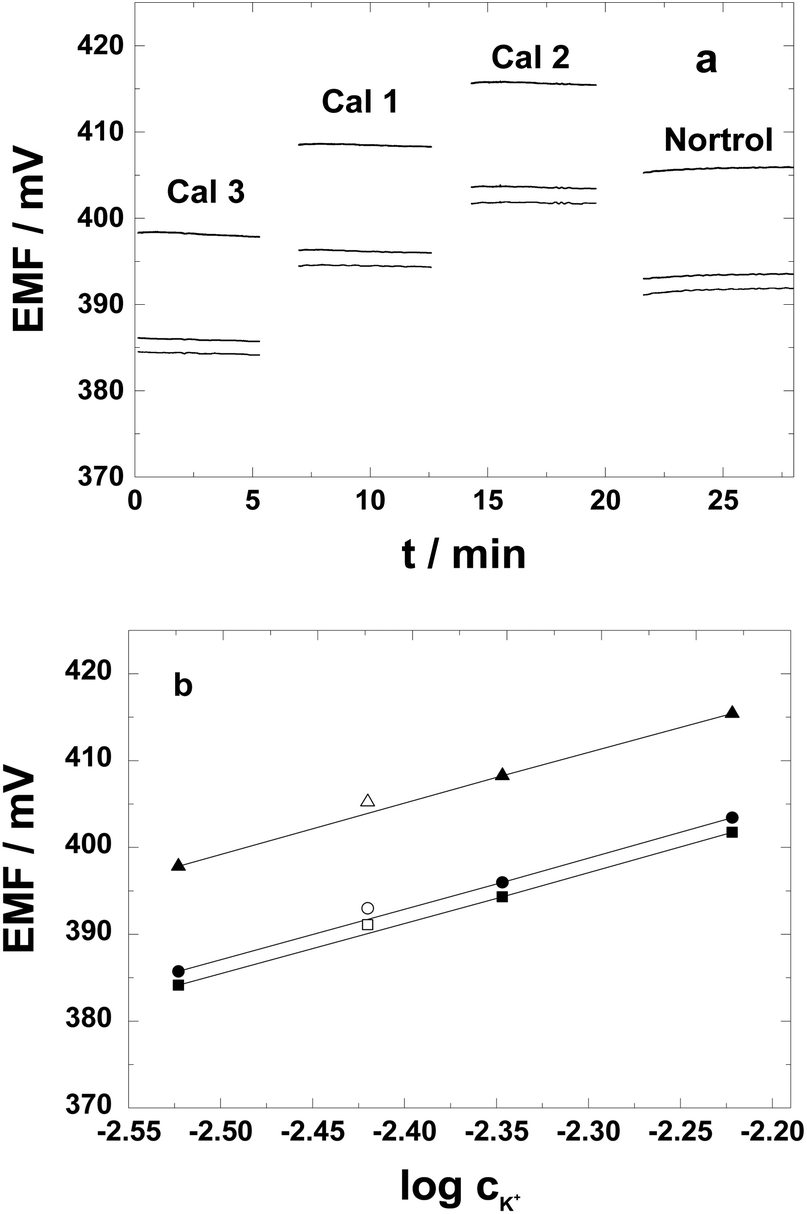 | ||
| Fig. 7 (a) Potential responses and (b) calibration graphs for three different PAz-based SC-ISEs (filled symbols) and the respective measured potentials in the Nortrol artificial serum solution plotted against their nominal concentration (hollow symbols). | ||
Conclusions
The results indicate that the PAz-based SC may confer unique advantages to SC-ISEs. The pre-polarization of the solid contact prior to the deposition of the ISM membranes at a rationally chosen potential, which provides both thermodynamic stability and hydrophobicity to the polymer layer, reduced the E0 variation of the respective ISEs in the worst case to ±7.9 mV (n = 4). While various solid contacts based on carbonaceous materials that are combined with redox buffers were reported to achieve an even better E0 reproducibility,55 the convenience and simplicity of the PAz fabrication may compensate for this minor deficiency in many applications. The complete lack of aqueous layer formation beneath the ISM even when using a plasticized PVC-based membrane with a conventional composition, which has notoriously high water uptake, makes PAz a robust alternative to the state-of-the-art SCs. Its insignificant light, oxygen and carbon dioxide sensitivity and excellent potential stability upon initial contact with the aqueous samples seem to surpass the best ECP-based SCs (e.g. POT). Finally, the preliminary results obtained using prepolarized PAz-based SC-ISEs for measuring the concentration of K+ ions in artificial serum are very promising in terms of achieving the required accuracy and coefficient of variation.Acknowledgements
The financial support from the Academy of Finland (project numbers 260036, 130588 and 263656), the Lendület program of the Hungarian Academy of Sciences (LP2013-63/13) and ERA-Chemistry (2014, 61133; OTKA NN117637) is gratefully acknowledged. Thermo Scientific is also gratefully acknowledged for providing the calibrator and artificial serum solutions. Moreover, the authors thank Prof. Carita Kvarnström, Dr Beatriz Meana-Esteban, Dr Anna Österholm and Dr Rose-Marie Latonen for directing our attention to polyazulene.Notes and references
- J. Bobacka, A. Ivaska and A. Lewenstam, Electroanalysis, 2003, 15, 366–374 CrossRef CAS.
