
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChip-based platform for dynamic analysis of NK cell cytolysis mediated by a triplebody†
Elisavet I.
Chatzopoulou
a,
Claudia C.
Roskopf
b,
Farzad
Sekhavati
a,
Todd A.
Braciak
b,
Nadja C.
Fenn
c,
Karl-Peter
Hopfner
c,
Fuat S.
Oduncu
b,
Georg H.
Fey
d and
Joachim O.
Rädler
*a
aFaculty of Physics and Graduate School of Quantitative Biosciences (QBM), Ludwig-Maximilians-Universität, Munich, Germany. E-mail: raedler@lmu.de; Fax: +0049-(0)2180-3182; Tel: +0049-(0)89-2180-2438
bDivision of Hematology and Oncology, Medizinische Klinik und Poliklinik IV, Klinikum der Universität München, Munich, Germany
cDepartment of Biochemistry and Gene Center, Ludwig-Maximilians-Universität, Munich, Germany
dDepartment of Biology, Friedrich-Alexander-University Erlangen-Nürnberg, Erlangen, Germany
First published on 9th March 2016
Abstract
Cancer therapy via redirected lysis mediated by antibodies and antibody-derived agents relies on the availability of substantial numbers of sufficiently active immune effector cells. To monitor antitumor responses before and during therapy, sensitive methods are needed, capable of quantitating specific lysis of target cells. Here we present a chip-based single-cell cytometric assay, which uses adherent human target cells arrayed in structured micro-fields. Using a fluorescent indicator of cell death and time-lapse microscopy in an automated high-throughput mode, we measured specific target cell lysis by activated human NK cells, mediated by the therapeutic single chain triplebody SPM-2 (33-16-123). This antibody-derived tri-specific fusion protein carries binding sites for the myeloid antigens CD33 and CD123 and recruits NK cells via a binding site for the Fc-receptor CD16. Specific lysis increased with increasing triplebody concentration, and the single-cell assay was validated by direct comparison with a standard calcein-release assay. The chip-based approach allowed measurement of lysis events over 16 hours (compared to 4 hours for the calcein assay) and required far smaller numbers of primary cells. In addition, dynamic properties inaccessible to conventional methods provide new details about the activation of cytolytic effector cells by antibody-derived agents. Thus, the killing rate exhibited a dose-dependent maximum during the reaction interval. In clinical applications ex vivo monitoring of NK activity of patient's endogenous cells will likely help to choose appropriate therapy, to detect impaired or recovered NK function, and possibly to identify rare subsets of cancer cells with particular sensitivity to effector-cell mediated lysis.
Introduction
Several antibody-derived proteins used in cancer therapy function in conjunction with cytolytic effector cells, such as NK-cells and cytotoxic T-cells (T-CTLs). Examples are the CD20 antibody Rituximab (Rituxan™) and the CD19-directed bispecific agent Blinatumomab (Blincyto™), as well as the bispecific proteins AMG330 and MGD006, developed for the treatment of Acute Myeloid Leukemia (AML;1–7). The cytolytic activity of several therapeutic antibodies in the classic IgG format largely depends on the Fcγ RIII-receptor CD16 present on NK-cells, monocytes and macrophages,8–12 while the activity of the mentioned bispecific agents depends on T-CTLs. Therapeutic use of these agents requires that the patient harbors the respective effector cells in substantial numbers and in a sufficiently active state. For AML originating in the bone marrow (BM), this requirement is often not met during the early stages of treatment. At diagnosis and during the first cycle of induction therapy of AML patients, NK- and T- cells are usually reduced by about 10- to 20-fold in numbers relative to a healthy BM, and the remaining cells are functionally impaired.13–16 To determine a suitable therapeutic agent and time point for the start of treatment, it would therefore help, if investigators could monitor the capacity of a patient's NK- and/or T-cells to mediate cancer cell lysis in conjunction with the therapeutic agent. This requires the availability of reliable functional assays permitting a quantitative assessment of the cytolytic potential of a patient's NK- and/or T-cells by using only small numbers of these cells, which are available in limited supply.Chromium-51 and Calcein-release assays have been used extensively to measure cytotoxicity induced by therapeutic agents.17,18 However, they cannot be used for reaction periods in excess of approximately 4 hours due to increasing spontaneous release of the label from target cells.19 They also do not provide further information regarding the intrinsic aspects of the investigated cellular interaction. On the other hand, imaging techniques traditionally offered a more detailed insight into the interrogated phenomenon, however at low-throughput. This led to the concomitant use of microscopy to visualize processes that have previously been hypothesized based on data averaged over large cell populations and produced with the standard assays.20 Nevertheless, time-lapse microscopy has evolved significantly over the past decade and now permits us to follow the death of individual cells at high throughput and for observation periods of up to 15 hours and beyond.21 These newly developed microscopy-based approaches have uncovered detailed features of NK physiology and their innate ability to direct lysis of tumor cells, the “natural killing mode of NK cells”. Furthermore, different types of NK cells have been used in these studies, including both NK-cell lines and primary NK cells activated by IL-2 as well as unstimulated NK cells isolated from healthy donors. As a result, many different approaches have been implemented to uncover new aspects of the complex physiology of NK cells, such as the kinetics of their natural killing mode22 and their “kinetic boosting” by Fc-engineered antibodies.23 Moreover, the NK population was classified based on migratory behavior and cytotoxic response of individual cells.24,25 Finally, altered NK cell cytotoxicity, migratory behavior and contact dynamics have been reported between IL-2 stimulated and non-stimulated NK cells.26
In oncology Antibody Dependent Cellular Cytotoxicity (ADCC) makes use of antibodies to enhance the natural killing mode of NK cells.27,28 This process more generally is called “redirected lysis; RDL” when the mediator protein is not an antibody, but an antibody-derived agent. Although unmodified antibodies in the classic immunoglobulin (IgG) format have been successful in the treatment of selected types of cancer, in particular of hematologic malignancies, their broader use for the treatment of solid tumors is limited. Antibody engineering has led to the development of more broadly applicable derived proteins.29–32 Some of these new molecular formats no longer carry the full antigen-binding-domains of classic IgGs (Fv-domains) but employ antibody fragments, termed “single-chain Fragment variables (scFvs)”, as recognition domains. Bispecific tandem diabodies are a class of recombinant fusion proteins based on scFv-recognition domains, and Blinatumomab, the prototype of this class of agents, is approved for the treatment of certain types of lymphomas and leukemias.3,4 An extension of the molecular format of tandem diabodies are the “single-chain tandem triplebodies” (“triplebodies” for simplicity), which carry two scFv recognition domains for target antigens on the cancer cell plus an scFv module specific for a trigger molecule on an effector cell, arranged in tandem in a single polypypeptide chain.33–35 They can be designed to bind either two copies of the same target antigen or one copy each of two different antigens on the surface of the same cancer cell. The latter “dual-targeting” mode of binding leads to an increased selectivity of cancer cells bearing both antigens in high combined density.18,36 Triplebody SPM-2 (33-16-123) was designed for the elimination of AML cells. It carries one binding site for CD16 and two binding sites for the tumor-antigens CD123 and CD33, present on AML cells.16,34 Triplebodies can bind their target cells either mono-valently with only one of the two target binding modules, leaving the other module non-engaged, or bi-valently, employing both target binding modules simultaneously.
Here we use single-cell cytometry (SCC) to assess the efficiency of SPM-2-mediated killing of malignant target cells by NK cells in a time-lapse mode. We use a design where target cells are arranged in arrays of microstructured adhesion sites, allowing for a facilitated assessment of the fraction of cells specifically lysed by NK cells, which are added to the assay. We recorded time-lapse movies over a period of 16 hours and followed the action of SPM-2 by the apoptosis marker propidium iodide (PI). Our main objective was to implement a single-cell assay for time-resolved studies of the triplebody and NK cell-mediated lysis in accordance with existing standard assays. The platform presented herein facilitated a dynamic analysis of the cytotoxic response of the NK cell population triggered by the triplebody. As model target cells, HEK293.123 cells were used. Our results open the possibility for future use of the assay to study not only the quality of primary effector cells from human donors, but also to study mechanistic details of the mode of action of the therapeutic agent on different subsets of target cells.
Results and discussion
To quantitate the extent of re-directed lysis of tumor cells by NK cells mediated by the triplebody SPM-2, we developed a single-cell assay based on fluorescence microscopy and micro-structured arrays. Arrays of adhesion sites spaced by 60 μm were fabricated as previously reported37,38 and further described in the Methods section. The 60 μm spacing of the adhesion sites was found to be the optimal distance for generating a single-cell array of the adherent HEK293.123 cells, after having tested smaller and larger distances in preliminary tests (data not shown). Triplebody SPM-2 (33-16-123) was synthesized as previously described,16 following rearrangement, humanization, and stabilization of the DNA sequences of an earlier prototype of this agent.34 Target cells (the established human cell line HEK293.123) were seeded on the microstructured topas surfaces using ibidi® channel slides. Fig. 1 illustrates the basic experimental imaging platform (Fig. 1A) and a typical image of a HEK-cell array after addition of NK cells (Fig. 1B). NK cells were derived from a healthy donor and activated as previously described.18,39 Image acquisition started immediately after NK cell addition. Images were acquired in 12 min intervals over a time course of 16 hours. The time-lapse sequences were analyzed using custom-made image-analysis software integrating the fluorescence intensities from each individual adhesion site at each time-point. | ||
| Fig. 1 Experimental set-up of the single-cell cytometry (SCC) assay. Chemically modified patterned surfaces (arrays) were prepared on 6-channel microscope slides (A). Protein-coated arrays were generated by plasma-induced patterning. (B): Squares with a side-length of 30 μm were coated with fibronectin (here fibronectin labeled with Alexa Fluor 488) and the backfilling (black) area with PLL(20k)-g(3.5)-PEG(2k) (PEG(2)). Each channel on the carrier surface, shown in (A), contained 4400 adhesive squares for cell attachment. In (B) a part of an overview microscopic scan of one coated channel of the slide is shown. (C): Arrays of adherent HEK293.123 target cells were prepared on the chemically modified surfaces (surfaces seeded with cells have adhesive squares with unlabeled fibronectin). NK cells were added and squares occupied by single target cells (framed in blue) were selected and tracked in a time-lapse mode. NK cells were identified cinematographically by their size and motility. The distance between the squares was 60 μm in our experiments, but arrays with different spacing can be produced. (D): The cell-impermeable red fluorescent marker PI (propidium iodide) was used to identify dead cells. Overlay of brightfield and PI emission is presented for the first and last frame of a measurement. (E): The mean fluorescence intensity of 40 cells over time is plotted (in the presence of 10 nM SPM-2 triplebody). Each track (“fate plot”) represents the fluorescence intensity of one cell in the PI channel. Tracks exceeding an intensity threshold (defined by a supervised selection process as described under Methods) represent lysed cells. Two exemplary time-courses of two target cells that were lysed are highlighted in red. | ||
We determined the fraction of specifically lysed cells (% specific lysis), representing the incremental lysis mediated by NK cells in the presence of the triplebody SPM-2 over the background of natural killing by NK cells alone according to the formula given in Methods section. Lysed cells were accounted for based on a threshold in the PI fluorescence signal. Fig. 2 shows the percentage of lysed cells as a function of triplebody dose. For clarity the percentage of total lysed cells and the percentage of natural killing by NK-cells are shown together with the calculated extent of specific lysis.
 | ||
Fig. 2 Validation of the SCC assay by direct comparison with the bulk assay. A: Data obtained with the SCC assay. Red open bars: percentage of dead target cells relative to the total number of target cells analyzed (“overall lysis”). Red filled bars: percentage of specific lysis induced by SPM-2 after subtraction of natural killing by NK cells alone (the numbers shown for 0 nM concentration). Black open bars: overall lysis produced by addition of control triplebody SPM-1 (19-16-19), a triplebody in the same molecular format as SPM-2, but recognizing the target antigen CD19, which is absent from HEK293.123 cells. This control was not performed for 50 and 100 nM concentrations of the control protein. Medium control: without added NK cells and triplebodies; this control measures the extent of spontaneous death of target cells over the measurement interval. The averaged value of all the dose-dependent measurements is shown. (B) Comparison of data obtained with the SCC assay (red circles) over a 16 hour measurement and the bulk assay (calcein release assay; blue circles) over a 4 hour period. Data points of the calcein assay represent the mean value of the percentage of specific lysis averaged over triplicate reaction wells on the same microtiter plate, and error bars represent the SEM (standard error of the mean). Effector cells were MACS-purified NK cells from a healthy donor, pre-stimulated with IL-2 (LAK cells), and seeded at an E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :T ratio of 2:1. :T ratio of 2:1. | ||
Dose-dependence measurements and comparison with calcein release assay
Dose-dependence measurements were also performed with the established calcein release assay (Fig. 2B). For these measurements the effector-to-target cell (E:T) ratio was kept constant at 2:1 and the concentration of the SPM-2 triplebody varied from 0–100 nM for the SCC assay and from 0–10 nM for the calcein release assay. As effector cells immunomagnetically (MACS) purified pre-stimulated NK cells from the same healthy donor were used. The average percentage of NK cells (CD56brightCD16bright) after the MACS purification was 83.3% ± 4.6%. This percentage was taken into account in order to achieve an actual NK-to-target ratio of 2:1 as mentioned above. The total number of target cells analyzed in this set of measurements is given in Table 1. Over a 16 hour measurement period, a maximum of about 35% of the cells analyzed were lysed in the SSC assay in the presence of triplebody SPM-2 (Fig. 2A, red open bars). The fraction of specifically lysed cells (% specific lysis) steadily increased with triplebody concentration and reached a plateau at about 25% for concentrations from 1 nM upwards (Fig. 2A, red bars). As a negative control the triplebody SPM-1 (19-16-19) was used, a protein in the same molecular format as SPM-2 and carrying the same scFv binding site for CD16, but with specificity for the target antigen CD19, which is absent from the surface of HEK293.123 cells. In separate experiments with CD19-positive target cells this control triplebody mediated cytolysis by NK cells,33 but it did not produce specific lysis of HEK293.123 targets in the SCC assay (Fig. 2A, black open bars). The fraction of specific lysis induced by SPM-2 showed comparable dose-dependence in the SSC and the calcein-release assays (Fig. 2B). In both cases specific lysis reached plateau values for concentrations from 1 nM upwards, and the shape of the curves was similar (isometric). The EC50 values (half-maximum effector concentrations; the concentrations, at which half of the maximum lysis was reached) derived from both curves were (10.2 ± 8.0) and (12.2 ± 0.1) pM for the calcein and the SCC assays, respectively. The errors represent the standard deviation computed by statistical modeling of the two assays (see Methods section). While the EC50 values derived from both data sets were very similar, the maximum fractions of specifically lysed cells were (48 ± 2)% for the calcein assay and (25 ± 1)% for the SCC assay. This difference most likely is explained by the fact that in the calcein assay both effector and target cells were present in the fluid phase, whereas in the SSC assay the targets were anchored to the substrate and were therefore less accessible to the effector cells. In addition, in the SCC assay the density of cells per unit volume was less than half of the density reached in the calcein assay. However, this quantitative difference in the maximum levels of specific lysis recorded with both assays does not affect the key conclusion that the SSC assay is validated by this comparison, as it captured the dose-dependence of the triplebody's lytic potential in the same qualitative manner as the calcein assay.
| nM | SPM-2 | Control triplebody |
|---|---|---|
| 0 | 3099 | 465 |
| 0.01 | 1648 | 303 |
| 0.1 | 1888 | 507 |
| 1 | 3482 | 524 |
| 10 | 3254 | 344 |
| 50 | 1528 | — |
| 100 | 1364 | — |
| Medium | 3075 | 489 |
Dependence of the extent of cytolysis on the E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) :T ratio
:T ratio
Next we measured the efficiency of lysis as a function of the effector-to-target cell (E:T) ratio (Fig. 3). For measurements of the dependence of this variable on the E:T ratio, the concentration of the SPM-2 triplebody was kept constant at 1 nM, while the E:T ratios tested were 0.2:1, 1:1, 2:1 and 5:1. The total number of target cells analyzed in this set of measurements is given in Table 2. The effector cells used were MACS-purified NK cells. The mean final percentage of the NK cells (CD56brightCD16bright) after the enrichment was 90.2% ± 3.0%. This percentage was taken into account in order to reach the actual NK to target cell ratio for each of the conditions mentioned above. The specifically lysed fraction increased as a function of the E:T ratio (Fig. 3). When the number of effector cells increased the probability for cell encounters increased too, explaining the increase in lytic events. Furthermore, in the case of the E:T = 5:1 the number of effector cells attacking one single target cell was also increased. In this case, we observed that more than one effector cells were able to form a synapse with one specific target cell at the same time. The maximum extent of specific lysis was achieved at the E:T = 5:1 condition.
 | ||
| Fig. 3 Dependence of specific lysis measured with the SCC assay on the E:T ratio. Lysis induced over a range of different Effector to Target (E:T) cell ratios by SPM-2 at a 1 nM saturating concentration. Data points are fitted to an exponential curve. Effector cells were MACS-purified NK cells from a healthy donor, pre-stimulated with IL-2 (LAK cells). | ||
:T ratio
| E:T |
1 nM | 0 nM |
|---|---|---|
| 0.2:1 |
1061 | 1300 |
| 1:1 |
992 | 815 |
| 2:1 |
3482 | 744 |
| 5:1 |
1309 | 867 |
| Medium | 1909 | 1909 |
Dynamic analysis of the lytic events
The automated SCC assay allowed us to monitor cellular lysis over a long period of time with high numeric precision. Focusing on the target cells enabled us to track the activity of the NK cell population, regarding their cytotoxic activity and how the SPM-2 agent affected it. In the following, we quantified the killing rate i.e. the number of lytic events that occurred per hour. This percentage corresponds to the number of lysed cells versus the total number of target cells analyzed. In Fig. 4B, the mean fluorescence intensity of 3 arbitrary single target cells is illustrated, while for one of these cells the corresponding time-lapse frames of selected time points are presented (Fig. 4A). We determined the individual time points of target cell lysis using the maximum slope of the fluorescence signal increase as an indicator. From such data, the cumulative percentage of lysed cells was computed and plotted for 3 different concentrations of SPM-2: 0, 1 and 100 nM (Fig. 4C). The variation of the killing rate over the course of 16 hours for various SPM-2 concentrations and for constant numbers of NK cells is plotted in Fig. 4D. For comparison, the first plot shows the rate of background lysis rate in the absence of NK cells and the SPM-2 agent (labeled “medium”). When the SPM-2 agent was absent or present in low concentration, the killing rate of the NK cells was almost constant, 0.5 ± 0.2 target cells were killed per hour at 0 nM and 1.3 ± 0.5 at 0.01 nM (Fig. 4D, 2nd and 3rd panel from the left). The killing rate increased until a maximum was reached after several hours into the measurement and then gradually decreased. The maximum rate was greater for higher concentrations of SPM-2 (50 and 100 nM; last 2 panels to the right in Fig. 4D) than for intermediate concentrations (0.1 to 10 nM; central panels in Fig. 4D), and therefore, the agent clearly influenced the maximum killing rate of the NK cells. The maximum rate also occurred earlier at high concentrations of the agent than at lower concentrations (Fig. 4D; 2 panels at right). Importantly, the rates of lysis were elevated in the 1st hour of the reaction (highlighted in yellow in Fig. 4D) and dropped strongly in the 2nd hour for all tested concentrations of the mediator protein, even without added mediator protein (Fig. 4D, 2nd panel from the left). Thereafter the rates increased until maximum rates were reached between 5–10 hours into the reaction, and then the rates declined again. The log normal distribution was fitted to the data points. In all cases, the first data point, indicated in the yellow region (Fig. 4D) was excluded from the fitting, because it consistently was an outlier. We explain this excessively high lytic rate as being probably owed to the previous stimulation of the NK cells by long-term culture in the presence of IL-2.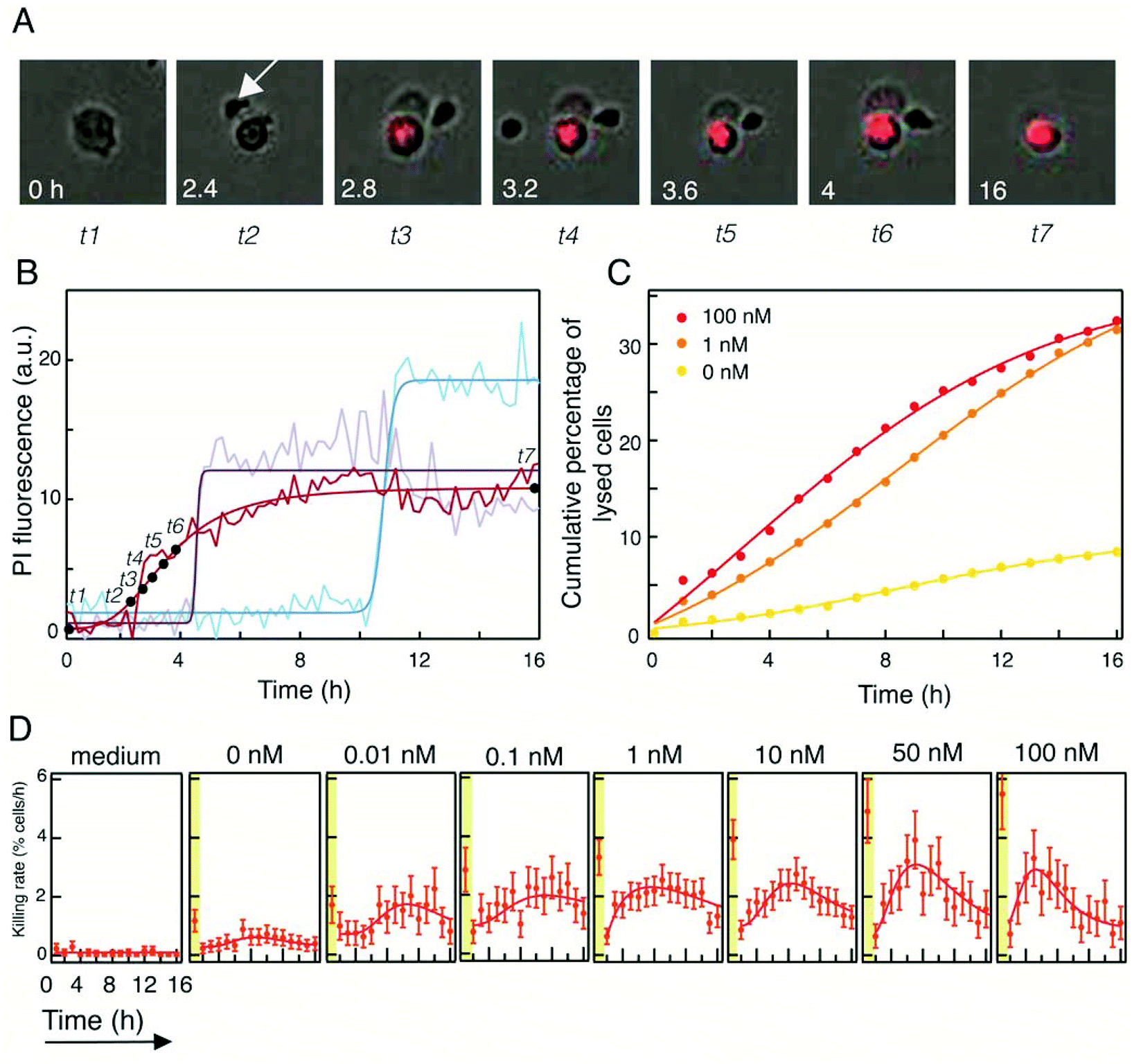 | ||
| Fig. 4 Dynamic analysis of lytic events with the SCC assay. (A) Time-lapse images of an exemplary target cell being killed by an NK cell (arrow). The progressively increased PI fluorescence intensity reflects progressive nuclear membrane disintegration (irreversible apoptosis). (B) PI fluorescence intensity of 3 exemplary target cells over the course of 16 hours, including the cell shown in (A) (red curve). Raw intensities were fitted with the Hill Equation. (C) Cumulative percentage of lysed target cells for 3 different concentrations of SPM-2: 0, 1 and 100 nM. Data points were fitted with the sigmoid function. (D) Killing rate as a function of time for increasing concentrations of SPM-2 by a constant number of NK cells. The left panel (“medium”) shows the dynamic of spontaneous cell death events, in the absence of NK cells. Data points were fitted with a log normal distribution curve. Data points for the first hour of the reaction (highlighted in yellow) represent natural killing by the NK cells but not specific lysis mediated by the triplebody. These events also occurred in the absence of added triplebody (2nd panel from the left), and were therefore excluded from the fitting. | ||
NK cells are “ready-to-kill” cells and the most common way to kill is through perforin/granzyme granule-mediated exocytosis.40 In this study, LAK (Lymphokine Activated Killer cells) NK cells, pre-stimulated with IL-2 from a healthy donor, were used. These cells exhibit greater cytotoxicity due to increased intracellular concentrations of effector molecules such as perforin and granzymes.41 They form a synapse with the target cell and then degranulation occurs which leads to apoptotic death of the target. At the beginning of the measurement the cytosol of the NK cells is full of granzymes, which then degranulate to lyse the large number of target cells at the beginning of the measurement. These distinctly large killing rates during the first hour of the measurement were further boosted by the triplebody in a dose-dependent manner. Subsequently, the NK cells were exhausted and gradually resumed their cytotoxic activity by producing again new granzymes. The triplebody played a role in the early hours of the reaction interval, either in the replenishment process or the subsequent lytic events or both, because the maximum rates of lysis were clearly augmented by the triplebody in a dose-dependent manner (Fig. 4D). The observed increase in lytic rates with time suggests an influence of the triplebody on the speed of replenishment or on other metabolic processes preparing the NK cell for the next degranulation event and on an acceleration of the cadence of lytic bursts. Furthermore, the initial increases in the rate during the first few hours of the reaction may also be a consequence of the lytic events themselves; either the NK cells or the targets or both may have released soluble mediators (e.g. cytokines and others) which favored the lytic process in a positive feedback manner (a paracrine loop). At the same time, the medium was progressively exhausted and cellular debris from dead target and effector cells accumulated, which must have inhibited the lytic activity. The system was a closed system in our set-up and the medium was not renewed. Therefore, these inhibitory influences eventually may have outpaced the positive feedback mechanisms, an optimum was passed, and beyond this point the reaction rates began to decline. Moreover, it has been previously shown42 that NK cells isolated from healthy donors and stimulated with IL-2 do not have a uniform cytotoxic activity. In essence a small subset of the NK cells are responsible for the majority of kills. Finally, taking into consideration that tumor cells can also induce the apoptosis of IL-2 activated NK cells43,44 we propose that the decrease of the reaction rates after a certain time point most likely was due to a partial apoptosis of the active cytotoxic subpopulation in combination with the exhaustion of the medium.
Natural killing dynamic analysis against non-adherent AML target cells
Many samples of primary cancer cells, especially those derived from hematologic malignacies, consist of non-adherent cells. To address this matter and to render the proposed assay suitable for non-adherent cells we developed a second version of the chip, in which the patterns of squares covered with fibronectin were replaced by squares covered with an antibody of IgM isotype. To test this variant of our assay, we measured the natural killing potency of primary NK cells from another healthy donor for the human AML-derived target cell line MOLM-13.A suitable antibody candidate for anchoring MOLM-13 cells to the micro-patterns, without interfering with the NK cells, is a CD15-specific antibody. The myeloid marker CD15 is present on the surface of the MOLM-13 cells45 but not on the primary NK cells (Fig. S1†). Both IgG1- and IgM-types were tested and MOLM-13 cell arrays of greater occupancy and greater stability for longer periods of time (up to 24 h) were achieved with the IgM antibody, probably due to more favorable stereochemical properties of the IgM relative to the IgG1 isotype. Patterns of squares with 25 μm side-length were used for the MOLM-13 cell line, because these cells were smaller than the adherent HEK-cells used so far. As a result, smaller square patterns led to a greater percentage of single-cell occupied patterns. Even though we could have chosen smaller distances between the squares for the non-adherent MOLM-13 cells, we chose to maintain the 60 μm for better comparability with the measurements of the adherent target cells and to avoid introducing a new variable into the experimental setup. An array of MOLM-13 cells in the presence of NK cells is shown in Fig. 5A. Size and morphology of MOLM-13 and NK cells are very similar, therefore MOLM-13 cells were stained with fluorescent tracker dye Green CMFDA to permit the distinction from the NK cells.
 | ||
| Fig. 5 Variation of the SCC assay adapted to non-adherent target cells. Arrays of antibody coated square patterns were generated with the same procedure as the fibronectin arrays, by substituting fibronectin with an anti-human CD15 antibody. Arrays of stained MOLM-13 cells (CellTracker™ Green CMFDA) were prepared on the chemically modified surfaces and then NK cells (unstained) were added (A). (B) Time-lapse images of an exemplary MOLM-13 target cell (green) being killed by an NK cell (unstained). The progressively increased PI fluorescence intensity reflects progressive nuclear membrane disintegration (irreversible apoptosis). (C) Data obtained with the SCC assay showing the natural killing potency of NK cells against MOLM-13 cells. The bars represent the percentage of dead target cells relative to the total number of target cells analyzed. (D) Dynamic analysis of the natural killing mode of NK cells against the MOLM-13 target cells. The right panel (“medium”) shows the dynamic of spontaneous cell death events, in the absence of NK cells. | ||
To test this version of the assay for non-adherent cells we measured the natural killing of MOLM-13 targets by NK cells alone without addition of a mediating protein. For the measurements with the non-adherent cells, NK cells were derived from a second healthy donor and activated by long-term culture in the presence of IL-2 as previously described.18,39 Preparation of the samples, image acquisition and analysis were performed as described for the measurements with the adherent HEK293.123 cells. The E:T ratio was 2:1 and after a 16 hour measurements period 2.6% of the cells analyzed (Table 3) were lysed (Fig. 5C). This percentage was lower than the average natural killing observed for the NK cells from the first donor at the same E:T ratio (8.4%) (Fig. 2A), which probably reflects donor-to-donor variability and the different types of target cells used. Time-lapse frames of an NK cell progressively killing a MOLM-13 cell are presented in Fig. 5B. The variation of the killing rate over the course of 16 hours for the natural killing of the NK cells measured is presented in Fig. 5D. A slightly greater killing rate during the 1st hour was also observed in this case and probably reflects the preceding stimulation of the NK cells with IL-2 as was discussed above.
| E:T = 2:1 |
Medium |
|---|---|
| 1775 | 2276 |
Compared with existing methods, single-cell cytometry allows for time-resolved studies of NK cell activity. Chromium-51 and calcein release assays offer a statistically valid measurement of cytotoxity, but they are restricted in duration due to spontaneous release of the label, and they produce time- and population-averaged data. Flow cytometry and ELISPOT assays offer single-cell data for large number of cells, but also only for at a single time point. Moreover, they measure cytotoxicity in an indirect manner, as with flow cytometry usually target cells that have survived are counted, while in ELISPOT assays usually IFN-γ secretion or degranulation of the effector cells is measured.46,47 The SCC assay presented here fills the gap between these two different approaches of measuring cytotoxicity. SCC is capable of following the progression of NK cytotoxic activity over an extended duration of 16 hours. Due to spatial ordering of the target cells, image analysis is feasible with limited computational means generating data that contain full information on the time course of killing events relevant to therapeutic applications.
To better understand the mode of action of novel therapeutics based on the recruitment of effector cells and the functional properties of the effector cells involved, it is essential to have the ability to study also the dynamics of these processes. Additional studies will likely produce suggestions for optimal dosing and administration schedules in clinical applications. Time-lapse methods are promising in this regard, since meta-analysis can be extended to the use of additional markers including biomarkers of therapy success, disease progression and impending relapse, and because they lend themselves to further automatization.
An additional advantage of single cell assays is that they generate reliable data with substantially smaller numbers of effector cells. For an SCC assay typically only 2 × 104 NK cells are needed per measurement point, 5-times fewer than the >105 cells needed for each measurement point in a calcein release assay. Patient-derived NK cells are a scarce resource, in particular to monitor disease status and therapy outcome for AML patients. Further miniaturization of the new method is possible, so that reliable measurements will likely become possible with as few as 5000–10000 NK cells. Furthermore, patterned arrays provide uniform micro-environments and spacing of target cells and hence potentially improve the standardization of cell–cell encounters. Hence, chip-based single cell assays are potentially valuable for clinical monitoring of the patients NK response and for the choice of personalized treatment for individual AML patients. They can also be applied to T-CTL as effector cells in combination with corresponding triplebodies and with other antibody-derived proteins recruiting T-cells as cytolytic effectors.4–7,48
Assays employing spatially arrayed target cells can also be useful to study questions regarding the timing in cell–cell recognition and immune response, such as for example the “memory” effect described for NK cells.22 In the experiments presented here the full potential of the dual-targeting triplebody SPM-2 has not yet been analyzed. Here we have so far only used the CD123-binding site of this agent. In the future the effect of simultaneous engagement of both binding sites by one copy each of CD33 and CD123 on the lytic activity of NK cells can be studied. Dual-targeting renders the SPM-2 agent particularly promising for the therapy of AML because virtually all patients expressed either one or the other of the two antigens.49 Indeed, in cell culture cytolysis assays with primary cells from a broad range of AML patients with different subtypes of AML and with a standard batch of NK cells from an unrelated healthy donor, all samples showed very effective lysis.50 This is an unusually high degree of responsiveness, considering that the response rate to the best antibody-derived agent available so far for the treatment of AML (Mylotarg™51) was in the range of 40% for blasts from patients with different subtypes of AML.52 Even blasts from patients with AML subtypes that typically show a poor response to conventional chemotherapy were lysed efficiently by SPM-2 plus NK cells.50 Moreover, the pair of CD33 plus CD123 is highly expressed on AML leukemia stem cells (AML-LSCs) but far less on normal hematopoietic stem cells (HSCs;52–54). Therefore, a therapeutic window appears to exist, which may permit a preferential elimination of the AML-LSCs over the normal HSCs of the patient and a reconstitution of the patient's hematopoietic system after the end of therapy from the patient's own HSCs, without the need for an allogeneic or autologous stem cell transplantation. If this could be achieved in the future, then this result would constitute major progress in the therapy of AML. Therefore, in the future it is important to study in detail not only how a patient's autologous NK cells in conjunction with this agent lyse the patient's bulk AML blasts, but also whether and how they lyse subsets of blasts progressively closer and closer to the leukemia initiating cells (LICs) and relapse initiating MRD cells, which are likely to be encompassed in the CD34posCD38negCD123high compartment of BM and peripheral blood cells, which comprises between 0.01 and 67% of all malignant cells for different AML patients.55 To this end cytolysis assays with rare subsets of patient-derived AML cells and NK- or T cells are required, which will be available in small numbers only. Finally, agents targeting the same pair of antigens, but recruiting T-cells as cytolytic effectors are also under development,56 and similar experiments as those outlined above will also need to be performed with the corresponding T-cell-recruiting agents in order to find the best suited agent for individual patients in the sense of a personalized medicine. Developments are well under way that personalized medicine will clearly become more prevalent in the future. Time-resolved SSC assays are promising means to further investigate the capabilities of Fc engineered antibodies and other bi- and tri-specific antibody-derived agents such as the triplebodies described here for enhanced target cell lysis. They therefore offer the potential to assist treatment decisions and monitoring of treatment success in cancer therapy.
Experimental
Cell culture
Human Embryonic Kindey 293 cells (HEK 293), obtained from the American Type Cell Culture Collection (ATCC, Manassas, VA, USA), were transfected with cDNA expression constructs for human CD123 and sublines were selected, which stably expressed CD123 over many passages in culture. The subline employed here has been in culture for several years and stably expresses approx. 360000 copies of CD123 per cell on the surface.57 It was cultured in RPMI 1640 medium (Biochrom; Merck Millipore, Berlin, Germany) supplemented with 10% (vol/vol) Fetal Bovine Serum (FBS; Gibco®, Life Technologies GmbH, Darmstadt, Germany) and 400 μg ml−1 Geneticin Selective Antibiotic G418 Sulfate (Roth, Karlsruhe, Germany). MOLM-13 cells were cultured in RPMI 1640 medium supplemented with 10% (vol/vol) FBS.
Ex vivo expansion of MNCs from healthy donors in the presence of IL-2 and immunomagnetic enrichment of NK cells
Mononuclear cells (MNCs) from peripheral blood (PBMCs) were expanded ex vivo in RPMI medium containing Interleukin-2 (IL-2) plus 5% (vol/vol) human serum (Life Technologies) for 20 days as described18,39 and were then frozen in aliquots for subsequent use. Prior to use in cytolysis experiments, the cells were thawed and cultured overnight in RPMI medium containing 5% (vol/vol) human serum plus 50 units per ml of penicillin and 50 μg ml−1 of streptomycin (PS; Life Technologies) respectively, but no additional IL-2. NK cells were then enriched by negative selection using the human NK Cell Isolation Kit (Miltenyi Biotec; Cat. No. 130-092-657; Bergisch Gladbach, Germany) according to the provider's instructions. The enriched batches contained 83–90% of CD56bright CD16bright NK cells.Surface patterning
Protein-coated arrays were prepared on polymer coverslips for 6-channel sticky slides (ibidi GmbH, Munich, Germany). First, coverslips were treated selectively with oxygen plasma (40 W for 3 min; Femto, Diener Electronic GmbH+Co. KG, Ebhausen, Germany). Selectivity was achieved using a polydimethylsiloxane (PDMS) stamp (cast from a master produced by photolithography) as a mask. The area exposed to plasma was passivated with PLL(20k)-g(3.5)-PEG(2k) (SuSoS AG; Dübendorf, Switzerland) at 1 mg ml−1 in aqueous buffer (10 mM HEPES pH 7.4 and 150 mM NaCl). Then the PDMS stamp was removed and the remaining hydrophobic areas (squares 30 × 30 μm) were exposed to fibronectin (50 μg ml−1; YO Proteins AB, Huddinge, Sweden) for 1 hour. Finally, channels were rinsed thoroughly with PBS and the slides were stored at 4 °C for a maximum of 5 days. Before cell seeding, PBS was exchanged with culture medium (RPMI with 10% (vol/vol) FBS) and kept for 1 hour at 37 °C.The same procedure was followed for the antibody-coated arrays with the following alterations: arrays were prepared on uncoated 8-well μ-slides (ibidi GmbH, Munich, Germany). After treatment with plasma, the PDMS stamp was removed and the remaining hydrophobic areas (squares 25 × 25 μm) were exposed to purified anti-human CD15 (SSEA-1) antibody (15 μg ml−1; BioLegend®, San Diego, CA USA) for 1 hour at room temperature.
Time-resolved fluorescence microscopy
000 cells per channel) and incubated at 37 °C, in culture medium for approx. 4 hours, until the cells were deposited in an array. MOLM-13 cells were stained in 1 μM CellTracker™ Green CMFDA dye (Thermo Fischer Scientific, Waltham, MA USA) for 15 min in serum-free media in 37 °C, followed by 1 h of recovery in complete media. Then stained cells were seeded into the wells (10000 cells per well), and after 2 hours the culture medium was exchanged to Leibovitz's L15 medium with GlutaMAX (Gibco®, Life Technologies) supplemented with 10% (vol/vol) FBS. Meanwhile, final solutions with the appropriate number of MACS enriched NK cells and the desired concentration of the SPM-2 triplebody or control agents were prepared and added to the channels/wells. A trypan blue exclusion test of the NK cells was performed immediately before the preparation of the final solutions.
Imaging was performed under an inverted Nikon Ti eclipse microscope with a motorized stage, a Plan Apochromat 4×/0.2 N.A. objective, an Andor Clara-E camera, and a Lumencor SOLA LED lamp. For detection of PI fluorescence a filter cube with 540/25 nm (excitation) and 630/60 nm (emission) filters was used. For the stained MOLM-13 cells a filter cube with 470/40 nm (excitation) and 525/50 nm (emission) filters was used. Images were taken with constant exposure times of 10 and 300 ms in the brightfield and the PI/Green CMFDA channels respectively at 12 min intervals for 16 hours. A pre-determined X–Y position list was used for the automated time-lapse recording of 84 positions for each measurement. During the recording samples were kept at a constant temperature of 37 °C using an ibidi heating system (ibidi GmbH).
Image and data analysis
Raw images were pre-processed with ImageJ (http://imagej.nih.gov/ij/). Using an in-house plugin, Microwell Analysis, an orthogonal grid was aligned over cells and single-cell occupied grid positions were selected. The mean intensities over the selected positions of the grid were extracted and exported to a data file. Custom Matlab scripts (MATLAB version R2014b Natick, Massachusetts: The MathWorks Inc., 2014) were employed to quantify the number of dead target cells and to find their lysis time-points based on the maximum slope of the signal increase in the PI channel. In the representative set of data, a threshold was chosen as a maximum value that can separate the weakest signal of a dead target cell from the background signals e.g. from healthy cells. Further analysis confirmed that the threshold was at least 4 times higher than the background fluorescence intensity of the healthy cells in the PI channel. Specific lysis was calculated as:
Redirected lysis (RDL) assays using calcein release
Target cells HEK293.123 were pre-labeled with Calcein AM (Life Technologies) and mixed with MACS-purified NK cells in RPMI 1640 GlutaMAX medium supplemented with 10% (vol/vol) FBS at E:T = 2:1. SPM-2 triplebody was added at the desired concentration to a 200 μL reaction volume in round-bottom 96-well plates. Reactions were incubated at 37 °C with 5% CO2 for 4 hours. Calcein release was quantitated by measuring the fluorescence intensity (relative light units, RLU) in the supernatant using a fluorimeter/ELISA plate reader at 485/535 nm. Maximum lysis was achieved by addition of 50 μL of a solution containing 10% Triton X-100 in RPMI 1640 GlutaMAX medium supplemented with 10% (vol/vol) FBS and 1% (vol/vol) PS. Specific cellular cytotoxicity was expressed as overall lysis minus the background of spontaneous lysis mediated by the NK cells alone, in the absence of added antibody-reagents. Specific lysis was evaluated by the formula:
Statistical analysis
In the case of the single-cell cytometry (SCC) assay we hypothesized that data follow the binomial distribution and error bars were calculated with a confidence level of 95%.Statistical modeling of the calcein assay and single cell assay results
We hypothesized that the killing events in every condition can be modeled using a binomial distribution. The metrics of the binomial distributions were calculated using the binofit() function in Matlab. Then, these binomial distributions were sampled with replacement to generate 1000 random measurements (in terms of dead cells) per condition (concentration). From this random sampled number of dead cells we subtracted the number of dead cells observed for the 0 nM condition and divided by the corresponding number of total cells, having subtracted the number of spontaneous cell death events. This number corresponds to the specific killing ratio. Dose response curves in the form of a sigmoid curve were fitted for every set of random killing ratios, yielding 1000 random sampled dose response curves. The fit of the sigmoid curves was performed using the nlinfit() function in Matlab. From the sigmoid curves the parameters corresponding to the EC50 values were extracted and averaged across all 1000 random samplings yielding a mean value and a standard deviation.
Conclusions
Single cell arrays combined with time-resolved fluorescence microscopy were used to study the interaction of primary human NK cells with human target cells mediated by triplebody SPM-2, an antibody-derived protein, which recruits NK cells for target cell lysis. The arrayed pattern of target cells allowed for highly efficient and automated counting of lytic events under standardized conditions. Lysis depended on the dose of the agent and the E:T ratio in a manner typical for standard cytolysis assays, and therefore this new assay was validated relative to existing standard procedures (calcein release assay). Use of the new SCC assay revealed so far unreported changes in the killing rate over long-term reaction periods (16 hours). Finally, a variation of the assay employing surface-coated antibodies demonstrated the feasibility to array non-adherent target cells in the chip-based single-cell cytometric assay. The proposed platform facilitates testing the susceptibility of many tumor-derived cell types to lysis by NK cells with or without an added mediator protein and can become a useful tool for the design of personalized therapies.
Disclosure of potential conflict of interest
The authors declare no conflict of interest.Acknowledgements
Financial support by the Deutsche Forschungsgemeinschaft within the Excellence Graduate School Quantitative BioSciences (QBM) and the program project grant (Sonderforschungsbereich) SFB 1032 (project B01) is gratefully acknowledged. Drs. Oduncu, Hopfner and Fey were funded by an M4 Excellence Award for Personalized Medicine from the Bavarian Ministry for Trade and the Economy. We thank Dr. Christoph Stein, Fraunhofer Institute, Aachen, Germany, for the kind gift of the HEK293.123 cell line. We are grateful to Max Albert for wafer and chip fabrication, and to Peter J. Röttgermann for microscopic pictures of the fibronectin-coated arrays.References
- L. Weiner, J. Murray and C. Shuptrine, Cell, 2012, 148, 1081–1084 CrossRef CAS PubMed.
- P. McLaughlin, A. J. Grillo-López, B. K. Link, R. Levy, M. S. Czuczman, M. E. Williams, M. R. Heyman, I. Bence-Bruckler, C. A. White, F. Cabanillas, V. Jain, A. D. Ho, J. Lister, K. Wey, D. Shen and B. K. Dallaire, J. Clin. Oncol., 1998, 16, 2825–2833 CAS.
- T. Dreier, G. Lorenczewski, C. Brandl, P. Hoffmann, U. Syring, F. Hanakam, P. Kufer, G. Riethmuller, R. Bargou and P. A. Baeuerle, Int. J. Cancer, 2002, 100, 690–697 CrossRef CAS PubMed.
- R. Bargou, E. Leo, G. Zugmaier, M. Klinger, M. Goebeler, S. Knop, R. Noppeney, A. Viardot, G. Hess, M. Schuler, H. Einsele, C. Brandl, A. Wolf, P. Kirchinger, P. Klappers, M. Schmidt, G. Riethmüller, C. Reinhardt, P. Baeuerle and P. Kufer, Science, 2008, 321, 974–977 CrossRef CAS PubMed.
- M. Aigner, J. Feulner, S. Schaffer, R. Kischel, P. Kufer, K. Schneider, A. Henn, B. Rattel, M. Friedrich, P. A. Baeuerle, A. Mackensen and S. W. Krause, Leukemia, 2013, 27, 1107–1115 CrossRef CAS PubMed.
- M. Friedrich, A. Henn, T. Raum, M. Bajtus, K. Matthes, L. Hendrich, J. Wahl, P. Hoffmann, R. Kischel, M. Kvesic, J. W. Slootstra, P. A. Baeuerle, P. Kufer and B. Rattel, Mol. Cancer Ther., 2014, 13, 1549–1557 CrossRef CAS PubMed.
- C. go. identifier NCT02152956.
- T. Takai, M. Li, D. Sylvestre, R. Clynes and J. V. Ravetch, Cell, 1994, 76, 519–529 CrossRef CAS PubMed.
- G. Cartron, L. Dacheux, G. Salles, P. Solal-Celigny, P. Bardos, P. Colombat and H. Watier, Blood, 2002, 99, 754–758 CrossRef CAS PubMed.
- W.-K. K. Weng and R. Levy, J. Clin. Oncol., 2003, 21, 3940–3947 CrossRef CAS PubMed.
- M. Albanesi, D. A. Mancardi, L. E. Macdonald, B. Iannascoli, L. Zitvogel, A. J. Murphy, M. Daëron, J. H. Leusen and P. Bruhns, J. Immunol., 2012, 189, 5513–5517 CrossRef CAS PubMed.
- F. Nimmerjahn and J. V. Ravetch, Nat. Rev. Immunol., 2008, 8, 34–47 CrossRef CAS PubMed.
- R. T. T. Costello, S. Sivori, E. Marcenaro, M. Lafage-Pochitaloff, M.-J. J. Mozziconacci, D. Reviron, J.-A. A. Gastaut, D. Pende, D. Olive and A. Moretta, Blood, 2002, 99, 3661–3667 CrossRef CAS PubMed.
- C. Fauriat, S. Just-Landi, F. Mallet, C. Arnoulet, D. Sainty, D. Olive and R. T. Costello, Blood, 2007, 109, 323–330 CrossRef CAS PubMed.
- E. Lion, Y. Willemen, Z. N. Berneman, V. F. Van Tendeloo and E. L. Smits, Leukemia, 2012, 26, 2019–2026 CrossRef CAS PubMed.
- T. Braciak, S. Wildenhain, C. Roskopf, I. Schubert, G. Fey, U. Jacob, K.-P. Hopfner and F. Oduncu, J. Transl. Med., 2013, 11, 289 CrossRef PubMed.
- K. T. Brunner, J. Mauel, J. C. Cerottini and B. Chapuis, Immunology, 1968, 14, 181–196 CAS.
- I. Schubert, D. Saul, S. Nowecki, A. Mackensen, G. H. Fey and F. S. Oduncu, mAbs, 2014, 6, 286–296 CrossRef PubMed.
- D. L. Nelson, C. C. Kurman and D. E. Serbousek, Curr. Protoc. Immunol., 2001, Ch. 7, Unit 7.27 Search PubMed.
- R. Bhat and C. Watzl, PLoS One, 2007, 2, e326 Search PubMed.
- J. Albeck, J. Burke, B. Aldridge, M. Zhang, D. Lauffenburger and P. Sorger, Mol. Cell., 2008, 30, 11–25 CrossRef CAS PubMed.
- P. Choi and T. Mitchison, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6488–6493 CrossRef CAS PubMed.
- G. Romain, V. Senyukov, N. Rey-Villamizar, A. Merouane, W. Kelton, I. Liadi, A. Mahendra, W. Charab, G. Georgiou, B. Roysam, D. A. Lee and N. Varadarajan, Blood, 2014, 124, 3241–3249 CrossRef CAS PubMed.
- E. Forslund, K. Guldevall, P. E. Olofsson, T. Frisk, A. E. Christakou, M. Wiklund and B. Onfelt, Front. Immunol., 2012, 3, 300 Search PubMed.
- Unknown.
- P. E. Olofsson, E. Forslund, B. Vanherberghen, K. Chechet, O. Mickelin, A. R. Ahlin, T. Everhorn and B. Onfelt, Front. Immunol., 2014, 5, 80 Search PubMed.
- C. Shuptrine, R. Surana and L. Weiner, Semin. Cancer Biol., 2012, 22, 3–13 CrossRef CAS PubMed.
- A. Scott, J. Wolchok and L. Old, Nat. Rev. Cancer, 2012, 12, 278–287 CrossRef CAS PubMed.
- P. J. Carter, Nat. Rev. Immunol., 2006, 6, 343–357 CrossRef CAS PubMed.
- R. E. Kontermann, mAbs, 2012, 4, 182–197 CrossRef PubMed.
- R. Kontermann and U. Brinkmann, Drug Discovery Today, 2015, 838–847 CrossRef CAS PubMed.
- C. Spiess, Q. Zhai and P. J. Carter, Mol. Immunol., 2015, 95–106 CrossRef CAS PubMed.
- C. Kellner, J. Bruenke, J. Stieglmaier, M. Schwemmlein, M. Schwenkert, H. Singer, K. Mentz, M. Peipp, P. Lang, F. Oduncu, B. Stockmeyer and G. H. Fey, J. Immunother., 2008, 31, 871–884 CrossRef CAS PubMed.
- M. Kügler, C. Stein, C. Kellner, K. Mentz, D. Saul, M. Schwenkert, I. Schubert, H. Singer, F. Oduncu, B. Stockmeyer, A. Mackensen and G. H. Fey, Br. J. Haematol., 2010, 150, 574–586 CrossRef PubMed.
- H. Singer, C. Kellner, H. Lanig, M. Aigner, B. Stockmeyer, F. Oduncu, M. Schwemmlein, C. Stein, K. Mentz, A. Mackensen and G. H. Fey, J. Immunother., 2010, 33, 599–608 CrossRef CAS PubMed.
- J. M. Rowe and B. Löwenberg, Blood, 2013, 121, 4838–4841 CrossRef CAS PubMed.
- P. J. Röttgermann, A. P. Alberola and J. O. Rädler, Soft Matter, 2014, 10, 2397–2404 RSC.
- M. Ferizi, C. Leonhardt, C. Meggle, M. Aneja, C. Rudolph, C. Plank and J. Rädler, Lab on a Chip, 2015, 15, 3561–3571 RSC.
- E. Alici, T. Sutlu, B. Björkstrand, M. Gilljam, B. Stellan, H. Nahi, H. C. Quezada, G. Gahrton, H.-G. G. Ljunggren and M. S. Dilber, Blood, 2008, 111, 3155–3162 CrossRef CAS PubMed.
- M. J. Smyth, E. Cretney, J. M. Kelly, J. A. Westwood, S. E. Street, H. Yagita, K. Takeda, S. L. van Dommelen, M. A. Degli-Esposti and Y. Hayakawa, Mol. Immunol., 2005, 42, 501–510 CrossRef CAS PubMed.
- M. Cheng, Y. Chen, W. Xiao, R. Sun and Z. Tian, Cell. Mol. Immunol., 2013, 10, 230–252 CrossRef CAS PubMed.
- B. Vanherberghen, P. E. Olofsson, E. Forslund, M. Sternberg-Simon, M. A. Khorshidi, S. Pacouret, K. Guldevall, M. Enqvist, K.-J. J. Malmberg, R. Mehr and B. Önfelt, Blood, 2013, 121, 1326–1334 CrossRef CAS PubMed.
- A. Poggi, A.-M. Massaro, S. Negrini, P. Contini and M. Zocchi, J. Immunol., 2005, 174, 2653–2660 CrossRef CAS.
- G. M. Spaggiari, P. Contini, A. Dondero, R. Carosio, F. Puppo, F. Indiveri, M. R. Zocchi and A. Poggi, Blood, 2002, 100, 4098–4107 CrossRef CAS PubMed.
- Y. Matsuo, R. A. MacLeod, C. C. Uphoff, H. G. Drexler, C. Nishizaki, Y. Katayama, G. Kimura, N. Fujii, E. Omoto, M. Harada and K. Orita, Leukemia, 1997, 11, 1469–1477 CAS.
- A. Malyguine, S. Strobl, K. Dunham, M. Shurin and T. Sayers, Cells, 2012, 1, 111–126 CrossRef CAS PubMed.
- E. Ranieri, I. Popescu and M. Gigante, Methods Mol. Biol., 2014, 1186, 75–86 CAS.
- C. C. Roskopf, C. B. Schiller, T. A. Braciak, S. Kobold, I. A. Schubert, G. H. Fey, K.-P. P. Hopfner and F. S. Oduncu, OncoTargets Ther., 2014, 5, 6466–6483 CrossRef PubMed.
- A. Ehninger, M. Kramer, C. Röllig, C. Thiede, M. Bornhäuser, M. von Bonin, M. Wermke, A. Feldmann, M. Bachmann, G. Ehninger and U. Oelschlägel, Blood Cancer J., 2014, 4, e218 CrossRef CAS PubMed.
- T. A. Braciak, C. C. Roskopf, S. Wildenhain, N. Fenn, C. Schiller, I. A. Schubert, U. Jacob, A. Honegger, C. Krupka, M. Subklewe, K. Spiekermann, K.-P. Hopfner, G. H. Fey, M. Aigner, S. Krause, A. Machensen and F. S. Oduncu, OncoTargets Ther. Search PubMed , in press.
- A. Burnett, M. Wetzler and B. Löwenberg, J. Clin. Oncol., 2011, 29, 487–494 CrossRef PubMed.
- C. T. Jordan, D. Upchurch, S. J. Szilvassy, M. L. Guzman, D. S. Howard, A. L. Pettigrew, T. Meyerrose, R. Rossi, B. Grimes, D. A. Rizzieri, S. M. Luger and G. L. Phillips, Leukemia, 2000, 14, 1777–1784 CrossRef CAS PubMed.
- A. W. Hauswirth, S. Florian, D. Printz, K. Sotlar, M.-T. T. Krauth, G. Fritsch, G.-H. H. Schernthaner, V. Wacheck, E. Selzer, W. R. Sperr and P. Valent, Eur. J. Clin. Invest., 2007, 37, 73–82 CrossRef CAS PubMed.
- R. B. Walter, F. R. Appelbaum, E. H. Estey and I. D. Bernstein, Blood, 2012, 119, 6198–6208 CrossRef CAS PubMed.
- F. Vergez, A. S. Green, J. Tamburini, J.-E. E. Sarry, B. Gaillard, P. Cornillet-Lefebvre, M. Pannetier, A. Neyret, N. Chapuis, N. Ifrah, F. Dreyfus, S. Manenti, C. Demur, E. Delabesse, C. Lacombe, P. Mayeux, D. Bouscary, C. Recher and V. Bardet, Haematologica, 2011, 96, 1792–1798 CrossRef CAS PubMed.
- C. Arndt, A. Feldmann, M. von Bonin, M. Cartellieri, E.-M. M. Ewen, S. Koristka, I. Michalk, S. Stamova, N. Berndt, A. Gocht, M. Bornhäuser, G. Ehninger, M. Schmitz and M. Bachmann, Leukemia, 2014, 28, 59–69 CrossRef CAS PubMed.
- C. Stein, C. Kellner, M. Kügler, N. Reiff, K. Mentz, M. Schwenkert, B. Stockmeyer, A. Mackensen and G. Fey, Br. J. Haematol., 2010, 148, 879–889 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5an02585k |
| This journal is © The Royal Society of Chemistry 2016 |