- A. Lewenstam, M. Maj-Zurawska and A. Hulanicki, Electroanalysis, 1991, 3, 727–734 CrossRef CAS.
- E. Bakker and E. Pretsch, in Electroanalytical Chemistry: A Series of Advances, vol 24, ed. A. J. Bard and C. Zoski, CRC Press-Taylor & Francis Group, Boca Raton, 2012, vol. 24, pp. 1–73 Search PubMed.
- E. Lindner, R. E. Gyurcsányi and E. Pretsch, in Applications of supramolecular chemistry for 21st century technology, ed. H.-J. Schneider, Taylor & Francis, Boca Raton, 2012, pp. xi, 441 p Search PubMed.
- R. Paciorek, P. D. van der Wal, N. F. de Rooij and M. Maj-Żurawska, Electroanalysis, 2003, 15, 1314–1318 CrossRef CAS.
- R. E. Gyurcsányi, N. Rangisetty, S. Clifton, B. D. Pendley and E. Lindner, Talanta, 2004, 63, 89–99 CrossRef PubMed.
- F. Sundfors, R. Bereczki, J. Bobacka, K. Tóth, A. Ivaska and R. E. Gyurcsányi, Electroanalysis, 2006, 18, 1372–1378 CrossRef CAS.
- C. Zuliani and D. Diamond, Electrochim. Acta, 2012, 84, 29–34 CrossRef CAS.
- M. Novell, M. Parrilla, G. A. Crespo, F. X. Rius and F. J. Andrade, Anal. Chem., 2012, 84, 4695–4702 CrossRef CAS PubMed.
- W. J. Lan, X. U. Zou, M. M. Hamedi, J. B. Hu, C. Parolo, E. J. Maxwell, P. Bühlmann and G. M. Whitesides, Anal. Chem., 2014, 86, 9548–9553 CrossRef CAS PubMed.
- J. Hu, K. T. Ho, X. U. Zou, W. H. Smyrl, A. Stein and P. Bühlmann, Anal. Chem., 2015, 87, 2981–2987 CrossRef CAS PubMed.
- A. J. Bandodkar, V. W. S. Hung, W. Jia, G. Valdes-Ramirez, J. R. Windmiller, A. G. Martinez, J. Ramirez, G. Chan, K. Kerman and J. Wang, Analyst, 2013, 138, 123–128 RSC.
- B. Schazmann, D. Morris, C. Slater, S. Beirne, C. Fay, R. Reuveny, N. Moyna and D. Diamond, Anal. Methods, 2010, 2, 342–348 RSC.
- J. Szücs and R. E. Gyurcsányi, Electroanalysis, 2012, 24, 146–152 CrossRef.
- J. Szücs, E. Pretsch and R. E. Gyurcsányi, Analyst, 2009, 134, 1601–1607 RSC.
- E. Lindner and R. E. Gyurcsányi, J. Solid State Electrochem., 2009, 13, 51–68 CrossRef CAS.
- A. Cadogan, Z. Gao, A. Lewenstam, A. Ivaska and D. Diamond, Anal. Chem., 1992, 64, 2496–2501 CrossRef CAS.
- J. Sutter, A. Radu, S. Peper, E. Bakker and E. Pretsch, Anal. Chim. Acta, 2004, 523, 53–59 CrossRef CAS.
- M. Vazquez, J. Bobacka, A. Ivaska and A. Lewenstam, Sens. Actuators, B, 2002, 82, 7–13 CrossRef CAS.
- T. Lindfors and A. Ivaska, Anal. Chem., 2004, 76, 4387–4394 CrossRef CAS PubMed.
- M. Guzinski, J. M. Jarvis, B. D. Pendley and E. Lindner, Anal. Chem., 2015, 87, 6654–6659 CrossRef CAS PubMed.
- J. Dumańska and K. Maksymiuk, Electroanalysis, 2001, 13, 567–573 CrossRef.
- A. Michalska and K. Maksymiuk, J. Electroanal. Chem., 2005, 576, 339–352 CrossRef CAS.
- T. Lindfors, J. Solid State Electrochem., 2009, 13, 77–89 CrossRef CAS.
- M. Fibbioli, W. E. Morf, M. Badertscher, N. F. De Rooij and E. Pretsch, Electroanalysis, 2000, 12, 1286–1292 CrossRef CAS.
- Z. Li, X. Li, S. Petrović and D. J. Harrison, Anal. Chem., 1996, 68, 1717–1725 CrossRef CAS.
- F. Sundfors, T. Lindfors, L. Höfler, R. Bereczki and R. E. Gyurcsányi, Anal. Chem., 2009, 81, 5925–5934 CrossRef CAS PubMed.
- T. Lindfors, F. Sundfors, L. Höfler and R. E. Gyurcsányi, Electroanalysis, 2009, 21, 1914–1922 CrossRef CAS.
- R. De Marco, J.-P. Veder, G. Clarke, A. Nelson, K. Prince, E. Pretsch and E. Bakker, Phys. Chem. Chem. Phys., 2008, 10, 73–76 RSC.
- J.-P. Veder, R. De Marco, G. Clarke, S. P. Jiang, K. Prince, E. Pretsch and E. Bakker, Analyst, 2011, 136, 3252–3258 RSC.
- N. He and T. Lindfors, Anal. Chem., 2013, 85, 1006–1012 CrossRef CAS PubMed.
- C.-Z. Lai, M. A. Fierke, A. Stein and P. Bühlmann, Anal. Chem., 2007, 79, 4621–4626 CrossRef CAS PubMed.
- G. A. Crespo, S. Macho and F. X. Rius, Anal. Chem., 2008, 80, 1316–1322 CrossRef CAS PubMed.
- R. Hernández, J. Riu, J. Bobacka, C. Vallés, P. Jiménez, A. M. Benito, W. K. Maser and F. X. Rius, J. Phys. Chem. C, 2012, 116, 22570–22578 Search PubMed.
- J. Ping, Y. Wang, J. Wu and Y. Ying, Electrochem. Commun., 2011, 13, 1529–1532 CrossRef CAS.
- E. Jaworska, W. Lewandowski, J. Mieczkowski, K. Maksymiuk and A. Michalska, Talanta, 2012, 97, 414–419 CrossRef CAS PubMed.
- F. Li, J. Ye, M. Zhou, S. Gan, Q. Zhang, D. Han and L. Niu, Analyst, 2012, 137, 618–623 RSC.
- J. Szűcs, T. Lindfors, J. Bobacka and R. E. Gyurcsányi, Electroanalysis, 2015 DOI:10.1002/elan.201500465.
- J. Sutter, E. Lindner, R. E. Gyurcsányi and E. Pretsch, Anal. Bioanal. Chem., 2004, 380, 7–14 CrossRef CAS PubMed.
- Z. Boeva and T. Lindfors, Sens. Actuators, B, 2016, 224, 624–631 CrossRef CAS.
- N. He, L. Höfler, R.-M. Latonen and T. Lindfors, Sens. Actuators, B, 2015, 207, 918–925 CrossRef CAS.
- R. Raj, S. C. Maroo and E. N. Wang, Nano Lett., 2013, 13, 1509–1515 CrossRef CAS PubMed.
- R. W. Burnett, A. K. Covington, N. Fogh-Andersen, W. R. Külpmann, A. Lewenstam, A. H. J. Maas, O. Müller-Plathe, C. Sachs, O. Siggaard-Andersen, A. L. Van Kessel and W. G. Zijlstra, Clin. Chem. Lab. Med., 2000, 38, 1065 CAS.
- N. Blomqvist-Kutvonen, J. Öst and A. Lewenstam, in Comprehensive Analytical Chemistry, ed. S. Alegret and A. Merkoçi, Elsevier, 2007, vol. 49, pp. e5–e11 Search PubMed.
- A. Österholm, A. Petr, C. Kvarnström, A. Ivaska and L. Dunsch, J. Phys. Chem. B, 2008, 112, 14149–14157 CrossRef PubMed.
- L. Robinson, J. Isaksson, N. D. Robinson and M. Berggren, Surf. Sci., 2006, 600, L148–L152 CrossRef CAS.
- N. Rubinova, K. Chumbimuni-Torres and E. Bakker, Sens. Actuators, B, 2007, 121, 135–141 CrossRef CAS.
- T. Lindfors, L. Höfler, G. Jágerszki and R. E. Gyurcsányi, Anal. Chem., 2011, 83, 4902–4908 CrossRef CAS PubMed.
- E. Bakker, E. Pretsch and P. Bühlmann, Anal. Chem., 2000, 72, 1127–1133 CrossRef CAS PubMed.
- B. Paczosa-Bator, Talanta, 2012, 93, 424–427 CrossRef CAS PubMed.
- J. Sutter and E. Pretsch, Electroanalysis, 2006, 18, 19–25 CrossRef CAS.
- H. Freiser, Ion-selective electrodes in analytical chemistry, Plenum Press, New York, 1978 Search PubMed.
- J. Bobacka, Anal. Chem., 1999, 71, 4932–4937 CrossRef CAS PubMed.
- U. Oesch, D. Ammann and W. Simon, Clin. Chem., 1986, 32, 1448–1459 CAS.
- X. U. Zou, X. V. Zhen, J. H. Cheong and P. Bühlmann, Anal. Chem., 2014, 86, 8687–8692 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |